

Abstract
Background
Breast cancer is the most common cancer in women. 12–15% of all tumors are triple-negative breast cancers (TNBC). So far, TNBC has been mainly associated with mutations in BRCA1. The presence of other predisposing genes seems likely since DNA damage repair is a complex process that involves several genes. Therefore we investigated if mutations in other genes are involved in cancer development and whether TNBC is an additional indicator of mutational status besides family history and age of onset.
Methods
We performed a germline panel-based screening of 10 high and low-moderate penetrance breast cancer susceptibility genes (BRCA1, BRCA2, ATM, CDH1, CHEK2, NBN, PALB2, RAD51C, RAD51D and TP53) in 229 consecutive individuals affected with TNBC unselected for age, family history or bilateral disease. Within this cohort we compared the number of mutation carriers fulfilling clinical selection criteria with the total number of carriers identified.
Results
Age at diagnosis ranged from 23 to 80 years with an average age of 50.2 years. In 57 women (24.9%) we detected a pathogenic mutation, with a higher frequency (29.7%) in the group manifesting cancer before 60 years. Deleterious BRCA1 mutations occurred in 14.8% of TNBC patients. These were predominantly recurrent frameshift mutations (24/34, 70.6%). Deleterious BRCA2 mutations occurred in 5.7% of patients, all but one (c.1813dupA) being unique.
While no mutations were found in CDH1 and TP53, 10 mutations were detected in one of the six other predisposition genes. Remarkably, neither of the ATM, RAD51D, CHEK2 and PALB2 mutation carriers had a family history. Furthermore, patients with non-BRCA1/2 mutations were not significantly younger than mutation negative women (p = 0.3341). Most importantly, among the 57 mutation carriers, ten (17.5%) would be missed using current clinical testing criteria including five (8%) with BRCA1/2 mutations.
Conclusions
In summary, our data confirm and expand previous studies of a high frequency of germline mutations in genes associated with ineffective repair of DNA damage in women with TNBCs. Neither age of onset, contralateral disease nor family history were able to discern all mutation positive individuals. Therefore, TNBC should be considered as an additional criterion for panel based genetic testing.
Keywords: Breast cancer, TNBC, Next-generation sequencing, Gene panel, Mutational spectrum
Background
Breast cancer is the most common cancer in women. 12–15% of all tumors lack immunohistochemical expression of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) [1] and are therefore termed triple-negative breast cancers (TNBC). Patients with TNBC are characterized by a high risk of relapse and poor prognosis [2] and are likely to receive chemotherapy since receptor specific therapies are ineffective. TNBC is more common among African Americans and western sub-Saharan Africans compared with White/Caucasian Americans and Europeans [3]. Besides, TNBC disproportionately affects young women [4].
Overall, women are more likely to be tested for breast/ovarian cancer susceptibility genes if they fulfill certain criteria. According to current guidelines, genetic testing in Germany is recommended in BC patients, who have at least a 10% prior probability of carrying a BRCA1/2 mutation based on clinical criteria as age of manifestation, family history and contralateral disease [5] and, only since end of 2016, considering receptor triple negativity before age 50. In the years 2007 and 2008, when gene testing was restricted to BRCA1 and BRCA2 several studies have demonstrated that BRCA1-mutation carriers are more likely to be diagnosed with TNBC than non-carriers [6]. Consequently testing in TNBC patients, if at all performed, was often restricted to BRCA1 only. However, though TNBCs constitute nearly 80% of BRCA1-associated breast cancers [7], BRCA1 mutations have only been found in a subset of TNBC patients [8]. Therefore a BRCA1/2-centered perspective may ignore the significance of other predisposing genes. Their presence seems likely since DNA damage repair by homologous recombination is a complex process that involves several other genes besides BRCA1 and BRCA2.
Recent technological advances such as high-throughput sequencing technologies have enabled cost-effective simultaneous interrogation of multiple risk genes. Though most familial breast cancer studies grouped all cancer subtypes together, few studies like Couch et al. (2015) evaluated genetic predisposition to a specific subtype like TNBC. The identification of mutation carriers is clinically of increased importance. As management conventions [9–11] are becoming established, more breast cancer patients will request testing to get access to specific therapies, regardless of their age and a priori lower risk. This requires appropriate selection criteria for genetic testing.
Therefore, we now investigated all 229 female individuals with TNBC at a single center over a period of 54 months to assess whether a diagnosis of TNBC is an additional indicator for a germline mutation in breast cancer predisposing genes.
Methods
All patients with TNBC, newly diagnosed or in aftercare, treated in the Department of Gynecology and Obstetrics between 01/2012 and 06/2016 were referred for diagnostic purposes to our interdisciplinary outpatient clinic.
Clinical data (including a three-generation pedigree) and informed consent for diagnostic testing were collected. All 229 unrelated females received genetic testing. One hundred thirty-eight patients were tested immediately after TNBC diagnosis. Ninety-one patients were in aftercare during the selection process (> 1–20 years after cancer diagnosis). Two hundred twenty-three women were of German origin (97.4%), four from Russia, one from Hungary and one from Thailand. 110 (48%) women had at least one relative with breast cancer. Family history was defined as at least one affected relative regardless of kinship degree. Cancers occurring through two unaffected females at > 60 years of age were discounted.
ER and PR receptor status was determined by immunohistochemistry and classified as negative if less than 1% of cells showed stained nuclei in tumor cells. HER2 status was considered as negative, if IHC scores were 0 and + 1. IHC scores of + 2 were also classified as negative in case of negative fluorescence in situ hybridization results for HER2. FISH was considered positive if the Her2/CEN 17 ratio was > = 2.0 or the majority of tumors cells showed at least 6 Her2 gene signals.
Genomic DNA was extracted according to standard procedures with an automated chemagic MSM I system (Perkin Elmer, Baesweiler, Germany). A targeted resequencing kit, the TruSight Cancer Sequencing Panel, was used for library preparation and sequencing on a MiSeq platform (Illumina, San Diego, CA, USA). All procedures were performed according to the manufacturers’ instructions. Library preparation with the TruSight Rapid Capture was done using 50 ng of genomic DNA per sample. For sequencing the prepared library was applied to MiSeq Flowcell. Paired sequences obtained were mapped to human genome reference GRCh37/hg19 using BWA-MEM version 0.7.7 [12]. Ten genes (BRCA1/2, ATM, CHEK2, PALB2, RAD51C, RAD51D, NBN, CDH1 and TP53) were analyzed with the SeqNext module of the Sequence Pilot software (JSI medical systems GmbH, Kippenheim, Germany).
For detection of copy number variants (CNVs), the SeqNext CNV analysis module from the Sequence Pilot software package was used. All coding exons of the analyzed genes served both as control and targets using the analysis mode “all versus all”. Identified CNVs were confirmed by multiplex ligation-dependent probe amplification (MLPA) analyses using the appropriate SALSA MLPA kits (BRCA1: P002; RAD51C: P260; CHEK2: P190) (MRC Holland, Amsterdam, The Netherlands) according to the manufacturer’s instructions.
To predict the potential impact of the identified nonsynonymous germline variants on protein function we used 5 web-based algorithms: UMD-Predictor [13], SIFT [14], Polyphen-2 [15], Mutation Taster [16] and additionally, ALIGN-GVGD [17, 18]. Suspected splice site mutations were tested by three different web-based splicing effect prediction tools, Splice Site Prediction by Neural Network [19], NetGene2 Server [20, 21], and Human Splice Finder (HSF 3.0) algorithm [22] to correlate splicing probabilities for wild type and mutated sequences. Frequencies of variants were compared with European-American and African-American control samples from the Exome Variant Server online database [23, 24]) and with 60,706 unrelated individuals sequenced as part of various disease-specific and population genetic studies from the Exome Aggregation Consortium (ExAC) [25] to exclude rare polymorphisms. All p-values were calculated using a Mann-Whitney Rank sum test; boxplots and p-value calculations were done in R version 2.15.3.
Results
We screened a total of 229 TNBC patients regarding mutations in one of the following breast cancer susceptibility genes: BRCA1, BRCA2, ATM, CDH1, CHEK2, NBN, PALB2, RAD51C, RAD51D and TP53. In 57women (24.9%) we detected a pathogenic mutation, with a higher frequency (29.7%) in the group manifesting cancer before 60 years (Table 1).
Table 1.
Age distribution of patients and their mutation status in relation to family history
| Overall | Family History | No family history | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Age | Number | Mutation | % | Number | Mutation | % | Number | Mutation | % |
| < 36 | 40 | 17 | 42.5% | 28 | 16 | 57.2% | 12 | 1 | 8.3% |
| 36–60 | 142 | 37 | 26% | 64 | 29 | 45.3% | 78 | 8 | 10.2% |
| > 60 | 47 | 3 | 6.3% | 10 | 1 | 10% | 37 | 2 | 5.4% |
| overall | 229 | 57 | 24.9% | 102 | 46 | 45% | 127 | 11 | 8.7% |
BRCA1 (34 cases, 56.6%) followed by BRCA2 (13 cases, 21.6%) represented the most frequently mutated genes. While no mutations were found in CDH1 and TP53, 10 mutations (17.5%) were detected in one of the 6 other predisposition genes (Table 2, Table 3). No individual presented more than one mutation.
Table 2.
Mutations in breast cancer risk genes and number of positive diagnostic criteria for gene testing
| Mutated gene | Number of diagnostic criteria | ||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | Total | |
| BRCA1 | 3 | 18 | 11 | 2 | 34 |
| BRCA2 | 2 | 7 | 3 | 1 | 13 |
| CHEK2 | 1 | 1 | 0 | 0 | 2 |
| NBN | 0 | 1 | 0 | 0 | 1 |
| RAD51C | 0 | 2 | 1 | 0 | 3 |
| RAD51D | 1 | 0 | 0 | 0 | 1 |
| PALB2 | 2 | 0 | 0 | 0 | 2 |
| ATM | 1 | 0 | 0 | 0 | 2 |
| Overall | 10 | 29 | 15 | 3 | 57 |
Each column delineates the number (0–3) of fulfilled diagnostic criteria for gene testing (0–3) for each of the risk-genes. One point was given each for diagnosis before age 35 years, for bi- or contralateral breast cancer/ ovarian cancer and for family history. Ten women out of 57 (17.5%) did not fulfill any of these criteria and would thus otherwise go untested
Table 3.
Mutations in BC/OC susceptibility genes identified and clinical presentation in respective patients
| # | Exon | cDNA-change | Predicted AA-change | BIC | GnomAD | Age at diagnosis | Contra-lateral disease | Other cancers | Family BC/OC history |
| BRCA1 | |||||||||
| 1 | 2 | c.68_69delAG | p.(Glu23Valfs*17) | 185delAG | 1.987e−4 | 46 | – | 34 y lymphoepithelial cancer Repeated skin cancer of unknown origin |
BC |
| 2 | 5 | c.181 T > G | p.(Cys61Gly) | C61G | 3.255e−5 | 38 | – | – | BC |
| 3 | 5 | c.181 T > G | p.(Cys61Gly) | C61G | 3.255e−5 | 43 | – | 36 y/ 42 y basalioma | OC |
| 4 | 11 | c.904delG | p.(Ala302Leufs*12) | 1023delG | 0 | 30 | – | – | BC, OC |
| 5 | 11 | c.962G > A | p.(Trp321*) | W321X | 8.237e−6 | 29 | – | – | BC 3rd degree relative |
| 6 | 11 | c.1127delA | p.(Asn376Ilefs*18) | 1246delA | 0 | 23/23 | yes | – | BC |
| 7 | 11 | c.1396delC | p.(Arg466Glyfs*9) | – | 0 | 45 | – | – | BC, OC |
| 8 | 11 | c.1504_1508delTTAAA | p.(Leu502Alafs*2) | 1623del5 | 8.135e−6 | 40/54 | yes | – | BC, OC |
| 9 | 11 | c.1504_1508delTTAAA | p.(Leu502Alafs*2) | 1623del5 | 8.135e−6 | 54 | – | – | BC, OC |
| 10 | 11 | c.1916 T > A | p.(Leu639*) | L639X | 0 | 34 | – | – | BC, OC |
| 11 | 11 | c.1916 T > A | p.(Leu639*) | L639X | 0 | 42 | – | – | BC, OC |
| 12 | 11 | c.2411_2412delAG | p.(Gln804Leufs*5) | 2530delAG | 0 | 37 | – | – | – |
| 13 | 11 | c.2411_2412delAG | p.(Gln804Leufs*5) | 2530delAG | 0 | 33 | – | – | BC, OC |
| 14 | 11 | c.2411_2412delAG | p.(Gln804Leufs*5) | 2530delAG | 0 | 33 | – | – | – |
| 15 | 11 | c.2411_2412delAG | p.(Gln804Leufs*5) | 2530delAG | 0 | 45/52 | yes | – | BC |
| 16 | 11 | c.2411_2412delAG | p.(Gln804Leufs*5) | 2530delAG | 0 | 53 | – | – | BC |
| 17 | 11 | c.3481_3491del | p.(Glu1161Phefs*3) | 3600del11 | 0 | 40 | – | – | BC, OC |
| 18 | 11 | c.3481_3491del | p.(Glu1161Phefs*3) | 3600del11 | 0 | 46 | – | – | BC, unk. Abd. canc. |
| 19 | 11 | c.3481_3491del | p.(Glu1161Phefs*3) | 3600del11 | 0 | 65 | – | – | – |
| 20 | 11 | c.3627dupA | p.(Glu1210Asnfs*11) | 3746ins A | 0 | 59 | – | – | BC |
| 21 | 11 | c.3700_3704delGTAAA | p.(Val1234Glnfs*8) | 3819del5 | 0 | 43/47 | yes | – | BC |
| 22 | 11 | c.3700_3704delGTAAA | p.(Val1234Glnfs*8) | 3819del5 | 0 | 50 | – | – | BC, OC |
| 23 | 11 | c.4035delA | p.(Glu1346Lysfs*20) | 4154delA | 4.066e−6 | 43 | – | – | BC |
| 24 | 11 | c.4676-1G > A | IVS15-1G > A | 0 | 30/30 | yes | – | BC 4th degree relative | |
| 25 | 13–15 | Deletion exon 13–15 | – | 0 | 58 | – | – | BC | |
| 26 | 20 | c.5194_5277del | p.(His1732_Lys1759del) | Exon20del | 0 | 32/36 | yes | – | BC |
| 27 | 20 | c.5266dupC | p.(Gln1756Profs*74) | 5382insC | 0 | 33 | – | – | – |
| 28 | 20 | c.5266dupC | p.(Gln1756Profs*74) | 5382insC | 0 | 33 | – | – | BC, OC |
| 29 | 20 | c.5266dupC | p.(Gln1756Profs*74) | 5382insC | 0 | 30/48 | yes | – | – |
| 30 | 20 | c.5266dupC | p.(Gln1756Profs*74) | 5382insC | 0 | 35 | – | – | BC, unk. Abd. canc. |
| 31 | 20 | c.5266dupC | p.(Gln1756Profs*74) | 5382insC | 0 | 39 | – | – | BC, unk. Abd. canc. |
| 32 | 21–24 | c.5278-?_5592 +?del | Deletion exon 21–24 | – | 0 | 33 | – | – | BC, unk. Abd. canc. |
| 33 | 21 | c.5291 T > C | p.(Leu1764Pro) | L1764P | 0 | 42 | – | – | BC |
| 34 | 24 | c.5492delC | p.(Pro1831Leufs*3) | 6511delC | 0 | 45 | – | – | BC |
| BRCA2 | |||||||||
| 1 | 5 | c.516 + 2 T > A | IVS6 + 2 T > A | 0 | 41 | – | – | BC | |
| 2 | 10 | c.1813dupA | p.(Ile605Argfs*9) | 2041ins A | 0 | 29 | – | – | BC |
| 3 | 10 | c.1813dupA | p.(Ile605Argfs*9) | 2041ins A | 0 | 29 | – | – | BC |
| 4 | 11 | c.2244_2245del | p.(Tyr748*) | 2472delCA | 0 | 31 | – | – | – |
| 5 | 11 | c.5303_5304delTT | p.(Leu1768Argfs*5) | 5531delTT | 4.075e−6 | 42 | – | – | OC |
| 6 | 11 | c.5616_5620delAGTAA | p.(Lys1872Asnfs*2) | 5844del5 | 4.126e−6 | 42 | – | – | – |
| 7 | 11 | c.6486_6489del4 | p.(Lys2162Asnfs*5) | 6714del4 | 8.785e−6 | 29/33 | yes | – | BC 3rd degree relative |
| 8 | 15 | c.7180A > T | p.(Arg2394*) | R2394X | 0 | 51/64 | yes | – | BC |
| 9 | 15 | c.7617 + 2 T > G | 7829 + 2 T > G | 0 | 53 | – | – | BC | |
| 10 | 17 | c.7878G > C | p.Trp2626Cys | W2626C | 4.064e−6 | 63 | – | – | BC |
| 11 | 20 | c.8560delT | p.(Tyr2854Metfs*9) | 8788delT | 0 | 38/52 | yes | – | BC |
| 12 | 22 | c.8755-1G > A | IVS21-1G > A | 0 | 68 | – | – | BC, OC | |
| 13 | 23 | c.8992_9025del34 | p.(Ser2998Ilefs*19) | – | 0 | 60 | – | – | – |
| # | Exon | cDNA-change | Predicted AA-change | ClinVar (number of reported individuals) | GnomAD frequency | Age at diagnosis | Contra-lateral disease | Other cancers | Family BC/OC history |
| RAD51C | |||||||||
| 1 | 7 | c.955C > T | p.(Arg319*) | Pathogenic (5) | 8.128e−6 | 30 | – | – | BC |
| 2 | 8 | c.1026 + 5_ + 7delGTA | Likely pathogenic (5) Pathogenic (1) |
0 | 43 | – | 64 y non-Hodgkin-lymphoma | BC | |
| 3 | 8 | c.1026 + 5_ + 7delGTA | Likely pathogenic (5) Pathogenic (1) |
0 | 51 | – | – | BC, unk. Abd. canc. | |
| RAD51D | |||||||||
| 1 | 7 | c.577-2A > G | Likely pathogenic (1) | 0 | 52 | – | – | – | |
| CHEK2 | |||||||||
| 1 | 4 | c.470 T > C | p.(Ile157Thr) | Likely pathogenic (4) Pathogenic (7) Unknown (3) |
5.061e−3 | 50 | – | – | BC 3rd degree relative |
| 2 | 4 | c.470 T > C | p.(Ile157Thr) | Likely pathogenic (4) Pathogenic (7) Unknown (3) |
5.061e−3 | 47 | yes | – | – |
| NBN | |||||||||
| 1 | 6 | c.657_661del5p | p.(Lys219Asnfs*16) | Pathogenic (12) Risk factor (1) |
1.988e−4 | 56 | – | 66 y OC | BC |
| ATM | |||||||||
| 1 | 26 | c.3802delG | p.(Val1268Terfs*1) | Pathogenic (5) | 3.972e−5 | 40 | – | – | – |
| PALB2 | |||||||||
| 1 | 4 | c.1227_1231delTGTTA | p.(Tyr409*) | – | 0 | 44 | – | 25 y melanoma | BC 3rd degree relative |
| 2 | 11 | c.3165C > A | p.(Tyr1055*) | Pathogenic (1) | 0 | 47 | – | – | – |
BC breast cancer, OC ovarian cancer, unk. Abd. canc abdominal cancer of unknown origin
Deleterious BRCA1 mutations in 14.8% of TNBC patients (Table 2, Table 3). These were predominantly frameshift mutations (24/34, 70.6%). The most frequent mutations both among them and in total were the founder mutations c.5266dupC and c.2411_2412delAG. Of the 20 different mutations identified 17 were previously reported as disease causing (International Database of Breast Cancer, https://research.nhgri.nih.gov/bic/), the remaining three comprised large deletions affecting exons 13–15 and 21–24 respectively and the frameshift mutation c.1396delC.
Deleterious BRCA2 mutations occurred in 5.7% of patients, all but one (c.1813dupA) being unique, including 11 truncating mutations (seven frameshift, three splice mutations and one nonsense) and one missense mutation (c.7878G > C). Despite the frameshift mutation c.8992_9025del34 all other alterations were listed in the International Database of Breast Cancer.
Mutations in non-BRCA1/2 predisposition genes were identified in 22% of mutation carriers (13/60) (Table 2, Table 3). In total, mutations in NBN and RAD51D were found in 3 individuals each, mutations in ATM, CHEK2 and PALB2 in 2 individuals each and one mutation in RAD51C (Table 2). Overall, 13 out 229 tested individuals (5.7%) carried a non-BRCA1/2 mutation.
Of the patients examined, 79.5% were younger than 60 years. Only 3 mutation carriers developed cancer beyond 60 years of age, which represents 5% of mutation carriers and 6.3% of their age group. Almost one third of all deleterious mutations (29.8%) were detected in very young women aged 35 years or less (Table 1). The median age at diagnosis was significantly younger for BRCA1 (40 years) and BRCA2 (41.5 years) carriers compared to patients without a mutation (50 years, p = 2.286e-05; Mann-Whitney) or compared to non-BRCA1/2 mutation carriers (50 years, p = 0.3341) (Fig. 1). In contrast, patients with non-BRCA1/2 mutations were not significantly younger than mutation negative women (p = 0.5288) (Fig. 1).
Fig. 1.
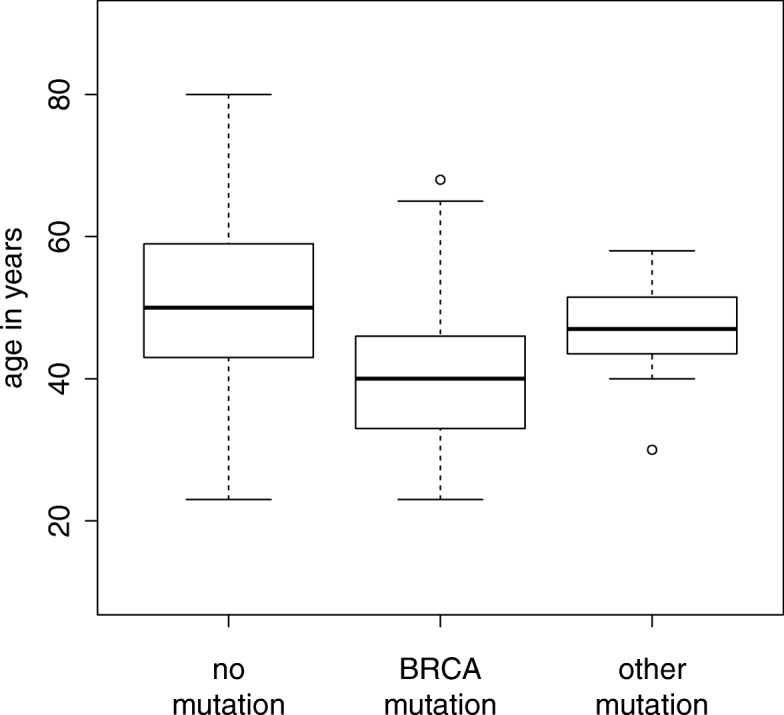
Age at first manifestation in relation to the mutation status. Box-plot diagram of age at first manifestation of breast cancer reveals that women with a BRCA1/2 mutation manifest significantly earlier than those without a mutation or a mutation in one of the other genes
Eighteen individuals of the entire group developed bi- or contralateral disease at latest 18 years after initial diagnosis. While in the unilateral group only 21.8% (n = 46) had a mutation, the majority of the bi- or contralateral affected was mutation positive (n = 11; 61.1%) 12 women had a family history (66.7%), but interestingly only nine of them were mutation positive. Overall, women with bi- or contralateral disease developed cancer at significantly younger age with a difference of median age of 8.5 years compared to those with unilateral disease (p = 0.01215; Mann-Whitney) (Fig. 2).
Fig. 2.
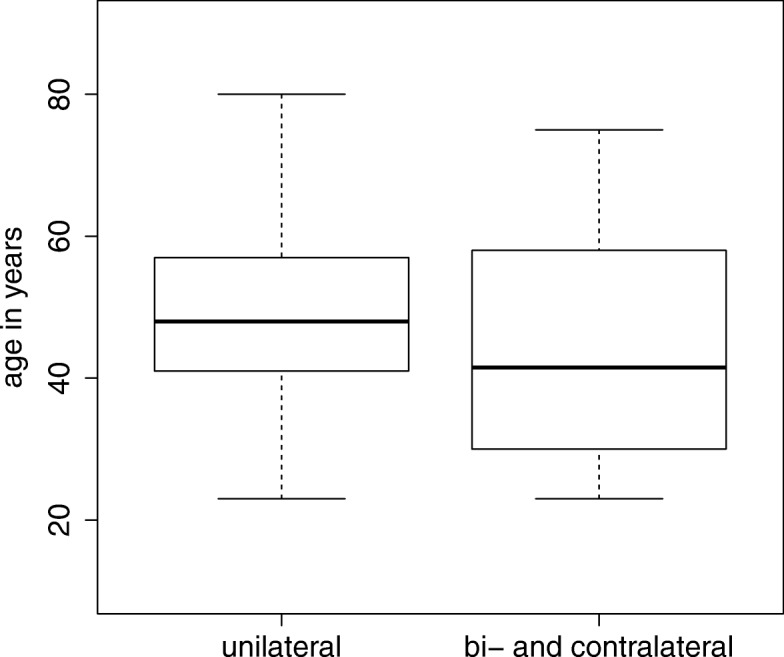
Age at first manifestation in dependence of unilateral or bi- and contralateral manifestation. Box-plot diagram of age at first manifestation of breast cancer reveals that women with bi-or contralateral manifestation (n = 18) manifest earlier than those with unilateral disease (n = 211)
A considerable number of women with TNBC had a family history (44.5%) of whom a mutation was found in 45% (Table 1). Among the BRCA1/2 mutation carriers, even 86.7% had at least one affected relative. Interestingly, family history had an independent influence on age at diagnosis (Fig. 3). Taken as a whole, women with family history had a median age at diagnosis 6 years earlier than those without (p = 0.00057). This difference was lost in mutation carriers (Fig. 3, middle) while it remained in cases without a detected mutation. Remarkably, neither of the ATM, RAD51D, CHEK2 and PALB2 mutation carriers had a family history, when considering first and second degree family members.
Fig. 3.

Age at first manifestation in relation to mutation status and family history. Box-plot diagram of age at first manifestation reveals that overall women with a positive family history manifest earlier than those without and that mutation positive women manifest earlier when compared to mutation negative women of their respective family history group
Discussion
We identified a germline mutation in 24.9% of 229 TNBC patients. 17.5% of individuals were diagnosed before age 36 and 79.4% before the age of 60 (Table 1). As reported previously, the mutation detection rate decreased with higher age at diagnosis (Sharma et al., 2014). More than 95% of patients with a germline mutation were diagnosed with breast cancer before age 60. Among the 47 cases with age > 60, only three (6.3%) were mutation positive.
Overall, we found a significant earlier disease manifestation in those women who have a family history (mean 45 vs. 51 years; p = 0.00057) (Fig. 3, left). Also 29% of women (37/127) without family history were aged > 60 years as compared to only 9.8% (10/102) of women with a family history (Table 1). Interestingly, this difference is lost in cases with a detected mutation (Fig. 3, middle) while it remains in those without a mutation in any of the investigated genes (Fig. 3 right). This is in agreement with the hypothesis that other genetic factors at single or multiple loci segregating in mutation-negative families predispose to BC/OC [26, 27]. In the mutation-positive individuals the penetrance of the identified mutation apparently overrides this background effect.
Regarding the mutational spectrum, BRCA1 (34 cases, 56.6%) followed by BRCA2 (13 cases, 21.6%) represented the most frequently mutated genes. The biological reason for the BRCA1 preponderance remains unclear, given that both genes collaborate in the homology-directed DNA repair pathway, but BRCA1 may exert a particular role in hormone receptor expression [28].
Most BRCA1/2 mutations were truncating, while only four individuals carried a missense mutation (Table 2 and Table 3). By comprehensively investigating genomic alterations using MLPA we found a deletion in an additional 1.3%. Altogether, we identified mutations in 20.5% of individuals, a higher incidence than the 14.6% reported by the largest study to date [29], but in line with other German studies reporting mutations in 21% [30] and 17.5% [28] respectively. In all above mentioned studies large genomic deletions were not investigated. Differences in ancestral background and the use of different genotyping methods may have further contributed to deviating BRCA1/2 mutation frequencies. Nevertheless, in all studies the prevalence of BRCA mutations exceeded 10%, a number often used as an a priori probability threshold in current testing guidelines.
The incidence of mutations varies widely among different populations; some present a wide spectrum of different mutations, while in some particular ethnic groups specific mutations show a higher frequency probably due to founder effects [31]. Three BRCA1 mutations identified in 34 women (Table 3) were particularly frequent indicating such founder effects. Mutation c.3481_3491del, although found in many geographical areas, is common in France where it accounts for up to 37% of all BRCA1/2 mutations [32]. The relatively high frequency in our study group may be related to the arrival of large numbers of French Huguenot refugees in the area in the late seventeenth century. Similarly, a second mutation, c.2411_2412delAG, is much more frequent in our study group than in patients from all across Germany [5]). Finally, the mutation c.5266dupC is a known founder mutation and not only the most common BRCA1 alteration in the German population [5] but also frequently found in multiple, apparently diverse populations [33]. In contrast, for BRCA2 all but one mutation (c.1813dupA) were unique, in agreement with previous studies [5].
DNA damage repair involves interactions of several specific proteins to restore genomic integrity. Therefore we extended our study also to the respective genes associated with low-moderate penetrance (Table 2) and identified a deleterious germline mutation in six out of eight genes in 10 women (4.3%). From these, one variant (CHEK2 p.(Ile157Thr) is currently classified as mutation, although we would suggest to reclassify it as a low penetrance risk factor due to its high frequency in large variant databases. This variant is described as associated with BC [34, 35], was found in 0.5% of Europeans in GnomAD [36, 37] (Table 3).
Arguably, CHEK2 is one of the most important breast cancer susceptibility genes after BRCA1/BRCA2. However, besides the variant c.470 T > C no other CHEK2 alteration was identified, suggesting that variants in this gene are not frequently associated with TNBC. Accordingly, on the basis of a study of 47 early-onset CHEK2-positive patients, it was concluded that CHEK2-positive patients are equally likely to be estrogen receptor (ER) positive as are patients with nonhereditary disease (Cibulsky et al., 2009). The same is true regarding CDH1, associated with lobular BC and gastric cancer [38, 39].
Besides CHEK2, mutations were identified in NBN, RAD51C, RAD51D, PALB2 and ATM. Mutations in RAD51D were mainly associated with ovarian cancer. However, it was suggested that mutations in RAD51D could confer a risk of TNBC [40]. PALB2 is a BRCA2-interacting protein that is crucial for key BRCA2 genome caretaker functions. Nevertheless, recent studies have shown that PALB2 also interacts with BRCA1 [41, 42]. Overrepresentation of triple negative status in PALB2-related breast cancers was suggested in studies performed in European cohorts [43, 44].
Not unexpectedly for low-moderate penetrance genes, the majority of carriers had no cancer family history. Median age at disease onset was also comparable to that of women without a mutation (Fig. 1). While 91% of women with a BRCA1/2 mutation show additional clinical signs associated with positive mutation status e.g. age of onset < 50 years, contralateral disease or a positive family history, none of the women with a RAD51D, PALB2 or ATM mutation showed any of these signs (Table 2). They would thus have been escaped genetic testing when relying only on criteria developed for high-penetrance genes such as BRCA1/2. When grouping patients by criteria used for assessing test eligibility, 10 of the women aged > 35 did not fulfil any of them (Table 2). Thus neither age of onset < 50 years, contralateral disease nor family history are able to discern all mutation positive individuals, suggesting that TNBC should be considered as an independent criterion for genetic testing.
Besides family history, age at diagnosis and mutations in specific genes, hormone receptor status is considered as a risk factor of developing a new primary breast cancer in the contralateral breast [45, 46]. In our study, 18 women had bi- or contralateral disease in the further course. Of these, 10 women carried a BRCA1/2 mutation and one woman the CHEK2 variant p.(Ile157Thr). Thus, 11 (61.1%) were mutation positive, a much higher frequency when compared to 46/211 (21.8%) women with unilateral disease. On the other hand, in the mutation-negative fraction only 7 out of 169 (4.15%) patients developed a second breast cancer indicating a small risk after excluding mutations in the known genes. Women with bi- or contralateral disease were significantly younger (median 8.5 years) when compared to those with unilateral disease (Fig. 2). Of the 6 women without a mutation, 3 had a family history, suggesting an underlying genetic predisposition. It is possible that a mutation outside the coding area or in a gene not covered by this investigation is predisposing in these individuals.
Conclusion
In summary, our data confirm and expand previous studies of a high frequency of germline mutations in genes known to be associated with ineffective repair of DNA damage by homologous recombination in women with TNBCs. Many of these mutations would be missed using current restrictive testing criteria. Therefore, gene panel based mutation testing should be offered to all women diagnosed with TNBC, irrespective of age or family history.
Furthermore, genetic testing has become a compelling predictive tool, as advanced targeted therapeutic agents, such as poly(ADP-ribose) polymerase (PARP) inhibitors emerge, that selectively induce synthetic lethality in tumor cells deficient in homologous recombination repair [47]. Identifying mutations in genes associated with homologous recombination may expand the number of tumors eligible for PARP inhibitors. Furthermore, presence of such mutations may allow dosage reduction of chemotherapeutic agents e.g. platinum treatment, thereby minimizing the risk of associated severe hematologic toxicities [48]. Moreover, once the disease causing mutation is identified, predictive testing can also be offered to all adult family members and where appropriate, risk-reducing preventive medical interventions [49].
Acknowledgements
We thank Leonora Klassen, Nicole Kretschmann, Andrea Eberwein and Judith Zieroff for expert technical assistance.
Availability of data and materials
All data analysed during this study are included in this published article.
Authors’ contributions
JH contributed to the design of the study, collected and interpreted the data, and drafted the manuscript. GV participated in collecting the data and made important suggestions for manuscript revision. SU performed statistical analysis of the data. MW participated in data collection and made important suggestions for manuscript revision. CK participated in experiment design, experiment execution, data collection and interpretation. PAF participated in experiment design and made important suggestions for manuscript revision. CTT contributed to the interpretation of data. AH provided clinicopathologic information. MWB participated in experiment design and made important suggestions for manuscript revision. MPL participated in collecting data and made important suggestions for manuscript revision. AR contributed to the conception and design of the study, data interpretation and drafting of the manuscript. All authors read and approved the final manuscript.
Ethics approval
All procedures were in accordance with the ethical standards of the FAU Erlangen-Nürnberg and the Helsinki Declaration. All analyses beyond diagnostic procedures were part of the study “Aufklärung genetischer Ursachen für familiären Brust- und Eierstockkrebs” approved by the ethics committee of the Friedrich-Alexander-Universität Erlangen-Nürnberg (Ethik-Kommission der FAU, No. 46_15 B). Written informed consent for diagnostic procedure (§3 Nr. 7 GenDG) in accordance with the Gendiagnostik-Kommission (GEKO, https://www.rki.de/DE/Content/Kommissionen/GendiagnostikKommission/GEKO_inhalt.html) of the Bundesministerium für Gesundheit of the German Government was obtained from all participants in this study.
Consent for publication
Not applicable.
Competing interests
Peter A. Fasching received honoraria from Novartis, Pfizer, Roche and Celgene. Peter A. Fasching’s institution received research funding from Novartis. All other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Juliane Hoyer, Phone: +49 9131 8522318, Email: Juliane.Hoyer@uk-erlangen.de.
Georgia Vasileiou, Email: Georgia.Vasileiou@uk-erlangen.de.
Steffen Uebe, Email: Steffen.Uebe@uk-erlangen.de.
Marius Wunderle, Email: Marius.Wunderle@uk-erlangen.de.
Cornelia Kraus, Email: Cornelia.Kraus@uk-erlangen.de.
Peter A. Fasching, Email: Peter.Fasching@uk-erlangen.de
Christian T. Thiel, Email: Christian.Thiel@uk-erlangen.de
Arndt Hartmann, Email: Arndt.Hartmann@uk-erlangen.de.
Matthias W. Beckmann, Email: Matthias.Beckmann@uk-erlangen.de
Michael P. Lux, Email: Michael.Lux@uk-erlangen.de
André Reis, Email: Andre.Reis@uk-erlangen.de.
References
- 1.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Zaky SS, Lund M, May KA, Godette KD, Beitler JJ, Holmes LR, O'Regan RM, Yu ES, Yu DS, Landry JC. The negative effect of triple-negative breast cancer on outcome after breast-conserving therapy. Ann Surg Oncol. 2011;18(10):2858–2865. doi: 10.1245/s10434-011-1669-4. [DOI] [PubMed] [Google Scholar]
- 3.Jiagge E, Jibril AS, Chitale D, Bensenhaver JM, Awuah B, Hoenerhoff M, Adjei E, Bekele M, Abebe E, Nathanson SD, et al. Comparative analysis of breast Cancer phenotypes in African American, white American, and west versus east African patients: correlation between African ancestry and triple-negative breast Cancer. Ann Surg Oncol. 2016;23(12):3843–9. [DOI] [PubMed]
- 4.Turkman YE, Kennedy HP, Harris LN, Knobf MT. "An addendum to breast cancer": the triple negative experience. Support Care Cancer. 2016;24(9):3715–3721. doi: 10.1007/s00520-016-3184-4. [DOI] [PubMed] [Google Scholar]
- 5.Meindl A. Comprehensive analysis of 989 patients with breast or ovarian cancer provides BRCA1 and BRCA2 mutation profiles and frequencies for the German population. Int J Cancer. 2002;97(4):472–480. doi: 10.1002/ijc.1626. [DOI] [PubMed] [Google Scholar]
- 6.Comen EA, Kirchhoff T, Balistreri L, Hansen J, Kosarin K, Offit K, Robson ME. Prevalence of BRCA1 and BRCA2 mutations in Jewish women with triple negative breast cancer. J Clin Oncol. 2008;26(No15S (May 20 Supplement)):22002. doi: 10.1200/jco.2008.26.15_suppl.22002. [DOI] [Google Scholar]
- 7.Domagala P, Huzarski T, Lubinski J, Gugala K, Domagala W. Immunophenotypic predictive profiling of BRCA1-associated breast cancer. Virchows Archiv. 2011;458(1):55–64. doi: 10.1007/s00428-010-0988-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma P, Klemp JR, Kimler BF, Mahnken JD, Geier LJ, Khan QJ, Elia M, Connor CS, McGinness MK, Mammen JM, et al. Germline BRCA mutation evaluation in a prospective triple-negative breast cancer registry: implications for hereditary breast and/or ovarian cancer syndrome testing. Breast Cancer Res Treat. 2014;145(3):707–714. doi: 10.1007/s10549-014-2980-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robson M, Offit K. Clinical practice. Management of an inherited predisposition to breast cancer. N Engl J Med. 2007;357(2):154–162. doi: 10.1056/NEJMcp071286. [DOI] [PubMed] [Google Scholar]
- 10.Sung JS, Stamler S, Brooks J, Kaplan J, Huang T, Dershaw DD, Lee CH, Morris EA, Comstock CE. Breast cancers detected at screening MR imaging and mammography in patients at high risk: method of detection reflects tumor Histopathologic results. Radiology. 2016;280(3):716–722. doi: 10.1148/radiol.2016151419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villani Anita, Shore Ari, Wasserman Jonathan D, Stephens Derek, Kim Raymond H, Druker Harriet, Gallinger Bailey, Naumer Anne, Kohlmann Wendy, Novokmet Ana, Tabori Uri, Tijerin Marta, Greer Mary-Louise C, Finlay Jonathan L, Schiffman Joshua D, Malkin David. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. The Lancet Oncology. 2016;17(9):1295–1305. doi: 10.1016/S1470-2045(16)30249-2. [DOI] [PubMed] [Google Scholar]
- 12.Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30(20):2843–2851. doi: 10.1093/bioinformatics/btu356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salgado D, Desvignes JP, Rai G, Blanchard A, Miltgen M, Pinard A, Levy N, Collod-Beroud G, Beroud C. UMD-predictor: a high-throughput sequencing compliant system for pathogenicity prediction of any human cDNA substitution. Hum Mutat. 2016;37(5):439–446. doi: 10.1002/humu.22965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 15.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 17.Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43(4):295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34(5):1317–1325. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in genie. J Comput Biol. 1997;4(3):311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 20.Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220(1):49–65. doi: 10.1016/0022-2836(91)90380-O. [DOI] [PubMed] [Google Scholar]
- 21.Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24(17):3439–3452. doi: 10.1093/nar/24.17.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Backe J, Hofferbert S, Skawran B, Dork T, Stuhrmann M, Karstens JH, Untch M, Meindl A, Burgemeister R, Chang-Claude J, et al. Frequency of BRCA1 mutation 5382insC in German breast cancer patients. Gynecol Oncol. 1999;72(3):402–406. doi: 10.1006/gyno.1998.5270. [DOI] [PubMed] [Google Scholar]
- 24.Backe J, Mulfinger L. Possibilities of examination of familial breast cancers and ovarian cancers. Use of molecular-genetic analysis of the BRCA1 gene and the BRCA2 gene. Krankenpflege Journal. 1996;34(10):440–445. [PubMed] [Google Scholar]
- 25.Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org) June, 2016.
- 26.Ripperger T, Gadzicki D, Meindl A, Schlegelberger B. Breast cancer susceptibility: current knowledge and implications for genetic counselling. Eur J Hum Genet. 2009;17(6):722–731. doi: 10.1038/ejhg.2008.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turnbull C, Rahman N. Genetic predisposition to breast cancer: past, present, and future. Annu Rev Genomics Hum Genet. 2008;9:321–345. doi: 10.1146/annurev.genom.9.081307.164339. [DOI] [PubMed] [Google Scholar]
- 28.Pern F, Bogdanova N, Schurmann P, Lin M, Ay A, Langer F, Hillemanns P, Christiansen H, Park-Simon TW, Dork T. Mutation analysis of BRCA1, BRCA2, PALB2 and BRD7 in a hospital-based series of German patients with triple-negative breast cancer. PLoS One. 2012;7(10):e47993. doi: 10.1371/journal.pone.0047993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, Olson JE, Godwin AK, Pankratz VS, Olswold C, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 2015;33(4):304–311. doi: 10.1200/JCO.2014.57.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muendlein A, Rohde BH, Gasser K, Haid A, Rauch S, Kinz E, Drexel H, Hofmann W, Schindler V, Kapoor R, et al. Evaluation of BRCA1/2 mutational status among German and Austrian women with triple-negative breast cancer. J Cancer Res Clin Oncol. 2015;141(11):2005–2012. doi: 10.1007/s00432-015-1986-2. [DOI] [PubMed] [Google Scholar]
- 31.Ferla R, Calo V, Cascio S, Rinaldi G, Badalamenti G, Carreca I, Surmacz E, Colucci G, Bazan V, Russo A. Founder mutations in BRCA1 and BRCA2 genes. Ann Oncol. 2007;18(Suppl 6):vi93–vi98. doi: 10.1093/annonc/mdm234. [DOI] [PubMed] [Google Scholar]
- 32.Fricker JP, Muller D, Cutuli B, Rodier JF, Janser JC, Jung GM, Mors R, Petit T, Haegele P, Abecassis J. Germ-line mutations of the BRCA1 gene in northeastern France. Bull Cancer. 2000;87(10):739–744. [PubMed] [Google Scholar]
- 33.Szabo CI, King MC. Population genetics of BRCA1 and BRCA2. Am J Hum Genet. 1997;60(5):1013–1020. [PMC free article] [PubMed] [Google Scholar]
- 34.Han FF, Guo CL, Liu LH. The effect of CHEK2 variant I157T on cancer susceptibility: evidence from a meta-analysis. DNA Cell Biol. 2013;32(6):329–335. doi: 10.1089/dna.2013.1970. [DOI] [PubMed] [Google Scholar]
- 35.Gao P, Ma N, Li M, Tian QB, Liu DW. Functional variants in NBS1 and cancer risk: evidence from a meta-analysis of 60 publications with 111 individual studies. Mutagenesis. 2013;28(6):683–697. doi: 10.1093/mutage/get048. [DOI] [PubMed] [Google Scholar]
- 36.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.[genome Aggregation Database (gnomAD) browser, http://gnomad.broadinstitute.org].
- 38.Masciari S, Larsson N, Senz J, Boyd N, Kaurah P, Kandel MJ, Harris LN, Pinheiro HC, Troussard A, Miron P, et al. Germline E-cadherin mutations in familial lobular breast cancer. J Med Genet. 2007;44(11):726–731. doi: 10.1136/jmg.2007.051268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suriano G, Yew S, Ferreira P, Senz J, Kaurah P, Ford JM, Longacre TA, Norton JA, Chun N, Young S, et al. Characterization of a recurrent germ line mutation of the E-cadherin gene: implications for genetic testing and clinical management. Clin Cancer Res. 2005;11(15):5401–5409. doi: 10.1158/1078-0432.CCR-05-0247. [DOI] [PubMed] [Google Scholar]
- 40.Ollier M, Radosevic-Robin N, Kwiatkowski F, Ponelle F, Viala S, Privat M, Uhrhammer N, Bernard-Gallon D, Penault-Llorca F, Bignon YJ, et al. DNA repair genes implicated in triple negative familial non-BRCA1/2 breast cancer predisposition. Am J Cancer Res. 2015;5(7):2113–2126. [PMC free article] [PubMed] [Google Scholar]
- 41.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A. 2009;106(17):7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19(6):524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dansonka-Mieszkowska A, Kluska A, Moes J, Dabrowska M, Nowakowska D, Niwinska A, Derlatka P, Cendrowski K, Kupryjanczyk J. A novel germline PALB2 deletion in polish breast and ovarian cancer patients. BMC Med Genet. 2010;11:20. doi: 10.1186/1471-2350-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heikkinen T, Karkkainen H, Aaltonen K, Milne RL, Heikkila P, Aittomaki K, Blomqvist C, Nevanlinna H. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res. 2009;15(9):3214–3222. doi: 10.1158/1078-0432.CCR-08-3128. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Thompson W, Semenciw R, Mao Y. Epidemiology of contralateral breast cancer. Cancer Epidemiol Biomarkers Prev. 1999;8(10):855–861. [PubMed] [Google Scholar]
- 46.Malone KE, Begg CB, Haile RW, Borg A, Concannon P, Tellhed L, Xue S, Teraoka S, Bernstein L, Capanu M, et al. Population-based study of the risk of second primary contralateral breast cancer associated with carrying a mutation in BRCA1 or BRCA2. J Clin Oncol. 2010;28(14):2404–2410. doi: 10.1200/JCO.2009.24.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 48.Domagala P, Jakubowska A, Jaworska-Bieniek K, Kaczmarek K, Durda K, Kurlapska A, Cybulski C, Lubinski J. Prevalence of Germline mutations in genes engaged in DNA damage repair by homologous recombination in patients with triple-negative and hereditary non-triple-negative breast cancers. PLoS One. 2015;10(6):e0130393. doi: 10.1371/journal.pone.0130393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kauff ND, Domchek SM, Friebel TM, Robson ME, Lee J, Garber JE, Isaacs C, Evans DG, Lynch H, Eeles RA, et al. Risk-reducing salpingo-oophorectomy for the prevention of BRCA1- and BRCA2-associated breast and gynecologic cancer: a multicenter, prospective study. J Clin Oncol. 2008;26(8):1331–1337. doi: 10.1200/JCO.2007.13.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data analysed during this study are included in this published article.