

Abstract
Atypical teratoid rhabdoid tumor (ATRT) is a rare, highly malignant tumor of the central nervous system, commonly affecting children below 3 years of age, with around 300 cases reported in the literature. Suprasellar area is a very rare location for such tumor in the pediatric population, with technical difficulties in complete excision. Third ventricular ATRT is very rare. Here, we report the case of a 2-year-old male child who presented with lethargy and vomiting. He had features of raised intracranial pressure with reduced vision in both eyes. Magnetic resonance imaging of the brain revealed a heterogeneously enhancing lobulated giant lesion in the suprasellar location, occupying the third ventricle and hypothalamus with encasement of both carotids. He underwent pericoronal parasagittal craniotomy, interhemispheric transcallosal interforniceal approach and gross total excision of the lesion. Postoperatively, the child had altered sensorium and diabetes insipidus, both of which recovered over a span of 10 days. Histopathological examination of the specimen was consistent with the diagnosis of World Health Organization Grade IV ATRT. In spite of all our efforts, he succumbed to his illness 5 months postoperatively.
Keywords: Atypical teratoid rhabdoid tumor, diabetes insipidus, pediatric, suprasellar, third ventricle
Introduction
Atypical teratoid rhabdoid tumors (ATRTs) are rare, aggressive, and highly malignant embryonal tumors of the central nervous system (CNS), comprising approximately 3% of pediatric brain tumors and 20% of CNS tumors in children under the age of 3 years.[1] Suprasellar location accounts for 5% of ATRTs in adults[2] a few cases reported in the pediatric age group.[3,4] We report a tumor occurring in the suprasellar region with its imageological features and the difficulties faced in its management and surgical decision-making. Due to rarity, no specific guidelines have been formulated in the management of these lesions.
Case Report
A 2-year-old male child, the firstborn of nonconsanguineous marriage, presented with complaints of decreased activity, tiredness of 1 month, reduced vision of 2 weeks, and vomiting of 1-day duration. His birth was by cesarean section at term due to overweight. He had normal growth and development. He had a reversal of sleep rhythm although there was no history suggestive of drop attacks, seizures, polyuria, weight loss, or fever. He had a normal head circumference. There was bilateral papilledema with no other cranial nerve involvement or motor deficits. There were no neurocutaneous markers. Systemic examination revealed no abnormality. Preoperative Goggle-Visual Evoked Potential (GVEP) revealed prolonged P100 latencies bilaterally.
On magnetic resonance imaging (MRI), a lobulated mass lesion (measuring approximately 4.6 cm × 4.6 cm × 5.2 cm) with heterogeneous signal intensity was seen involving the suprasellar region and extending superiorly into the third ventricle and hypothalamus region. The lesion was extending to the frontal horns and bodies of bilateral lateral ventricles and abutting the inferior surface of the body of corpus callosum. Posteriorly, the lesion was extending to the interpeduncular cistern and was causing compression over the midbrain. Laterally, the lesion was abutting the medial temporal lobes bilaterally, and inferiorly, it was extending into suprasellar cistern. Lesion partially encased supraclinoid segments of bilateral internal carotid arteries. On postcontrast study, the lesion was showing contrast enhancement with central nonenhancing necrotic areas [Figure 1]. The optic chiasm, prechiasmatic segment of the optic nerves bilaterally, and oculomotor nerves were not separately visualized from the mass. Normal pituitary gland was visualized, and infundibulum was not separately seen from the mass. Magnetic resonance spectroscopy (MRS) showed elevated choline, reduced N-acetylaspartate (NAA) and small lactate peak [Figure 2]. The imaging features were in favor of germinoma or optic chiasmatic-hypothalamic glioma.
Figure 1.
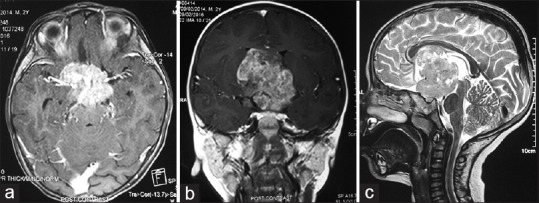
(a) Postcontrast axial T1-weighted magnetic resonance image showing the relation with brainstem. (b) Postcontrast coronal T1-weighted magnetic resonance image showing the relation with internal carotid artery. (c) Sagittal T2-weighted magnetic resonance image showing the vertical extent of the lesion
Figure 2.
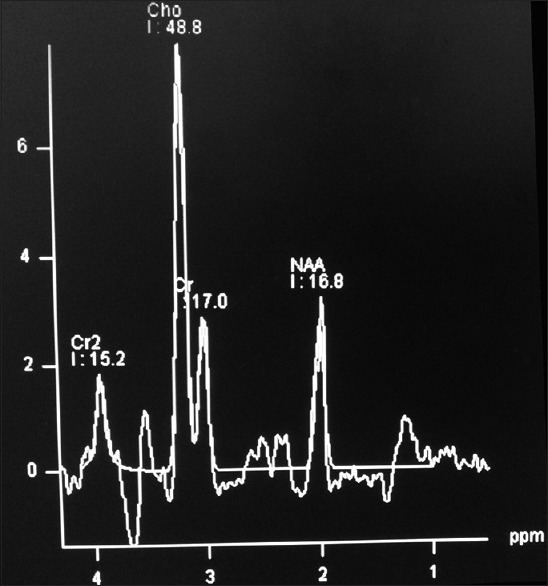
Magnetic resonance spectroscopy showing the choline peak
Hormonal workup and tumor markers in the serum (beta-human chorionic gonadotropin and alpha-fetoprotein) were normal.
The patient underwent right pericoronal parasagittal craniotomy, interhemispheric interforniceal approach to the third ventricle, and total excision of the tumor. Intraoperatively, the tumor was grayish-white, soft, suckable, and vascular. The optic and oculomotor nerves could be separately visualized. Bilateral internal carotid bifurcations and the basilar bifurcation intended the lesion. Postoperative computerized tomography scan showed no gross enhancing lesion [Figure 3].
Figure 3.
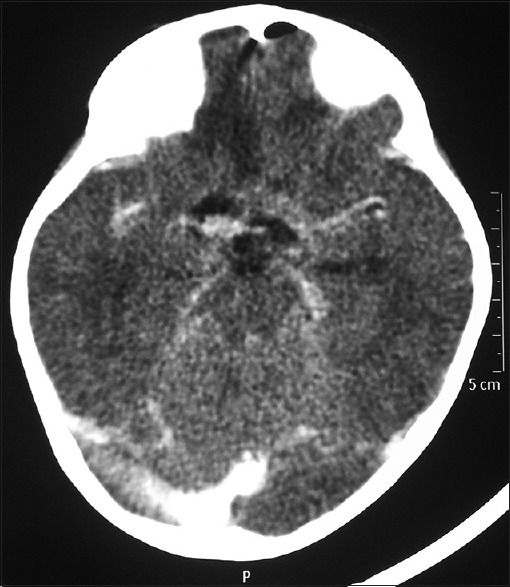
Postoperative contrast-enhanced axial computed tomogram of the brain showing the complete excision of the tumor
In initial postoperative period, the child was drowsy, with spikes of high-grade fever and diabetes insipidus with hypernatremia requiring intermittent doses of vasopressin. Postoperative GVEP study showed no consistent responses from left eye and prolonged P100 latency from right eye. At 2 weeks, the patient was conscious and taking food orally with no motor deficit or electrolyte imbalance although verbalization was reduced. Adjuvant therapy was deferred by the oncologist owing to the very poor prognosis, and he succumbed to his illness 5 months postoperatively.
The histopathological examination of the surgical specimen showed a highly cellular tumor composed of cells arranged in sheets and fascicles. Tumor cells had a moderate amount of eosinophilic cytoplasm with hyperchromatic nuclei in some of them exhibiting moderate nuclear atypia. Mitosis was brisk. At places, tumor cells showed eccentrically placed nuclei (rhabdoid morphology) with areas of necrosis. Immunohistochemistry showed loss of expression of nuclear integrase interactor 1 (INI1). Vimentin and epithelial membrane antigen were positive [Figure 4].
Figure 4.

(a) Microphotograph showing a densely cellular neoplasm (H and E, ×100). (b) Microphotograph showing small/primitive looking cells arranged in sheets (H and E, ×200). (c) Microphotograph showing predominantly rhabdoid cells (H and E, ×400). (d) Microphotograph showing neoplastic cells exhibiting marked pleomorphism and aberrant mitosis (H and E, ×400). (e) Microphotograph showing loss of nuclear staining of integrase interactor 1 with positively stained endothelial cells acting as internal control for the stain (integrase interactor 1 IHC, ×200, inset ×400). (f) Microphotograph showing diffuse and variable positivity for epithelial membrane antigen (IHC for epithelial membrane antigen × 200) and inset showing strong positive staining for vimentin (IHC for vimentin ×400)
Discussion
The first apparent case of a primary CNS rhabdoid tumor was reported in 1987, and the entity of ATRT suggested in the same year by Lefkowitz et al.[5] While ATRT represents 1%–2% of all pediatric CNS tumors, they account for up to 20% in children <36 months of age. Two-thirds of the children diagnosed with ATRT are <3 years of age at the time of diagnosis. Although there may be a slight predominance of tumor distribution in the supratentorial compartment, primary location in the posterior fossa is more common in children <24 months of age.[6]
MRI is the investigation of choice. In contrast to primitive neuroectodermal tumors (PNET), which is a close differential diagnosis, ATRTs have a propensity to occur off-midline and have readily visible calcification and more of cyst formation though all these features were lacking in our case. Cysts are usually eccentric with enhancing walls. MRS appearance mimics that of PNET, with marked choline elevation and low or absent NAA and creatine.[7]
Maximum safe resection is the best treatment modality. Due to the rarity of these tumors, there is no current standard of care for these tumors. However, there is strong evidence to show that complete tumor resection is associated with a more favorable outcome.[6]
Overall survival in ATRT is poor with median survival of around 17 months, with no standard curative chemotherapeutic regimen available.[8]
The presence of densely packed malignant round cells along with large, pale, eosinophilic, rhabdoid cells with oval nucleus and central nucleolus is the hallmark of ATRT. Mitoses, necrosis, and dystrophic calcification also may be present. Deletions and mutations of the hSNF5/INI1/SMARCB1 locus in chromosome band 22q11.2 have been documented in these tumors.[9]
Pediatric suprasellar ATRTs provide some unique challenges in the management:
Young age at presentation, which poses surgical risks and difficulty in providing adjuvant radiation
Encasement of blood vessels and proximity to brain stem and cranial nerves
Endocrinological dysfunction
Hypothalamic disturbance and postoperative diabetes insipidus
Infiltration of optic nerves, sometimes necessitating a subtotal decompression
Ventricular extension causing hydrocephalus.
Further prospective studies and trials are required to decide on the optimum management of such aggressive tumors which claim the lives of some of the very young members of our population.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
Authors sincerely thank the Department of Interventional Radiology, Sree Chitra Tirunal Institute for Medical Sciences and Technology, Medical College P.O., Thiruvananthapuram - 695 011, Kerala, India, for providing us with the radiological investigations and images.
References
- 1.Lau CS, Mahendraraj K, Chamberlain RS. Atypical teratoid rhabdoid tumors: A population-based clinical outcomes study involving 174 patients from the surveillance, epidemiology, and end results database (1973-2010) Cancer Manag Res. 2015;7:301–9. doi: 10.2147/CMAR.S88561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oka H, Scheithauer BW. Clinicopathological characteristics of atypical teratoid/rhabdoid tumor. Neurol Med Chir (Tokyo) 1999;39:510–7. doi: 10.2176/nmc.39.510. [DOI] [PubMed] [Google Scholar]
- 3.Darmoul M, Ben Nsir A, Chabchoub I, Hattab MN. Atypical teratoid rhabdoid tumor of the lateral ventricle. J Pediatr Neurosci. 2015;10:382–5. doi: 10.4103/1817-1745.174455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park HG, Yoon JH, Kim SH, Cho KH, Park HJ, Kim SH, et al. Adult-onset sellar and suprasellar atypical teratoid rhabdoid tumor treated with a multimodal approach: A case report. Brain Tumor Res Treat. 2014;2:108–13. doi: 10.14791/btrt.2014.2.2.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefkowitz IB, Rorke LB, Packer RJ. Atypical teratoid tumor of infancy: Definition of an entity. Ann Neurol. 1987;22:448–9. [Google Scholar]
- 6.Lafay-Cousin L, Strother DR, Chan JA, Torchia J, Huang A. Atypical teratoid rhabdoid tumors. In: Scheinemann K, Bouffet E, editors. Pediatric Neuro-Oncology. 1st ed. New York: Springer; 2015. pp. 163–71. [Google Scholar]
- 7.Biswas A, Goyal S, Puri T, Das P, Sarkar C, Julka PK, et al. Atypical teratoid rhabdoid tumor of the brain: Case series and review of literature. Childs Nerv Syst. 2009;25:1495–500. doi: 10.1007/s00381-009-0903-x. [DOI] [PubMed] [Google Scholar]
- 8.Ginn KF, Gajjar A. Atypical teratoid rhabdoid tumor: Current therapy and future directions. Front Oncol. 2012;2:114. doi: 10.3389/fonc.2012.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parwani AV, Stelow EB, Pambuccian SE, Burger PC, Ali SZ. Atypical teratoid/rhabdoid tumor of the brain: Cytopathologic characteristics and differential diagnosis. Cancer. 2005;105:65–70. doi: 10.1002/cncr.20872. [DOI] [PubMed] [Google Scholar]