


Abstract
3′-untranslated regions (UTRs) can vary through the use of alternative polyadenylation sites during pre-mRNA processing. Multiple publically available pipelines combining high profiling technologies and bioinformatics tools have been developed to catalog changes in 3′-UTR lengths. In our recent RNA-seq experiments using cells with hyper-activated mammalian target of rapamycin (mTOR), we found that cellular mTOR activation leads to transcriptome-wide alternative polyadenylation (APA), resulting in the activation of multiple cellular pathways. Here, we developed a novel bioinformatics algorithm, IntMAP, which integrates RNA-Seq and PolyA Site (PAS)-Seq data for a comprehensive characterization of APA events. By applying IntMAP to the datasets from cells with hyper-activated mTOR, we identified novel APA events that could otherwise not be identified by either profiling method alone. Several transcription factors including Cebpg (CCAAT/enhancer binding protein gamma) were among the newly discovered APA transcripts, indicating that diverse transcriptional networks may be regulated by mTOR-coordinated APA. The prevention of APA in Cebpg using the CRISPR/cas9-mediated genome editing tool showed that mTOR-driven 3′-UTR shortening in Cebpg is critical in protecting cells from endoplasmic reticulum (ER) stress. Taken together, we present IntMAP as a new bioinformatics algorithm for APA analysis by which we expand our understanding of the physiological role of mTOR-coordinated APA events to ER stress response. IntMAP toolbox is available at http://compbio.cs.umn.edu/IntMAP/.
INTRODUCTION
Eukaryotic gene expression involves multiple key steps of post-transcriptional RNA processing including 5′-capping, splicing, and polyadenylation. At the end of transcription by RNA polymerase II, polyadenylation occurs through the cleavage of a transcript downstream of the polyadenylation signal (PAS) in the 3′-UTR followed by the addition of poly(A) tails by poly(A) polymerase (1–3). Since 3′-UTR functions as a binding platform for regulatory miRNAs and RNA-binding proteins (RBPs), APA in the 3′-UTR of mRNAs serves as a key mechanism in eukaryotic gene expression regulation at the post-transcriptional level. Altered 3′-UTR length could affect various aspects of mRNA metabolism, such as mRNA localization and translation efficiency, and is linked to diverse disease contexts including cancer (3–9).
Many publically available algorithms have been developed for the identification and quantification of 3′-UTR APA on a genome-wide scale (10–17). If alternative poly(A) sites are known, 3′-UTR APA changes can be quantitatively profiled by comparing the RNA-seq read coverage before and after these sites. However, unannotated or novel alternative poly(A) sites cannot be accurately identified by analyzing RNA-seq data. To this end, various versions of 3′-end-targeted sequencing methods have been used to map out the location of poly(A) sites and to quantitate transcripts with different 3′-UTR lengths (6–19). Yet, the quantitative power of 3′-end-targeted sequencing methods is limited by several factors such as false priming on A-rich regions or inconsistent efficiency in library preparation (11,25,27).
mTOR, a serine/threonine kinase, is the major component of two multi-protein complexes known as mTOR Complex 1 (mTORC1) and 2 (mTORC2) (30–32). mTORC1 is negatively controlled by the heterodimer of tuberous sclerosis complexes (TSC1 and TSC2) and directly phosphorylates two substrates, p70S6 Kinase 1 (S6K1) and eIF4E Binding Protein (4E-BP), which increase protein synthesis (30). Through the activation of transcription networks, the mTOR signaling pathway plays a central role in regulating various cellular metabolisms such as lipid/mitochondria metabolism and stress response pathways including nutrient/growth factor deprivation, ER stress, hypoxia and oxidative stress; its dysregulation is also associated with many pathological conditions (31–34).
Several studies using polyribosome footprinting profiling identified that the translation of mRNAs containing the 5′-terminal oligopyrimidine tract (TOP) or TOP-like elements in their 5′-UTRs is highly affected by mTORC1 inhibition (30,35). These studies showed that mTORC1 activation led to the utilization of distinct molecular signatures in the 5′-UTR of mRNA for protein synthesis. Recently, we found that the activation of mTORC1 leads to genome-wide 3′-UTR shortening which in turn increases protein synthesis in genes belonging to certain pathways, such as ubiquitin-mediated proteolysis, thus establishing a connection between 3′-end processing of mRNA and mTORC1 (36). To better understand the dynamics of 3′-UTR length in the mTOR-hyperactivated transcriptome, we developed an algorithm named IntMAP (Integrative Model for Alternative Polyadenylation) that provides an improved analysis of transcriptome-wide APA events. This method combines the RNA-Seq data as well as the 3′-end-seq data to take advantage of the quantitative power of RNA-Seq and the precise mapping of poly(A) site usage by 3′-end-seq, allowing accurate profiling of 3′-UTR APA events. Interestingly, we were able to identify novel genes and pathways that are targeted by mTORC1-mediated 3′-UTR APA using IntMAP. Among these genes is the C/EBPγ (CCAAT/enhancer binding protein gamma) gene, a C/EBP family member that acts as a negative regulator of other C/EBP members’ transcriptional activities by forming heterodimers (37,38). Studies have shown that C/EBPγ promotes cell proliferation and inhibits senescence by interacting with C/EBPβ (39). C/EBPγ is also crucial for cellular redox homeostasis and attenuates cellular stress by forming heterodimer with ATF4, a bZIP transcription factor (40). In this study we show that mTORC1 activation up-regulates the protein expression of C/EBPγ via 3′-UTR APA to enable cells to respond to ER stress, making an unexpected link between mTOR signaling and C/EBPγ pathway and demonstrating the advantage of IntMAP in mRNA post-transcriptional regulation studies.
MATERIALS AND METHODS
Cell lines and cell culture
WT and Tsc1−/− MEF cells were obtained from Dr. Kwiatkowski at Harvard University (41). WT and Tsc1–/– MEFs were cultured in DMEM (Gibco, USA) supplemented with 10% (v/v) FBS (Gibco, USA), 100 μg/ml streptomycin, and 100 U/ml penicillin at 37°C.
Real-time quantitative PCR (RT-qPCR) analysis and primer sequences
Total cellular RNAs were isolated by Trizol method according to manufacturer's protocol. Reverse transcription reaction was performed using Oligo-d(T) or random hexamer priming and superscript III (Thermo Fischer Scientific) according to the manufacturer's protocol. SYBR Green was used to detect and quantitate PCR products in real-time reactions via the comparative Ct method. We normalized the Ct values to total RNAs for quantitation of total or long transcripts. The PCR primers for quantitative analyses are as follows: Cth forward 5′-AGCAATCATGACCCATGCCT-3′, Cth reverse 5′-CTCTAGGCCCACAGAAAGTCG-3′, GPt2 forward 5′-GCAGCGAGAAGGCACTTAC-3′, GPt2 reverse 5′-TGGAGCACGGTCTTCAGTTTA-3′, Mthfd2 forward 5′-ATATCACTCCCGTCCCTGGT-3′, Mthfd2 reverse 5′-TCTTAGCACTTTCTTTGCGGC-3′, Slc1a5 forward 5′-GAGCCCGAATTGATCCAGGT-3′, Slc1a5 reverse 5′-TGGTACTGTTTCAGGAGGGGA-3′, Slc7a11 forward 5′-CACCGGGGTCGGTTTTCTTA-3′, Slc7a11 reverse 5′-TCGTCTGAACCACTTGGGTTT-3′, Mtdh total forward 5′-GACCAAGTCTGAAACTAACTGGGA-3′, Mtdh total reverse 5′-TTCACGTTTCCCGTCTGG-3′, Mtdh long forward 5′-TGTCAACTAGGAAAGCTAAAATGGT-3′, Mtdh long reverse 5′-GAGGAAAGCTGTCCATTAATAAGGC-3′, Appl1 total forward 5′-TCATTTCCCTGGGATGTGGC-3′, Appl1 total reverse 5′-GCTGAAGCACACTACTGTAAAGC-3′, Appl1 long forward 5′-TTTCTGTGTAGTCCTGGGAGC-3′, Appl1 long reverse 5′-GACAGAGGCAAGCGGGTATG-3′, Cebpg total forward 5′-ACACTGCAAAGAGTAAACCAGC-3′, Cebpg total reverse 5′-GTGCGCATGCTCAAGAAACA-3′, Cebpg long forward 5′-TGTAGAGTGCTCCTGATGCC-3′, Cebpg long reverse 5′-GGCAGATCTGATTAGCTGTGGA-3′, Agfg1 total forward 5′-TTCAGCCCCAGACTACAGGT-3′, Agfg1 total reverse 5′-TTCCAAAATCAGCACTGGAGGA-3′, Agfg1 long forward 5′-CCCCTACCTGTGACCCTAAAG-3′, Agfg1 long reverse 5′-CATTCATTCCAAGCTCGTGGG-3′, Clip1 total forward 5′-CTGAGAAGCAAGCTCGACCA-3′, Clip1 total reverse 5′-ACCTTCAGCTCCTCCATTGC-3′, Clip1 long forward 5′-AACTGAAGAGTTGACCCCGC-3′, Clip1 long reverse 5′-GGAAGTTCTCGTCCTGCACA-3′, Crtc1 total forward 5′-GCTGCCACCACTCTCAGTTA-3′, Crtc1 total reverse 5′-AGTGGCATCGATCCCCATTG-3′, Crtc1 long forward 5′-CAACCTCCTGGTCAGGGTA-3′, Crtc1 long reverse 5′-TGATGGATCCCTAGCCCCAA-3′, Cdk5 total forward 5′-CATCGACATGTGGTCAGCC-3′, Cdk5 total reverse 5′-GTGTCCCTAGCAGTCGGAAG-3′, Cdk5 long forward 5′-AAGGCGAGACACCAGTTTGT-3′, Cdk5 long reverse 5′-ATTGCACCAACGGTGGAGAG-3′, Ctdsp2 total forward 5′-ACCAGGGCTGCTATGTCAA-3′, Ctdsp2 total reverse 5′-CTGCACAGGCACTGCATTTT-3′, Ctdsp2 long forward 5′-TGCCCCATCCTTTAGTTCGG -3′, Ctdsp2 long reverse 5′-TCCCCTCCAAATGGCATCAC-3′, Eya3 total forward 5′-TCGCTACCGAAAAGTGAGGG-3′, Eya3 total reverse 5′-CAGCACCTCGATCTCTGCTC-3′, Eya3 long forward 5′-CCTTGAGTAAGCGTGTGCT-3′, Eya3 long reverse 5′-GAACGCCAACCTGGCATCTA-3′, Eif4b total forward 5′-TCTCCAACCTCTAAGCCTCCT-3′, Eif4b total reverse 5′-TAGAGCTTCGCTTCACCCAC-3′, Eif4b long forward 5′-GGGTTGACTTATGTCCCGCT-3′, Eif4b long reverse 5′-ACATCTCAAGACATCTCCCAAGT-3′, Mapk9 total reverse 5′-TCAGCGGGGTCATACCAAAC-3′, Mapk9 long forward 5′-GGGTTTCCATTTCAAGCGCA-3′, Mapk9 long reverse 5′-GTTTTGTCTGCAGGCCCAATG-3′.
Measurement of relative shortening index (RSI)
A numerical presentation of 3′-UTR APA events was developed previously by calculating the Relative Shortening Index (RSI) (36). Briefly, a relative stoichiometry of long 3′-UTR transcripts vs. total (short+long) transcripts was measured by RT-qPCR analyses. The following equation was used to determine the RSI.
LI = [normalized expression of longer 3′UTR-containing transcript]/[normalized expression of total (long + short) transcript]
RSI = –log2(LI/[LI in reference cell line]), so RSI = 0 for a reference cell line.
If RSI is positive in a target cell line, then there is a 3′-UTR shortening. If RSI is negative in a target cell line, then there is a 3′-UTR lengthening. The RSI contains the information about the changes in the proportion of a longer 3′UTR-containing transcript in a given cellular context compared to a reference cell.
Luciferase assays
psiCheck1 containing various Cebpg 3′-UTR deletion mutants and pGL4.13 firefly reporter plasmid (obtained from Dr Aaron Goldstrohm laboratory at the University of Minnesota Twin Cities;(42)) were transfected into Tsc1−/− MEFs using Lipofectamine 3000 (Thermo Fisher Scientific). 24 hours after transfection, the luciferase activity was measured by Dual-Glo reagent using a Glomax Discover luminometer (Promega). To normalize to the transfection efficiency, a relative response ratio was calculated by dividing the relative light unit values of RnLuc by those for FfLuc for each individual well (42). Four technical replicates were conducted in each independent experiment. Fold changes were calculated relative to the parental vector psiCheck1 containing proximal PAS deletion mutants (Cebpg short 3′-UTR form).
Cellular reactive oxygen species (ROS) production assay
Cellular ROS production was measured using a DCFDA assay kit (Abcam) according to the manufacturer's protocol. DCFDA (2′,7′-dichlorofuorescein diacetate), a cell-permeable fuorogenic dye, is deacetylated to a non-fluorescent compound by cellular esterases and later oxidized into highly fluorescent DCF (2′,7′-dichlorofuorescein) by ROS, allowing measurement of hydroxyl, peroxyl and other ROS activities within the cell. Tsc1−/−, Tsc1−/- with Cebpg knockdown and CebpgΔPAS Tsc1−/- MEFs were grown on 96-well plates. Once cells are confluent, cells were washed with PBS twice and then incubated with 25 μM DCFDA at 37°C for 45minutes. Cellular fluorescence in the 96-well plates was measured by 488 nm excitation and 525 nm emission using a Victor3 V Multilabel Plate Counter (PerkinElmer, Inc.).
siRNAs and antibodies
Cells were transfected with siRNA oligos synthesized by IDT for each gene using RNAi Max (Thermo Fisher Scientific) according to the manufacturer's protocol. The following sequences were used for the knockdown (KD) of Cebpg transcript: 5′- GAT ACA CTG CAA AGA GTA AAC CAG C-3′ (Cebpg siRNA). The antibodies used for western analysis are as follows: anti-hnRNP A1 (4B10 from Abcam), anti-CPSF3 (sc-393001, Santa Cruz Biotechnology), anti-CPSF4 (sc-393316, Santa Cruz Biotechnology), CPSF7 (sc-393880, Santa Cruz Biotechnology), CstF1 (sc-393260, Santa Cruz Biotechnology), anti-CPSF6 (ab175237 from Abcam), anti-Tubulin (sc-53646, Santa Cruz Biotechnology), anti-C/EBPγ (sc-517003, Santa Cruz Biotechnology), anti-Lamin A/C (#4777 from Cell Signaling), anti-ATF4 (sc-390063, Santa Cruz Biotechnology), anti-Actin (ac-47778, Santa Cruz Biotechnology), anti-S6 (#2317 from Cell Signaling), and anti-pS6 (#2211 from Cell Signaling).
Trypan blue exclusion assay
Cells were incubated at room temperature with 0.2% trypan blue in PBS for 5 min. Trypan blue positive as well as total cell population were counted to calculate the percentage of viable cells.
Cloning and assembly of CRISPR-Cas9 targeting constructs
Cebpg ΔPAS Guide RNAs were identified using crispr.mit.edu. Guide sequences were cloned into Addgene plasmid #48138 using the following primers: Cebpg ΔPAS(1) forward 5′–CACCGCTTGTGACTTGAACATGAG–3′ Cebpg ΔPAS(1) reverse 5′-AAACCTCATGTTCAAGTCACAAGC-3′, Cebpg ΔPAS(2) forward 5′-CACCGTTTATACATCTATAAAATA-3′, Cebpg ΔPAS(2) reverse 5′-AAACTATTTTATAGATGTATAAAC-3′
Polysome isolation and analysis
Isolation of polysome fractions from total cell lysates using sucrose gradient was carried out as previously described (36). Briefly, cells were lysed in the polysome buffer (20 mM Tris pH 7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 100 mg/ml cycloheximide and 1% Triton X-100). Cell extracts were loaded onto sucrose gradient (5–45%). Fractionation was done by centrifugation at 190 000 × g for 2 h at 4°C. Twelve fractions were collected for the analysis. The percentage of mRNAs in each fraction was calculated as described previously (36). Five per cent (v/v) of total RNAs in each fraction were used for RT-qPCR. The signals of total transcripts for a gene in all fractions of Tsc1−/- were combined and set as 100%. Next, the percentage of a total transcript in each fraction was calculated and the distribution of total transcripts for a gene was shown by the fraction. In case of long 3′-UTR transcripts, the signals of long transcripts in all fractions were combined and the relative amounts of long transcripts were scaled down based on the relative differences between total and long transcript signals in input. The percentage and distribution of long transcripts in each fraction were calculated and presented. The same procedure was applied to CebpgΔPAS Tsc1−/− MEFs.
Short read alignments and peak identification
To evaluate the transcriptome features under mTOR-hyperactivation at single-nucleotide resolution, we performed RNA-Seq and Poly (A)-site sequencing (PAS-Seq) (43) analyses of poly(A+) RNAs isolated from WT and Tsc1−/− MEFs. In the RNA-Seq analysis, 63 742 790 paired-end 50 bp reads for WT and 74 251 891 paired-end 50 bp reads for Tsc1−/− MEFs were produced from Hi-Seq pipeline. The short reads were aligned to the mm10 reference genome by TopHat (44), with up to two mismatches allowed. The unmapped reads were first trimmed to remove poly-A/T tails (repeats of [A/N]s or [T/N]s) from read ends/starts and then aligned to the reference genome. Only the reads with at least 30 bp in both ends were retained after trimming. Finally, 87.1% of short reads from WT and 87.5% of sequence reads from Tsc1−/− MEFs were mapped to the reference genome by TopHat for APA analysis in the study. In the PAS-Seq analysis, the reads from WT and Tsc1−/− were preprocessed to trim A’s off the 3′-ends and then filtered by removing the reads of low-quality 3′-ends (Phred score < 30) and shorter than 25 bp. The remaining reads were aligned to the mm10 reference genome by Bowtie (45) without allowing any mismatches. In total, 6 186 893 aligned reads were aligned for WT and 5 382 111 reads were aligned for Tsc1−/−. All aligned reads from PAS-Seq were pooled together in order to identify peaks and the corresponding cleavage sites in the reference genome by the read coverage signals. In each read alignment ‘hill’, the location with the highest read coverage between two zero coverage positions was considered as the peak of the ‘hill’. The 3′-end of the peak is chosen as the potential corresponding cleavage sites where the read coverage at the peak quantifies the cleavage at the site. To remove false positives, only peaks with at least four supporting reads were analyzed.
IntMAP algorithm
To detect alternative PAS in the 3′-UTR of a gene, we propose an integrative model for alternative polyadenylation, IntMAP, by combining the RNA-seq read alignments and PAS-seq peak calling. IntMAP is built on a constrained probabilistic model with the probabilistic modeling of the RNA-seq read mapping uncertainty for estimating the abundance of all the short and long transcripts of a gene and the constraint on the abundance by the PAS peak callings.
Let 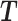 denote the set of the transcripts with different 3′UTR lengths in a gene with
denote the set of the transcripts with different 3′UTR lengths in a gene with 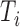 being the ith transcript in
being the ith transcript in 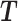 , and a set of reads r aligned to the 3′UTRs of the gene. Note that the complete list of the transcripts in a gene is combined from all the transcript variants with different cleavage sites predicted by the PAS-seq data or annotated in the reference genome. The probability of a read being generate by the transcript in T is modeled by a categorical distribution specified by parameter
, and a set of reads r aligned to the 3′UTRs of the gene. Note that the complete list of the transcripts in a gene is combined from all the transcript variants with different cleavage sites predicted by the PAS-seq data or annotated in the reference genome. The probability of a read being generate by the transcript in T is modeled by a categorical distribution specified by parameter 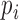 , where
, where 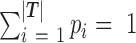 and 0
and 0 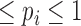 . We consider the likelihood that each read in r is sampled from one of the 3′-UTRs to which the read aligns. Specifically, for each read
. We consider the likelihood that each read in r is sampled from one of the 3′-UTRs to which the read aligns. Specifically, for each read  aligned to the 3′-UTRs in
aligned to the 3′-UTRs in  , the probability of obtaining
, the probability of obtaining 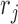 by sampling from
by sampling from 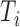 , namely Pr(
, namely Pr(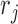 |
|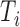 ) is
) is 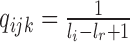 (46–48), where
(46–48), where 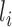 and
and 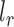 are the length of the 3′-UTR in
are the length of the 3′-UTR in 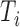 and the length of the read, respectively. Assuming each read is independently sampled from one transcript, the uncommitted likelihood function (49) to estimate the parameters P from the observed read alignments against the 3′-UTRs in T is
and the length of the read, respectively. Assuming each read is independently sampled from one transcript, the uncommitted likelihood function (49) to estimate the parameters P from the observed read alignments against the 3′-UTRs in T is
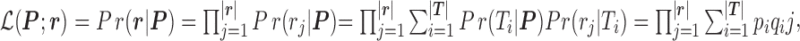 |
where 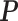 is the probability of a read being generated by the transcript T, specifically, P = [
is the probability of a read being generated by the transcript T, specifically, P = [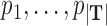 ]. The likelihood function is concave and an Expectation Maximization (EM) algorithm is applied to obtain the optimal
]. The likelihood function is concave and an Expectation Maximization (EM) algorithm is applied to obtain the optimal 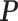 . With P estimated, the abundance of the ith transcript is derived as
. With P estimated, the abundance of the ith transcript is derived as 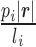 , where
, where 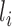 is the length of transcript
is the length of transcript 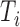 .
.
In the PAS-Seq analysis, the read coverage (the height of the peak in Figure 2(A)) also provides the measure of abundances of the transcript variants at 3′UTR. IntMAP adopts the following constrained log-likelihood function,
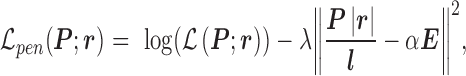 |
(1) |
where the vector 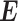 contains the expression values for the transcripts provided by PAS-seq,
contains the expression values for the transcripts provided by PAS-seq, 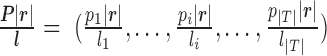 represents the transcripts’ expressions and
represents the transcripts’ expressions and 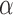 is a scaling factor between the expression values learned from RNA-seq data and PAS-seq data. There are two terms in the penalized log-likelihood function (Equation (1)). The first term is the likelihood of the observed the RNA-seq read alignment as in the original probabilistic model. The second term,
is a scaling factor between the expression values learned from RNA-seq data and PAS-seq data. There are two terms in the penalized log-likelihood function (Equation (1)). The first term is the likelihood of the observed the RNA-seq read alignment as in the original probabilistic model. The second term, 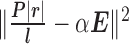 , encourages the consistency between the transcript expressions learned from RNA-seq data and PAS-seq data. The parameter
, encourages the consistency between the transcript expressions learned from RNA-seq data and PAS-seq data. The parameter 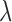 balances the two terms, where larger
balances the two terms, where larger 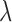 weights PAS-seq data more. By subtracting the second convex term, the penalized log-likelihood function is still concave and a similar EM algorithm is applied to obtain the optimal
weights PAS-seq data more. By subtracting the second convex term, the penalized log-likelihood function is still concave and a similar EM algorithm is applied to obtain the optimal 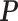 with the CVX package (50,51). The scaling factor
with the CVX package (50,51). The scaling factor 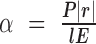 is updated with current
is updated with current 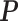 in each iteration in the EM algorithm. Then the chi-squared test is applied to the abundances of transcripts with different 3′-UTR lengths in WT and Tsc1−/− to detect 3′-UTR APA events. The detailed IntMAP algorithm is outlined in the supplementary document. The program is available at Github: https://github.com/kuanglab/IntMAP.
in each iteration in the EM algorithm. Then the chi-squared test is applied to the abundances of transcripts with different 3′-UTR lengths in WT and Tsc1−/− to detect 3′-UTR APA events. The detailed IntMAP algorithm is outlined in the supplementary document. The program is available at Github: https://github.com/kuanglab/IntMAP.
Figure 2.

Development of a novel algorithm IntMAP by integrating RNA-Seq and PAS-Seq dataset. (A) A schematic of the experimental workflow and algorithm to integrate RNA-Seq and PAS-Seq data. An algorithm integrating two datasets was developed for comprehensive profiling of 3′-UTR APA events. (B) A Venn diagram of 3′-UTR shortened transcripts in Tsc1−/− MEFs using IntMAP. The number of genes producing 3′-UTR shortened transcripts in each section of Venn diagram is indicated.
RESULTS
Comprehensive profiling of 3′-UTR length dynamics by an integration model, IntMAP
A number of profiling techniques that specifically enrich and sequence polyadenylated RNA fragments in a transcriptome is available to determine the transcriptome-wide changes in the 3′-UTR length (10,18–29,43). In addition, multiple algorithms are available to assess the quantities of mRNA isoforms with variable 3′-UTR length using RNA-Seq data (10,11,36). All of these approaches aim to assess global 3′-UTR shortening in a transcriptome by APA. Interestingly, one study showed that the outcome of APA analysis on 3′-UTR could vary by the choice of profiling methods (11). Thus, the profiling technique affects the APA analysis in a transcriptome and one profiling method may not provide complete information on 3′-UTR length dynamics.
Using RNA-seq experiments, we showed that the cellular mTORC1 activation leads to transcriptome-wide 3′-UTR shortening and enhances translation of those short 3′-UTR transcripts, which activates multiple biological pathways including ubiquitin-mediated proteolysis (36). To investigate whether other APA profiling methods could expand our understanding of mTOR-controlled biological pathways, we used the PAS-Seq method to map out experimentally proven polyadenylation sites and to quantitate APA events in the wild-type (WT) and Tsc1−/− mouse embryonic fibroblast (MEF) transcriptomes. Compared to 846 short 3′-UTR transcripts identified by the RNA-Seq method, we found 843 short 3′-UTR transcripts in the Tsc1−/− transcriptome using the PAS-Seq method (Figure 1(A) and Supplementary Table S1). Consistent with the previous report (11), our APA profiling using two separate methods also showed a limited overlap of 305 genes (36%) (Figure 1(A)). Although the number of overlapping short 3′-UTR transcripts between the two datasets is not considerably large, many KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways including protein processing in the endoplasmic reticulum (mmu04141) and ubiquitin-mediated proteolysis (mmu04120) exist in both datasets with a significant P-value (Figure 1(B)). In comparison, KEGG pathways that are unique to either dataset tend to have P-values that are less significant. (Figure 1(B) and Supplementary Table S1). By pooling the two datasets together, we were able to identify 3′-UTR APA events in 1,384 genes in the Tsc1−/− transcriptome (Figure 1(A)). Interestingly, the combined data enriched distinct KEGG pathways such as RNA transport (mmu03013) and adherens junction (mmu04520) which did not appear in either dataset (Figure 1(B) and Supplementary Table S1). Thus, two profiling methods increase the total number of genes with 3′-UTR APA events and broaden the list of biological pathways that are modulated by 3′-UTR APA in Tsc1−/− MEFs.
Figure 1.

Profiling of 3′-UTR APA events in the mTOR-activated transcriptome using Poly(A) Site sequencing (PAS-Seq). (A) A Venn diagram of 3′-UTR shortened transcripts in Tsc1−/− MEFs using two independent profiling methods. (B) KEGG pathway analysis of 3′-UTR shortened transcripts identified by PAS-Seq, RNA-Seq and the union of the two methods. The KEGG pathways only enriched after combining two profiling methods are highlighted in dark violet.
Even though the union of two datasets shows an advantage in expanding the APA profile of a transcriptome over using single APA profiling method, this approach does not fully complement limitations inherited from each profiling method. Thus, we developed a novel algorithm named IntMAP (Integrative Model for Alternative Polyadenylation), which integrates RNA-Seq and PAS-Seq data for exhaustive analysis of 3′-UTR APA events (Figure 2(A)). In IntMAP, first the positions of multiple polyadenylation sites in a 3′-UTR of a gene are defined and the 3′-UTR isoforms of the gene are accordingly deduced. Then the quantitative information of RNA-Seq and PAS-Seq data is integrated to calculate the expression level of inferred 3′-UTR isoforms. Two elements in IntMAP work systemically to help the quantitation of isoform expression. The first element promotes the isoform expression to comply with the observed read counts from RNA-Seq data. The second element encourages the consistency between the isoform expression learned from RNA-Seq and PAS-Seq data. After the quantitation by IntMAP, the calculated expression level of different 3′-UTR isoforms is applied to the chi-squared test to determine the 3′-UTR shortening of a gene in a given biological context compared to control (Figure 2(A)). Next, we applied IntMAP to our RNA-Seq and PAS-Seq data and found 975 genes with 3′-UTR APA events (Figure 2(B) and Supplementary Table S2). Among 975 genes, 592 and 370 genes overlapped with the PAS-Seq and RNA-Seq analyses, respectively. Of note, IntMAP could not entirely encompass the genes with 3′-UTR APA found in the PAS-Seq or RNA-Seq analysis (Figure 2(B)). Importantly, IntMAP identified 280 new 3′-UTR APA events, which were not reported in the union of two data sets (Figure 2(B) and Supplementary Table S2). By pooling genes showing APA altogether from RNA-Seq, PAS-Seq, and IntMAP analyses, we could determine 1658 APA events in the Tsc1−/− transcriptome (Figure 2(B)).
Examples of RNA-Seq and PAS-Seq read alignments from newly identified 280 genes show a marginal pattern for 3′-UTR shortening which was not significant enough to be determined as APA events in both analyses (Figure 3(A) and Supplementary Figure S1). But the RSI (Relative Shortening Index which measures the relative stoichiometry of long 3′-UTR over total transcripts from a gene) of those genes measured by qRT-PCR was positive, indicating the production of short 3′-UTR transcripts from those genes (Figure 3(B)). The KEGG pathway analysis using 975 genes identified by IntMAP showed distinct enriched pathways such as FoxO signaling pathway (mmu04068) and p53 signaling pathway (mmu04115) as well as pathways identified by RNA-Seq or PAS-Seq (Figure 3(C) and Supplementary Table S3). Thus, the newly developed IntMAP could comprehensively catalog 3′-UTR APA events in the Tsc1−/− transcriptome and reveal additional biological pathways that might be regulated by APA in Tsc1−/− MEFs. Using the list of genes combined altogether, we were able to expand the repertoire of 3′-UTR APA-modulated cellular pathways upon the activation of mTOR (Figure 3(D), Supplementary Figure S2, and Supplementary Table S3).
Figure 3.

Identification of novel 3′-UTR APA events by IntMAP. (A) Examples of RNA-Seq and PAS-Seq alignments for genes showing 3′-UTR APA events identified by IntMAP. Read alignments of the 3′-most exon or last two exons of RNA- and PAS-Seq are shown. The orientation of genes is presented from 5′ (left) to 3′ (right). (B) The relative shortening index (RSI) of genes based on the comparison between WT and Tsc1−/− MEFs was measured using qPCR to validate the analysis done by IntMAP. qPCR results are from four technical replicates. Data represent the mean ± SEM. Two-tailed student t-tests were performed for statistical significance. (C) KEGG pathway analysis using 975 genes identified by IntMAP is shown. The KEGG pathways only enriched in IntMAP analysis are highlighted in dark green. (D) KEGG pathway analysis of 3′-UTR APA events in the union of RNA-Seq, PAS-Seq and IntMAP analyses is shown. The complete list of KEGG pathways is shown in Supplementary Figure S2.
Alternative polyadenylation as a molecular link between cellular stress response and mTOR activation
To determine whether the APA events solely identified by IntMAP have significant physiological impacts, we set out to examine the newly identified 280 APA events more closely. We first performed the gene ontology (GO) analysis to search for enriched molecular functions in 280 genes. Because mTOR has been known to activate many cellular pathways through the modulation of transcriptional networks, we particularly looked for transcription-related functions in the enriched GO analysis (30,31). From this approach, we found that several transcription factors including Appl1, Mtdh and Cebpg showed 3′-UTR APA in the Tsc1−/− transcriptome (Supplementary Table S4). Consistent with IntMAP-identified APA events, the RSI of those transcription factors showed 3′-UTR shortening although the APA event was not apparent in the RNA-Seq read alignments and PAS-Seq counts (Figures 3A, B and 4A, Supplementary Figure S1). Notably, several of these transcription factors have not been linked to the mTOR signaling pathway extensively, including C/EBPγ. C/EBPγ is known to function in the integrated cellular stress responses to redox imbalance and nutrient deprivation stress by forming a heterodimer with ATF4 (40). In Tsc1−/− MEFs, the expression of C/EBPγ protein is highly upregulated compared to WT MEFs (Figure 4B and Supplemental Figure S3B). Because C/EBPγ is known to activate oxidative stress response-related transcription networks with ATF4 (40), we examined the expression level of C/EBPγ downstream target genes using qPCR in Tsc1−/− and WT MEFs. As anticipated, all tested target genes of C/EBPγ were significantly up-regulated in Tsc1−/− compared to WT MEFs (Figure 4C). To validate whether the up-regulation of these genes is specifically mediated by the increase of C/EBPγ in Tsc1−/− MEFs, we specifically knocked down C/EBPγ using RNAi (Figure 4D and Supplementary Figure S3A) and measured its effect on target gene expression. When C/EBPγ was down-regulated, the expression of all tested genes was decreased (Figure 4D), indicating that the transcriptional activation of oxidative stress response-related genes in Tsc1−/− MEFs is mediated by the upregulation of C/EBPγ.
Figure 4.

mTOR-coordinated cellular stress response network identified by IntMAP. (A) RNA- and PAS-Seq read alignments of Cebpg are shown. An absolute quantitation of long 3′-UTR and total (long+short 3′-UTR) Cebpg mRNAs was made using qPCR. The amounts of short versus long 3′-UTR transcripts are presented by copies/μg of total RNAs. The shortening index of Cebpg in the Tsc1−/− transcriptome was calculated based on absolute or normalized quantitation. qPCR results are from 4 technical replicates. Data represent the mean ± SEM. Two-tailed Student's t-tests were performed for statistical significance. (B) Western blotting for Cebpg, hnRNP A1 and Lamin A/C using WT and Tsc1−/− cell extracts. Relative amounts of Cebpg mRNA were measured by qPCR. qPCR results are from four technical replicates. Data represent the mean ± SEM. Two-tailed student t-tests were performed for statistical significance. *P = 7.97e–007. (C) Relative gene expression of Cebpg target genes in WT and Tsc1−/− MEFs was measured using qPCR. qPCR results are from 4 technical replicates. Data represent the mean ± SEM. Two-tailed Student's t-tests were performed for statistical significance. (D) RNAi knockdown (KD) of Cebpg in Tsc1−/− MEFs and the analysis of Cebpg target genes. Western blotting was conducted for Cebpg, Cpsf6, Tubulin, and hnRNP A1 using con or Cebpg KD Tsc1−/− cell extracts. Relative amounts of Cebpg mRNA in the KD cells compared to control cells were measured using qPCR. Expression of Cebpg target genes in the KD and control cells were quantitated using qPCR. qPCR results are from four technical replicates. Data represent the mean ± SEM. Two-tailed Student's t-tests were performed for statistical significance.
Mechanistic insight in the regulation of Cebpg 3′-UTR length and protein production
Interestingly, as opposed to the drastic increase in the protein level, the transcript level of Cebpg only increased by ∼2-fold from WT to Tsc1−/− MEFs (Figure 4A and B, and Supplementary Figure S3B). The fact that the protein level change cannot be fully explained by the transcript level change indicates that APA may play an important role in the regulation of C/EBPγ expression in WT and Tsc1−/− MEFs. Therefore, we next examined the role of 3′-UTR APA of Cebpg in its protein production. According to the PAS-Seq data from WT and Tsc1−/− MEFs, Cebpg contains 4 actionable polyadenylation signals (PASs) (Figures 4A and 5A). Among those PASs, the first and second PAS (PAS1 and PAS2) are highly used for polyadenylation and the other PASs (PAS3 and PAS4) are minimally used (Figure 4A). To test whether the APA event of Cebpg is crucial for the regulation of its protein production and function in cellular stress responses, the region of approximately 500 bp covering the most upstream proximal PAS in the 3′-UTR of Cebpg were deleted in Tsc1−/− MEFs (CebpgΔPAS Tsc1−/−) using the CRISPR/Cas9-mediated genome editing tool (Figure 5A). After CRISPR/Cas9 transfection, we isolated three clones for further characterization. Analysis of the genomic DNA confirmed that they are heterozygous clones (CebpgΔPAS#5, #11 and #12) (Supplementary Figure S4A and B). The effect of the PAS deletion on Cebpg APA event was examined by measuring the absolute amounts of total and long 3′-UTR containing Cebpg transcripts. While Cebpg transcripts in Tsc1−/− MEF predominately contain short 3′-UTR, CebpgΔPAS clones, with actionable PAS removed, significantly increased the expression of transcripts with long 3′-UTR (Figure 5B). The application of absolute quantitation results to the RSI calculation confirmed the lengthening of 3′-UTR in Cebpg in the cell lines with deleted actionable PAS (Figure 5B). We also calculated the RSI of Cebpg along with other genes in CebpgΔPAS#11 Tsc1−/- and control Tsc1−/− MEFs using qPCR and relative normalization. Consistent with the RSI by the absolute quantitation, the 3′-UTR length of Cebpg mRNA drastically increased in CebpgΔPAS#11 Tsc1−/− while other tested genes did not show substantial changes in their 3′-UTR length (Figure 5C). Western blot analysis using the whole cell extracts from control Tsc1−/− and CebpgΔPAS#11 Tsc1−/− MEFs showed that the C/EBPγ protein decreased significantly in CebpgΔPAS#11 Tsc1−/− MEFs even though the amount of Cebpg mRNA only decreased by ∼20% in CebpgΔPAS#11 Tsc1−/− MEFs (Figure 5B and D). Consistent with these observations, all tested CebpgΔPAS Tsc1−/− MEF clones showed more drastic changes in the amount of C/EBPγ protein compared to that of Cebpg transcripts (Figure 5B and Supplementary S5A and B). All these data provide evidence that the 3′-UTR APA of Cebpg is a key regulatory process in the cellular production of C/EBPγ protein.
Figure 5.

A regulatory role of 3′-UTR APA in Cebpg gene expression. (A) Schematic of proximal PAS knockout (KO) in Tsc1−/− MEFs. Using the CRISPR/Cas9-mediated genome editing tool, two juxtaposed proximal PASs in the 3′-UTR of Cebpg were deleted and CebpgΔPAS Tsc1−/− MEFs were established. (B) An absolute quantitation of total Cebpg mRNAs and Cebpg mRNA with long 3′-UTR was measured and presented by copies/μg total RNAs. The quantitation was made using three CebpgΔPAS Tsc1−/− MEF clones (#5, #11 and #12). The shortening index of Cebpg in these clones was calculated based on the absolute quantitation. qPCR results are from four technical replicates. Data represent the mean ± SEM. Two-tailed Student's t-tests were performed for statistical significance. (C) The RSI of selected genes in CebpgΔPAS#11 Tsc1−/− MEFs compared to control Tsc1−/− MEFs was measured using qPCR. qPCR results are from four technical replicates. Data represent the mean ± SEM. Two-tailed Student's t-tests were performed for statistical significance. (D) CebpgΔPAS Tsc1−/− MEFs were characterized using western blotting. Expression of C/EBPγ, Cpsf6, Tubulin and hnRNP A1 protein was analyzed using total cell extracts from CebpgΔPAS and control Tsc1−/− MEFs. qPCR using the same set of cell lines was performed to measure the relative level of Cebpg mRNA. qPCR results are from 4 technical replicates. Data represent the mean ± SEM. Two-tailed Student's t-tests were performed for statistical significance. (E) Total and long 3′-UTR Cebpg mRNAs were analyzed using polysome profiling. Cytoplasmic extracts from Tsc1−/− and CebpgΔPAS Tsc1−/− MEFs were fractionated by 5–45% sucrose gradients. Percentage of total and long 3′-UTR Cebpg mRNAs was calculated in each fraction using qPCR. 80S monosome and polysomes in the fractionation were indicated. qPCR results are from four technical replicates. Data represent the mean ± SEM. (F) A serial deletion of the Cebpg 3′-UTR was tested for the regulation of Renilla luciferase expression. 100 (Δ0.1), 250 (Δ0.25), 350 (Δ0.35) and 500 (Δ0.5) nucleotide deletion constructs were tested. Renilla luciferase activity was normalized to firefly luciferase activity which was used as a transfection control. The assay was conducted four technical replicates. Three independent experiments showed similar results. One representative result is shown. Data represent the mean ± SEM. Two-tailed Student's t-tests were performed for statistical significance. P-values were obtained by comparing Short versus each deletion construct. *P < 0.00054. **P > 0.05. (G) Western blot analysis of multiple polyadenylation factors using WT and Tsc1−/− MEFs extracts is shown (left panel). RNAi-mediated knockdown of polyadenylation factors in WT MEFs were conducted (middle panel). The knockdown effect of those polyadenylation factors on the RSI was measured by qRT-PCR (right panel).
Our previous study demonstrated that transcripts with a shorter 3′-UTR are more efficient in protein production than their longer 3′-UTR counterparts in mTOR-hyperactivated cells (36). To determine if this is the case for the Cebpg transcript, we performed polysome fractionation experiments using CebpgΔPAS#11 Tsc1−/- and control Tsc1−/− MEFs (Supplementary Figure S6(A)). Then we used each fractionated sample to quantitate the absolute amounts of total and long 3′-UTR Cebpg transcripts using qPCR. As shown in Figure 5E, in Tsc1−/− MEFs, the Cebpg transcripts with the long 3′-UTR distribute almost equally across the fractions, while the Cebpg transcripts with short 3′-UTR are highly enriched in polysome fractions, suggesting that the translation efficiency of Cebpg increased significantly in Tsc1−/− MEFs by the APA event in Cebpg. In contrast, the portion of Cebpg transcripts in heavier polysome fractions (such as fractions 10 and 12) decreased dramatically in CebpgΔPAS#11 Tsc1−/− MEFs compared to control Tsc1−/− MEFs, suggesting that the lengthening of the 3′-UTR in Cebpg impedes the formation of polysomes and decreases protein production (Figure 5E). These results also indicate that the 3′-UTR of Cebpg contains sequence elements that would interact with trans-acting element(s) to hamper the recruitment of ribosomes.
To understand how Cebpg expression is coordinated through the cis- and trans-regulatory networks, we first delineated sequence elements in the 3′-UTR that render the repression of polysome formation. To this end, we produced a series of reporter constructs by placing serial deletions of Cebpg 3′-UTR DNA fragment spanning two major polyadenylation sites downstream of Renilla luciferase (Supplementary Figure S6(B)). Transfection experiments with the Short or long-mut 3′-UTR containing constructs set up the reference luciferase activities for the formation of polysomes or the repression of polysome formation, respectively (Figure 5F and Supplementary Figure S6C). The calculation of normalized Renilla luciferase activities showed that the deletion of immediately upstream 100 nucleotides from the PAS2 of Cebpg 3′-UTR in the reporter construct (mtPAS1-longΔ0.1) is sufficient to rescue the luciferase activity from the suppression (Figure 5F and Supplementary Figure S6C). All tested reporter constructs expressed similar amounts of transcripts, confirming that the unknown sequence element(s) in the highly conserved 100 nucleotides renders the impairment of ribosome associations (Supplementary Figures S6D and E).
To investigate how the cellular mTOR activity leads to the APA events in Cebpg transcripts, we compared the expression levels of known 3′-end processing factors between WT and Tsc1−/− MEFs. Our RNA-Seq data showed that some of the polyadenylation factors changed their transcript levels upon the mTOR activation (Supplementary Figure SS7A). Notably, a subunit of the Cpsf (cleavage and polyadenylation specificity factor) complex, Cpsf6, decreased its mRNA level by >2-fold while the rest of them mostly increased their transcript levels by ∼2-fold or more (Supplementary Figure S7A). Interestingly, using WT and Tsc1−/− MEF extracts, western blot analyses showed that most of these polyadenylation factors appear to be similar in their tendency in mRNA and protein expression (Figure 5G and Supplementary Figure S7A). However, the Cpsf5 protein decreased its amount significantly in spite of the drastic upregulation of Cpsf5 mRNA in Tsc1−/− MEFs (Figure 5G and Supplementary Figure S7A). Thus, we conducted RNAi knockdown experiments to simulate the expression level changes of these polyadenylation factors between WT and Tsc1−/− MEFs and measured the RSI of Cebpg. While the knockdown of Cstf1, Cpsf3 and Cpsf4 in Tsc1−/− MEFs did not reverse the APA events of Cebpg significantly, the knockdown Cpsf5 or Cpsf6 led to the shortening of Cebpg 3′-UTR (Figure 5G), providing evidence that the downregulation of Cpsf5 and Cpsf6 is the major mechanism of the APA events in Cebpg upon mTOR activation.
Physiological relevance of the 3′-UTR APA events in Cebpg
C/EBPγ forms a heterodimer with ATF4 and activates transcriptional networks related to the metabolism of ROS (40). Indeed, ATF4 is upregulated in Tsc1−/− compared to WT MEFs along with C/EBPγ and their expressions are highly associated with the cellular mTOR activity (Supplementary Figure S7B). Thus, we asked whether the APA events in Cebpg followed by the changes in the amount of C/EBPγ protein are critical for the transcriptional activation of genes associated with the intracellular redox homeostasis. As shown in Figure 6A, the expression of all tested genes significantly decreased in CebpgΔPAS#11 Tsc1−/− compared to control Tsc1−/− MEFs, providing evidence that the APA of Cebpg is a molecular determinant for the activation of the C/EBPγ-modulated transcription network. These results also suggest that the Cebpg APA events are critical for the production of cellular ROS and cellular responses to related stresses. To examine the physiological relevance of Cebpg APA events in ROS metabolism, we measured the level of ROS in CebpgΔPAS Tsc1−/− MEF clones using DCF (2′,7′-dichlorofluorescein) staining. All tested CebpgΔPAS Tsc1−/− MEF clones and the Tsc1−/− MEFs with Cebpg knockdown showed an increase in DCF staining, providing evidence that the APA events in Cebpg are crucial in managing ROS levels in cells (Figure 6B and Supplementary Figure S7C).
Figure 6.

ER stress response network modulated by Cebpg 3′-UTR APA events. (A) Analysis of Cebpg target gene expression in CebpgΔPAS#11 Tsc1−/− MEFs compared to control Tsc1−/− MEFs was carried out using qPCR. qPCR results are from 4 technical replicates. Data represent the mean ± SEM. Two-tailed Student's t-tests were performed for statistical significance. (B) DCF staining (2′,7′-dichlorofluorescein) was conducted to measure the level of ROS in Tsc1−/− MEFs with control or Cebpg RNAi KD and CebpgΔPAS#11 Tsc1−/− MEFs (left panel). Relative amount of Cebpg mRNAs in control or Cebpg RNAi KD is shown (right panel). (C) Cellular response of CebpgΔPAS and control Tsc1−/− MEFs to ER stress-inducing drugs. CebpgΔPAS and control Tsc1−/− MEFs were treated with Brefeldin A (BA; 5 μg/ml), Calcium ionophore A23187 (CI; 2 μM), Thapsigargin (TG; 1 μM), Tunicamycin (TM; 1 μM), or Kifunensine (KF; 50 μg/ml) for 24 h. Cell death rate was measured using Trypan Blue staining followed by cell counting. The data are from three biological repeats in which each repeat contains 4 technical replicates. Data represent the mean ± SEM. Two-tailed Student's t-tests were performed for statistical significance. (D) Cellular response of CebpgΔPAS, Cebpg KD and control Tsc1−/− MEFs to ER stress-inducing drugs. Cells were treated with Tunicamycin (TM) or Kifunensine (KF) and cell death rate was measured as described in (A). The data are from three biological repeats in which each repeat contains four technical replicates. Data represent the mean ± SEM. Two-tailed Student's t-tests were performed for statistical significance.
In addition, a genome-wide survey for C/EBPγ-binding sites by ChIP-Seq suggested the role of C/EBPγ in cellular ER stress responses (40). To determine the physiological relevance of Cebpg APA in ER stress responses, we assessed the tolerance of CebpgΔPAS Tsc1−/− MEF clones against ER stresses. To this end, cells were treated widely used small molecules that induce ER stress through various cellular mechanisms and the cell survival was measured using the trypan blue assay (52,53). The exposure of control and CebpgΔPAS Tsc1−/− MEF clones to Brefeldin A, calcium ionophore A23187, and thapsigargin increased the cell death rate significantly compared to the mock treatment (Figure 6C and Supplementary Figures S8A and B). In contrast, the treatment of tunicamycin (a potent N-linked glycosylation inhibitor (53)) or kifunensine (a potent inhibitor of the mannosidase I enzyme (54,55)) impaired the viability of CebpgΔPAS Tsc1−/− MEF clones more significantly than control Tsc1−/− MEFs (Figure 6C). To test whether the vulnerability to cell death by increasing ER stress in CebpgΔPAS Tsc1−/− MEF clones is caused by the downregulation of C/EBPγ, we also knocked down Cebpg using RNAi and measured the cell death rate. As shown in Figure 6D and Supplementary Figure S8C, Tsc1−/− with Cebpg kockdown and CebpgΔPAS Tsc1−/− MEF clones increased the cell death rate in the presence of tunicamycin or kifunensine, providing evidence that the downregulation of C/EBPγ by the 3′-UTR lengthening renders cells more susceptible to ER stress. These data also suggest that the C/EBPγ-regulated transcription network is associated with the cellular response to ER stress caused by the lack of post-translational modification, illustrating C/EBPγ as a specificity factor in cellular ER stress responses.
DISCUSSION
The 3′-UTR length of mRNA is an important regulatory component in eukaryotic gene expression, and its dynamics are associated with diverse aspects of cellular mechanisms such as cellular signaling and disease pathogenesis (6–9,18–20,22,23,26,36). Thus, profiling the changes of 3′-UTR length is a critical aspect in the understanding of comprehensive gene expression. Multiple workflows adopting high profiling technologies and bioinformatics tools have been developed to catalog the dynamics of 3′-UTR length on a global scale (11–17). However, an exhaustive survey of 3′-UTR APA is still far from complete due to inherent weaknesses of current available profiling methods. For example, several versions of profiling methods were designed to specifically sequence the fragmented poly(A) tail region of mRNAs (19,21,22,26,27,29,43). Despite the advantage of acquiring high resolution peaks of polyadenylation sites transcriptome-wide, these methods suffer from false signals caused by internal priming on A-rich sequences instead of poly(A) tails and frequent unmanageable production of very short reads during the library preparation (11,25). In addition, a variable efficiency of library preparations between samples often complicates the quantitation of APA events. Alternatively, widely conducted RNA-Seq experiments can be used to assess APA events. In this case, a customized or current bioinformatics algorithm is needed to register the pattern of APA events in the RNA-Seq data (11–17). Depending on the parameters and considerations implanted in an algorithm, the outcome of the analysis could vary. For instance, the definition of the actionable proximal PAS in a 3′-UTR could vary by the algorithms, and the resolution on the usage of multiple proximal PASs in a 3′-UTR could be limited by the nature of RNA-Seq read alignments.
Our findings of prevalent 3′-UTR APA events in the Tsc1−/− transcriptome revealed APA-driven biological pathways upon mTOR activation and suggested APA as a critical molecular process coordinating mTOR downstream pathways (36). Thus, the resolution and depth of APA survey is critical in investigating mTOR-regulated pathways. Accordingly, we looked for any combinations of sequencing technologies and bioinformatics tools to help improve the APA survey in the Tsc1−/− transcriptome. One simple solution to this quest was simply combining RNA-Seq and PAS-Seq data. As shown in Figure 1, this approach expanded the number of APA events in the Tsc1−/− transcriptome compared to the APA analysis using only a single method. However, this union method gives equal weight to each profiling method regardless of the quality of the data. In addition, one method does not complement the data analysis of the other method in this approach. In contrast, newly developed IntMAP formulates one unified machine learning framework to integrate the quantitative information from RNA-Seq and PAS-Seq to estimate the expression level of different 3′-UTR isoforms based on a global optimization strategy. IntMAP could amplify the weak signals of 3′-UTR shortening from both data platforms to avoid false negative genes. Thus, this method improves isoform quantification and APA profiling compared to RNA-Seq or PAS-Seq method alone, or the simple union of both methods. In addition, the model can be tailored by the parameter λ to balance the contributions of RNA-Seq and PAS-Seq data. As such, the optimization of λ is critical, and the model can be trained by qRT-PCR validation experiments on a small set of genes (Figure 2(A)). Intriguingly, we noticed that the APA events characterized by IntMAP do not completely encompass the RNA-Seq or PAS-Seq data (Figure 2B). This may reflect the ability of IntMAP to filter out false positives. Some APA events falsely identified by the RNA-Seq method may not contain experimentally proven actionable PASs and thus are eliminated by IntMAP due to the lack of APA evidence in PAS-seq data. Other APA events falsely identified by the PAS-Seq method may come from the internal priming on A-rich regions, which can be eliminated by IntMAP based on insignificant changes in read density before and after the putative poly(A) sites. On top of this, each APA profiling method uses different cut-off values for the statistical significance of data analysis, which could cause an incomplete overlap among the datasets.
Characterization of the 3′-UTR APA events in the Tsc1−/− transcriptome by the RNA-Seq method highlighted the mTOR-regulated ubiquitin-mediated proteolysis (36). Consistent with these findings, the ubiquitin-mediated proteolysis was ranked second among the enriched pathways in the APA events profiled in this study (Figure 3D). Compared to the 27 genes previously identified, we found that 41 genes were enriched in the ubiquitin-mediated proteolysis pathway by APA in this study (Supplementary Table S3). These results again provide a link between the mTOR signaling pathway and ubiquitin-mediated proteolysis. Importantly, in contrast to the previous findings of ubiquitin-mediated proteolysis mainly targeting tumor suppressors or cell cycle regulators, the newly identified APA genes of E2 and E3 ligases regulate cellular pathways of hypoxia, p53, and other signaling cascades (Supplementary Table S3). Thus, our results suggest that the mTOR activation controls diverse cellular pathways through a much broader spectrum of APA-driven selective proteolysis.
Similarly, finding several novel transcription factors whose expressions are driven by 3′-UTR APA also expands the identification of cellular pathways regulated by mTOR-modulated transcription networks. mTOR is known to regulate diverse cellular mechanisms including lipid metabolism and stress responses through transcriptional activation or post-translational modification of transcription factors (30–33). Thus, the molecular basis of C/EBPγ activation through mTOR-driven APA and subsequent regulation of transcription network provide a new mechanistic insight into cellular stress responses at the level of post-transcriptional regulation. Delineation of the 3′-UTR in Cebpg using a luciferase reporter assay revealed that highly conserved immediately upstream 100 nucleotides from the second PAS is critical for the regulation of C/EBPγ protein synthesis (Figure 5F and Supplementary Figure S6E). A search for trans-acting element-binding sites using multiple bioinformatics resources suggested highly conserved multiple miRNAs and the QKI protein as potential interactors (Supplementary Figure S6E). However, the experiments using antisense oligonucleotides (anti-miRs) against these miRNAs did not rescue the luciferase activity (Supplementary Figure S9A). Moreover, the knockdown of Ago1 and Ago2, the components of RISC (RNA-induced silencing complex), or QKI did not rescue the luciferase activity (Supplementary Figure S9B), indicating that the regulation of C/EBPγ protein synthesis through the 3′-UTR is not likely mediated by miRNAs or QKI. Thus, it is possible that unknown RBP(s) that interacts with this highly conserved domain in the 3′-UTR suppresses the translation of C/EBPγ. Numerous 3′-end processing factors are known to play a role in APA (4). Particularly, downregulation of Cpsf5 or Cpsf6 leads to genome-wide APA events (7,29,56). Consistent with these findings, the expression of Cpsf5 and Cpsf6 protein is decreased in Tsc1−/− compared to WT MEFs (Figure 5G) although Cpsf5 mRNA is highly upregulated in Tsc1−/− MEFs (Supplementary Figure S7A). Because Cpsf5 and Cpsf6 protein form a heterodimer and affect each other's stability (7), it is possible that the decrease of Cpsf6 protein by downregulation of Cpsf6 mRNA could be a major reason for the decrease Cpsf5 protein. The mechanistic insight of how Cpsf6 mRNA is downregulated upon the mTOR activation needs further investigations.
C/EBPγ is a transcription factor and is known to activate differential gene expression for many metabolic pathways like glutathione metabolism and ROS metabolism (40). Genome-associated studies on C/EBPγ-binding sites indicated a potential link between C/EBPγ and ER stress responses since C/EBPγ binding is concentrated in regions around transcription start sites of many stress-related genes (40). Accordingly, the expression level of C/EBPγ likely determines the vulnerability of cells to ER stress. Many small molecules are available to induce ER stress in cells via various means (52–54,57). In our data, C/EBPγ did not seem to play a role in cellular response to ER stress that is induced by either the disruption of intracellular calcium homeostasis (by thapsigargin and ionophore A23187) or the interruption of protein transport to the Golgi apparatus (by Brefeldin A) (Figure 6). However, we found that tunicamycin and kifunensine could sensitize Cebpg KD and CebpgΔPAS Tsc1−/− MEFs to cell death (Figure 6). Tunicamycin blocks the biosynthesis of glycoprotein and causes the unfolded protein response in the ER, leading to ER stress (52,53). Kifunensine is a potent inhibitor of α1,2-mannosidase of the ER and inhibits the degradation of misfolded glycoprotein (54,55). Thus, C/EBPγ seems to play a role in the ER stress particularly caused by the cellular mechanisms related to protein glycosylation. Although the mechanistic insights of C/EBPγ’s function in this type of ER stress are elusive, these data implicate the existence of sophisticated transcription networks that regulate cellular responses to ER stress by protein unfolding.
Overall, the data shown in this study showcase IntMAP as a powerful bioinformatics tool for a comprehensive profiling of transcriptome-wide 3′-UTR APA events. As exemplified by the findings of C/EBPγ in ER stress response, newly identified cellular pathways and transcription networks activated by mTOR-driven APA events provide a broader list of downstream target pathways of mTOR signaling cascade. In this context, applications of IntMAP to other biological conditions or disease models will help elucidate new cellular pathways activated by the 3′-UTR APA events. Additionally, IntMAP is a new general approach for flexible integration of RNA-Seq short read alignments with other mRNA quantification platforms to detect gene transcript variances. For example, other than integration of RNA-seq and PAS-seq, IntMAP can also be applied to the integration of RNA-Seq data and Exon-array expressions or NanoString color-barcode counts for isoform detection/quantifications. In these applications, similar qRT-PCR validation experiments can be applied to optimize the tuning parameter λ, which balance the contributions of RNA-Seq and other mRNA quantification platforms.
DATA AVAILABILITY
The accession number for the RNA-seq data in this study is SRP056624. The accession number for the PAS-Seq data in this study is PRJNA436720.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Kwiatkowski at Harvard University for providing us Tsc1−/− MEFs and matching wild type MEFs. We also would like to thank Dr Aaron Goldstrohm and his lab members for providing us materials and technical supports for Renilla luciferase assay.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [1R01GM113952-01A1]; Department of Defense – Congressionally Directed Medical Research Programs [W81XWH-16-1-0135 to J.Y.]. Funding for open access charge: Department of Defense-Congressionally Directed Medical Research Programs.
Conflict of interest statement. None declared.
REFERENCES
- 1. Darnell J.E. Reflections on the history of pre-mRNA processing and highlights of current knowledge: a unified picture. RNA. 2013; 19:443–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bentley D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014; 15:163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manley J.L. Polyadenylation of mRNA precursors. BBA - Gene Struct. Expr. 1988; 950:1–12. [DOI] [PubMed] [Google Scholar]
- 4. Tian B., Manley J.L.. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem. Sci. 2013; 38:312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yeh H.-S., Yong J.. Alternative polyadenylation of mRNAs: 3′-Untranslated region matters in gene expression. Mol. Cells. 2016; 39:281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mayr C., Bartel D.P.. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009; 138:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Masamha C.P., Xia Z., Yang J., Albrecht T.R., Li M., Shyu A.-B., Li W., Wagner E.J.. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. 2014; 510:412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morris A.R., Bos A., Diosdado B., Rooijers K., Elkon R., Bolijn A.S., Carvalho B., Meijer G.A., Agami R.. Alternative cleavage and polyadenylation during colorectal cancer development. Clin. Cancer Res. 2012; 18:5256–5266. [DOI] [PubMed] [Google Scholar]
- 9. Jenal M., Elkon R., Loayza-Puch F., Van Haaften G., Kühn U., Menzies F.M., Vrielink J.A.F.O., Bos A.J., Drost J., Rooijers K. et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell. 2012; 149:538–553. [DOI] [PubMed] [Google Scholar]
- 10. Yeh H.S., Zhang W., Yong J.. Analyses of alternative polyadenylation: from old school biochemistry to high-throughput technologies. BMB Rep. 2017; 50:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xia Z., Donehower L.A., Cooper T.A., Neilson J.R., Wheeler D.A., Wagner E.J., Li W.. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun. 2014; 5:5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang W., Wei Z., Li H.. A change-point model for identifying 3′UTR switching by next-generation RNA sequencing. Bioinformatics. 2014; 30:2162–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grassi E., Mariella E., Lembo A., Molineris I., Provero P.. Roar: detecting alternative polyadenylation with standard mRNA sequencing libraries. BMC Bioinformatics. 2016; 17:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Le Pera L., Mazzapioda M., Tramontano A.. 3USS: A web server for detecting alternative 30UTRs from RNA-seq experiments. Bioinformatics. 2015; 31:1845–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim M.H., You B.H., Nam J.W.. Global estimation of the 3′ untranslated region landscape using RNA sequencing. Methods. 2015; 83:111–117. [DOI] [PubMed] [Google Scholar]
- 16. Birol I., Raymond A., Chiu R., Nip K.M., Jackman S.D., Kreitzman M., Docking T.R., Ennis C.A., Robertson A.G., Karsan A.. Kleat: cleavage site analysis of transcriptomes. Biocomput. 2014; 2015:347–385. [PMC free article] [PubMed] [Google Scholar]
- 17. Shenker S., Miura P., Sanfilippo P., Lai E.C.. IsoSCM: improved and alternative 3′ UTR annotation using multiple change-point inference. RNA. 2015; 21:14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jan C.H., Friedman R.C., Ruby J.G., Bartel D.P.. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature. 2011; 469:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lianoglou S., Garg V., Yang J.L., Leslie C.S., Mayr C.. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 2013; 27:2380–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma L., Pati P.K., Liu M., Li Q.Q., Hunt A.G.. High throughput characterizations of poly(A) site choice in plants. Methods. 2014; 67:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mangone M., Manoharan A.P., Thierry-Mieg D., Thierry-Mieg J., Han T., Mackowiak S.D., Mis E., Zegar C., Gutwein M.R., Khivansara V. et al. The landscape of C. elegans 3 UTRs. Science. 2010; 329:432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mata J. Genome-wide mapping of polyadenylation sites in fission yeast reveals widespread alternative polyadenylation. RNA Biol. 2013; 10:1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ozsolak F., Kapranov P., Foissac S., Kim S.W., Fishilevich E., Monaghan A.P., John B., Milos P.M.. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell. 2010; 143:1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang H., Lim J., Ha M., Kim V.N.. TAIL-seq: Genome-wide determination of poly(A) tail length and 3′ end modifications. Mol. Cell. 2014; 53:1044–1052. [DOI] [PubMed] [Google Scholar]
- 25. Routh A., Ji P., Jaworski E., Xia Z., Li W., Wagner E.J.. Poly(A)-ClickSeq: click-chemistry for next-generation 3′-end sequencing without RNA enrichment or fragmentation. Nucleic Acids Res. 2017; 45:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoque M., Ji Z., Zheng D., Luo W., Li W., You B., Park J.Y., Yehia G., Tian B.. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods. 2012; 10:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zheng D., Liu X., Tian B.. 3′READS+, a sensitive and accurate method for 3′ end sequencing of polyadenylated RNA. RNA. 2016; 22:1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanfilippo P., Miura P., Lai E.C.. Genome-wide profiling of the 3′ ends of polyadenylated RNAs. Methods. 2017; 126:86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martin G., Gruber A.R., Keller W., Zavolan M.. Genome-wide analysis of Pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor i in the regulation of 3′ UTR length. Cell Rep. 2012; 1:753–763. [DOI] [PubMed] [Google Scholar]
- 30. Ben-Sahra I., Manning B.D.. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017; 45:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Laplante M., Sabatini D.M.. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013; 126:1713–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saxton R.A., Sabatini D.M.. mTOR signaling in growth, metabolism, and disease. Cell. 2017; 168:960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aramburu J., Ortells M.C., Tejedor S., Buxadé M., López-Rodríguez C.. Transcriptional regulation of the stress response by mTOR. Sci. Signal. 2014; 7:re2. [DOI] [PubMed] [Google Scholar]
- 34. Appenzeller-Herzog C., Hall M.N.. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012; 22:274–282. [DOI] [PubMed] [Google Scholar]
- 35. Thoreen C.C., Chantranupong L., Keys H.R., Wang T., Gray N.S., Sabatini D.M.. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012; 485:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang J.-W., Zhang W., Yeh H.-S., de Jong E.P., Jun S., Kim K.-H., Bae S.S., Beckman K., Hwang T.H., Kim K.-S. et al. mRNA 3′-UTR shortening is a molecular signature of mTORC1 activation. Nat. Commun. 2015; 6:7218. [DOI] [PubMed] [Google Scholar]
- 37. Parkin S.E., Baer M., Copeland T.D., Schwartz R.C., Johnson P.F.. Regulation of CCAAT/enhancer-binding protein (C/EBP) activator proteins by heterodimerization with C/EBPγ (Ig/EBP). J. Biol. Chem. 2002; 277:23563–23572. [DOI] [PubMed] [Google Scholar]
- 38. Cooper C., Henderson A., Artandi S., Avitahl N., Calame K.. Ig/EBP (C/EBP gamma) is a transdominant negative inhibitor of C/EBP family transcriptional activators. Nucleic Acids Res. 1995; 23:4371–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huggins C.J., Malik R., Lee S., Salotti J., Thomas S., Martin N., Quiñones O.A., Alvord W.G., Olanich M.E., Keller J.R. et al. C/EBPγ suppresses senescence and inflammatory gene expression by heterodimerizing with C/EBPβ. Mol. Cell. Biol. 2013; 33:3242–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huggins C.J., Mayekar M.K., Martin N., Saylor K.L., Gonit M., Jailwala P., Kasoji M., Haines D.C., Quiñones O.A., Johnson P.F.. C/EBPγ is a critical regulator of cellular stress response networks through heterodimerization with ATF4. Mol. Cell. Biol. 2015; 36:693–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kwiatkowski D.J., Zhang H., Bandura J.L., Heiberger K.M., Glogauer M., el-Hashemite N., Onda H.. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum. Mol. Genet. 2002; 11:525–534. [DOI] [PubMed] [Google Scholar]
- 42. Van Etten J., Schagat T.L., Goldstrohm A.C.. A guide to design and optimization of reporter assays for 3′ untranslated region mediated regulation of mammalian messenger RNAs. Methods. 2013; 63:110–118. [DOI] [PubMed] [Google Scholar]
- 43. Shepard P.J., Choi E.-A., Lu J., Flanagan L.A., Hertel K.J., Shi Y.. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA. 2011; 17:761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pachter L. Models for transcript quantification from RNA-Seq. 2011; arXiv:1104.3889v2 [q-bio.GN] [Google Scholar]
- 47. Zhang W., Chang J.W., Lin L., Minn K., Wu B., Chien J., Yong J., Zheng H., Kuang R.. Network-based isoform quantification with RNA-Seq data for cancer transcriptome analysis. PLoS Comput. Biol. 2015; 11:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li B., Ruotti V., Stewart R.M., Thomson J.A., Dewey C.N.. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2009; 26:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xing Y., Yu T., Wu Y.N., Roy M., Kim J., Lee C.. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res. 2006; 34:3150–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grant M., Boyd S.. CVX: Matlab software for disciplined convex programming, version 2.1. 2013; http://cvxr.com/cvx
- 51. Grant M.C., Boyd S.P.. Graph implementations for nonsmooth convex programs. Lect. Notes Control Inf. Sci. 2008; 371:95–110. [Google Scholar]
- 52. Samali A., Fitzgerald U., Deegan S., Gupta S.. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int. J. Cell Biol. 2010; 2010:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Oslowski C.M., Urano F.. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2013; 490:71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tokunaga F., Brostrom C., Koide T., Arvan P.. Endoplasmic reticulum (ER)-associated degradation of misfolded N-linked glycoproteins is suppressed upon inhibition of ER mannosidase I. J. Biol. Chem. 2000; 275:40757–40764. [DOI] [PubMed] [Google Scholar]
- 55. Wang F., Song W., Brancati G., Segatori L.. Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J. Biol. Chem. 2011; 286:43454–43464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gruber A.R., Martin G., Keller W., Zavolan M.. Cleavage factor I m is a key regulator of 3′ UTR length. RNA Biol. 2012; 9:1405–1412. [DOI] [PubMed] [Google Scholar]
- 57. Cao S.S., Kaufman R.J.. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014; 21:396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data in this study is SRP056624. The accession number for the PAS-Seq data in this study is PRJNA436720.