

Abstract
Objective
To evaluate the impact of an initiative to increase hydroxyurea use among children with sickle cell anemia (SCA) who presented to the emergency department (ED).
Study design
This observational cohort study included children with SCA not taking hydroxyurea who presented to the ED with pain or acute chest syndrome (ACS) and then attended a Quick-Start Hydroxyurea Initiation Project (Q-SHIP) session. A Q-SHIP session includes a hematologist-led discussion on hydroxyurea, a video of patients talking about hydroxyurea, and a direct offer to start hydroxyurea.
Results
Over 64 weeks, 112 eligible patients presented to the ED and 59% (N=66) participated in a Q-SHIP session a median of 6 days (IQR 2, 20 days) after ED or hospital discharge; 55% of participants (N=36) started hydroxyurea. After a median follow-up of 49 weeks, 83% (N=30) of these participants continued hydroxyurea. Laboratory markers of hydroxyurea adherence were significantly increased from baseline: median mean corpuscular volume +8.6 fL (IQR 5.0, 17.7, P < .0001), median hemoglobin F +5.7% (IQR 2.5, 9.8, p=0.0001). Comparing Q-SHIP participants to non-participants, 12 weeks after ED visit, participants were more likely to have started hydroxyurea than non-participants (53% vs. 20%, p=0.0004) and to be taking hydroxyurea at last follow-up (50% versus 20%, p=0.001). Two years after the implementation of Q-SHIP the overall proportion of eligible patients on hydroxyurea presenting to our ED increased from 56% to 80%, p=0.0069.
Conclusions
Participation in a clinic to specifically address starting hydroxyurea after a SCA complication increases hydroxyurea use.
Keywords: Sickle cell disease, Hydroxyurea, Pediatrics, Patient Education
Hydroxyurea is a FDA-approved, daily oral medication that decreases SCA complications primarily by inducing fetal hemoglobin production.(1) Hydroxyurea treatment decreases pain and acute chest syndrome (ACS) events in children with SCA(2,3) and is associated with improved cerebrovascular health, growth, health-related quality of life, and survival.(4–15) In 2002, the National Heart, Lung and Blood Institute (NHLBI) recommended hydroxyurea for children with severe disease. In 2014, the NHLBI guidelines expanded the pediatric indication for hydroxyurea and now state that treatment with hydroxyurea should be offered to all children with SCA starting at 9 months of age.(16,17). The new recommendation was based primarily on evidence from the phase 3, randomized, placebo-controlled trial BABY HUG.(3) Despite evidence of clinical benefit, low cost, and few other available treatments, hydroxyurea remains underused among children and adults with SCA.(18–21)
Barriers to hydroxyurea treatment include patient, parent, provider and systems-level challenges. Physicians report not initiating hydroxyurea because of concerns about patient adherence.(21–24) Patients and families worry about side effects, often lack understanding of how hydroxyurea works, and cite insufficient discussion of their concerns as barriers to hydroxyurea initiation.(25, 26) However, treatment acceptance could be improved by discussing hydroxyurea with parents and patients in ways that define the indications for drug use, explain the potential benefits of therapy, acknowledge patient and family concerns, and reduce the burden of clinic attendance or obtaining medication refills.(26–29) In a multi-center survey study, hydroxyurea use in children with SCA was significantly associated with parents’ level of knowledge about the medication.(25) In a single-center study, most parents of children with SCA who received education about hydroxyurea concluded that hydroxyurea was safe, beneficial, and preferable to treatment with chronic transfusions or hematopoietic stem cell transplant.(30) Clinical events may also prompt initiation of therapy. Parents of children with SCA who never initiated hydroxyurea reported that acute events requiring emergency care or hospitalization would cause them to request a hydroxyurea prescription.(25)
Strategies for overcoming hydroxyurea treatment barriers are needed.(26) We hypothesized that providing intensive hydroxyurea education and the opportunity to initiate hydroxyurea to parents and their children with SCA shortly after an emergency department (ED) visit for pain or ACS would lead to increased treatment acceptance. Given this background, we implemented the Quick-Start Hydroxyurea Initiation Project (Q-SHIP) in February 2016 with a goal of increasing hydroxyurea use in children at our institution who are eligible for this treatment.
METHODS
We evaluated the effectiveness of our clinical program Q-SHIP, which was designed to increase hydroxyurea use by patients with SCA, among eligible patients who presented to the Children’s National Health System (CNHS) ED between February 1, 2016 and April 23, 2017. We attempted to reach all patients who met eligibility criteria (see below) to participate in Q-SHIP. This was not a controlled trial, but rather compares results for those who voluntarily participated in the program and those who did not.
Eligible patients had laboratory confirmed SCA (hemoglobin SS or Sβ0-thalassemia), presented with SCA-related pain or ACS during the study period, and were not already taking hydroxyurea. Patients were deemed ineligible if they were less than less than 9-months old, receiving chronic red cell transfusions, or pregnant, as hydroxyurea is not currently indicated for these patients. Patients who were not primarily followed at our center (pediatric hematology providers unaffiliated with CNHS refer patients to our ED) were also excluded because they could not follow up at CNHS for the required monitoring of hydroxyurea treatment.
Patients eligible for Q-SHIP were identified through a weekly chart review of all patients with sickle cell disease (SCD) evaluated in the CNHS ED, using an electronic ED clinical registry that includes all ED patient encounters. Clinical providers or a Q-SHIP team member attempted to contact all eligible patients to invite them to participate in a Q-SHIP session.
Participants attended a Q-SHIP session in the outpatient hematology clinic after ED discharge or during a hospitalization (if approved by the inpatient service attending). This session is held weekly and led by one pediatric hematologist (RSN). It is held separate from a routine clinic visit so that it focuses on hydroxyurea. Participants complete a brief survey (Appendix 1; available at www.jpeds.com) and spend approximately 45 minutes reviewing “Hydroxyurea for Sickle Cell Disease: A Guide for Starting Treatment,” a handbook for families developed by the interdisciplinary SCD team at CNHS, which is available at https://www.childrensnational.org/Hydroxyurea (Appendix 2; available at www.jpeds.com). Participants then watch a 15-minute video (https://www.youtube.com/watch?v=2a7FXibkubQ&feature=youtu.be) about hydroxyurea that merges publicly available footage from academic medical centers and patient advocacy group videos (Table I; available at www.jpeds.com). These videos feature patients and parents of children with SCA discussing their experiences with hydroxyurea. At the conclusion of the session, parents who are ready to start their child on hydroxyurea receive a prescription contingent on laboratory confirmation that their child meets institutional guidelines for treatment (APPENDIX 3; available at www.jpeds.com). Follow-up with the participant’s primary hematologist is arranged 2-4 weeks after starting therapy. Parents who are not interested in starting their child on hydroxyurea, or who want more time to review the presented information, are encouraged to contact RSN or their primary hematologist if they later decide to start hydroxyurea. Q-SHIP participants receive no additional special follow-up.
TABLE 1. online-only, Q-SHIP Video Citations.
The Q-SHIP video incorporates videos from these freely available sources listed here.
| Institution/Organization | Video Title | URL |
|---|---|---|
| Children’s Medical Center Dallas, University of Texas Southwestern | Hydroxyurea: The Patient Experience | https://www.youtube.com/watch?v=BhCrB3cKAAQ |
| Monroe Carell Jr Children’s Hospital at Vanderbilt | Hydroxyurea Treatment for Children with Sickle Cell Disease | https://www.youtube.com/watch?v=nx7MvMjeNJ0 |
| Sickle Cell Disease Association of America Philadelphia/Delaware Valley Chapter | Hydroxyurea “Ask Me Why” | https://www.youtube.com/watch?v=mgp5PI-DsDI |
| National Heart Lung and Blood Institute, National Institutes of Health | My Story: Living with Sickle Cell Disease | https://www.youtube.com/watch?v=qe59ar-GZmg |
| University of Florida | You Don’t Know Until You ASK! Is Hydroxyurea Your Hope for Better Days? | Division of Hematology/Oncology, Department of Medicine, University of Florida courtesy of Dr.Richard Lottenberg (lottenr@medicine.ufl.edu) |
| Central-Northern New Jersey Sickle Cell Network, Newark Beth Israel Medical Center and the Children’s Hospital of New Jersey | Hydroxyurea: The Best Hope for Sickle Cell Anemia Patients | https://www.youtube.com/watch?v=L_xSgQWjO7A |
Per institution hydroxyurea treatment guidelines (APPENDIX 3), SCD providers at CNHS routinely offer hydroxyurea treatment to all patients with SCA older than 9 months if there is no contraindication to its use. Eligible patients who do not participate in Q-SHIP all receive standard care that would typically include a discussion of hydroxyurea as a component of routine SCA care. The educational booklet (APPENDIX 2) developed for Q-SHIP is available for all providers for use outside of a formal Q-SHIP session.
The CNHS Institutional Review Board approved this study. A waiver of written informed consent was granted.
Statistical Analyses
For this analysis, patients were classified as “started hydroxyurea after Q-SHIP” if they had a clinic visit for hydroxyurea monitoring within three months of their participation in a Q-SHIP session. Three months was chosen as the time interval rather than just one month in order to account for possible delays in starting hydroxyurea due to the insurance authorization process, as well as to allow for additional time that some families may need to review the material discussed during a Q-SHIP session before making a decision to start hydroxyurea. Patients were classified as “taking hydroxyurea at recent follow-up” if hydroxyurea use was documented in a clinical encounter within the last three months of the evaluation period (April 1, 2017 - July 1, 2017). To evaluate hydroxyurea adherence, the most recent mean corpuscular volume (MCV) and hemoglobin F (%HbF) measurements were compared with baseline MCV and %HbF measurements. Q-SHIP participants who started hydroxyurea were compared to those who did not start hydroxyurea. Q-SHIP participants were also compared with eligible patients who did not participate in Q-SHIP (“non-participants”). In addition, the proportion of eligible SCA patients who were actually taking hydroxyurea was measured among those who presented to the ED for pain or ACS in the month of February in 2015, 2016, 2017, and 2018.
Clinical and demographic information was obtained by retrospective chart review. Patients were classified as “previously offered hydroxyurea” if a clinic note documented that hydroxyurea was recommended or that the patient or patient’s family declined hydroxyurea. If documentation in a clinic note stated that hydroxyurea was discussed but did not explicitly indicate that therapy was offered, the participant was classified as having “no previous hydroxyurea offer.” Categorical data was analyzed with the chi-square or Fisher exact test. Continuous data was analyzed using the Wilcoxon rank-sum test or the 2-sample t-test. MCV and %HbF measures were compared using the paired Wilcoxon signed-rank test. Statistical calculations were performed with SAS 9.3 (SAS Institute Inc, Cary, NC).
RESULTS
Over 64 weeks (2/1/2016 – 4/23/2017), there were a total of 2,309 ED encounters among 739 patients with SCD (all genotypes) in the CNHS ED (FIGURE 1). Initially, 297 patients were excluded: 164 did not have SCA and 133 were not established CNHS hematology patients. Among 442 patients with SCA followed at CNHS, an additional 330 patients were excluded: 213 were already taking hydroxyurea, 62 did not have an ED encounter for pain or ACS during the study period, 42 were receiving chronic red cell transfusions, and 13 were ineligible for hydroxyurea due to other reasons.
FIGURE 1. Flow diagram of Q-SHIP participants and differences in hydroxyurea initiation between Q-SHIP participants and non-participants at 12-weeks and at long-term follow-up.
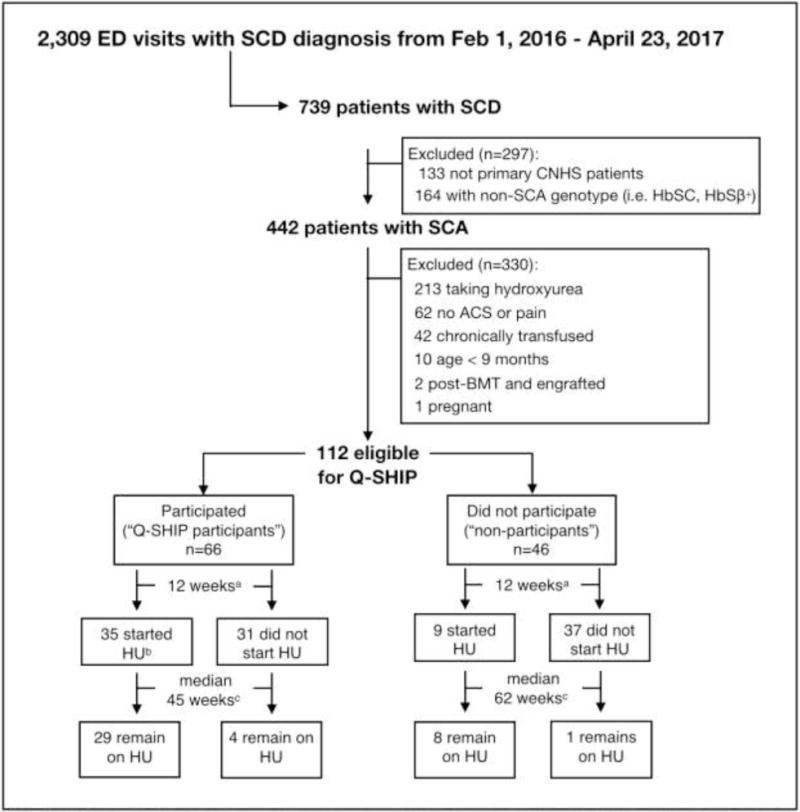
“Started hydroxyurea” is defined as clinic documentation of treatment initiation during the 12-week period and hydroxyurea use at the subsequent clinic visit. “Taking hydroxyurea at recent encounter” is defined as a medical encounter from 4/1/17-7/1/17 documenting hydroxyurea use. a12 weeks from ED encounter discharge. bOne participant started hydroxyurea 12 weeks after the ED encounter discharge but less than 12 weeks after the Q-SHIP session. cTotal follow-up time from ED encounter discharge to 7/1/2017.
Among 112 patients eligible for Q-SHIP, 59% (N=66) participated in Q-SHIP a median of 6 days (IQR 2, 20 days) after ED or hospital discharge for pain (N=42), ACS (N=17), or pain and ACS (N=7). There were no significant differences in the clinical and demographic information of Q-SHIP participants compared with non-participants including markers of prior disease severity (TABLE 2; available at www.jpeds.com). Fifty-three percent of participants attended a group Q-SHIP session with other families (median 2 families, range 2-5), and 21% participated in Q-SHIP while hospitalized. Among the 46 non-participants, only three families involving 4 non-participants (9%) explicitly declined to participate in Q-SHIP. The remaining families did not participate because they could not be reached to offer participation, could not come to clinic at the time of the weekly Q-SHIP session, or had already started hydroxyurea through a routine clinic visit before participation in Q-SHIP could be offered.
TABLE 2. online-only, Characteristics of Q-SHIP Participants Compared to Non-Participants.
There were no differences in age, sex, insurance type, frequency of hematology clinic follow-up, baseline hemoglobin and %HbF, ED visits in the past 2 years, previous pediatric intensive care unit (PICU) admission, and previous transfusions.
| Participated in Q-SHIP N=66 |
Not participated in Q-SHIP N=46 |
p-value | |
|---|---|---|---|
| Age, median year (IQR) | 8.5 (4.4, 16.0) | 9.0 (5.1, 16.5) | 0.38 |
| Male | 34 (52%) | 22 (48%) | 0.70 |
| Medicaid insurance | 49 (74%) | 32 (70%) | 0.59 |
| Hematology clinic visit in the last year | 54 (82%) | 35 (76%) | 0.46 |
| Baseline hemoglobin, mean g/dl (SD)a | 8.4 (1.1) | 8.7 (1.2) | 0.23 |
| Baseline %HbF, mean % (SD)b | 15.9 (9.4) | 15.7 (10.4) | 0.93 |
| Number of ED visits or hospitalizations for ACS or pain in prior 2 years, median (IQR) | 3 (2, 5) | 2 (1, 4) | 0.42 |
| Prior PICU admission or transfer | 17 (26%) | 14 (30%) | 0.59 |
| Number of prior transfusions, median (IQR) | 2 (0, 4) | 1 (0, 4) | 0.72 |
defined as hemoglobin at last regular hematology clinic visit, >3 months from any transfusion;
n=102, ten patients were excluded as they either did not have a %HbF after age 3 years or no %HbF in the last 6 months if age < 3 years.
Hydroxyurea offers before Q-SHIP
Pre-participation surveys from 65 of 66 participating families were analyzed (one participant did not complete a survey). Fifty-five percent (N=36) reported previously receiving an offer to start hydroxyurea. These participants reported previously declining hydroxyurea because of side-effect concerns (50%, N=18), not thinking their child’s disease was severe enough to warrant treatment (31%, N=11), wanting more information (28%, N=10), and the requirement of more frequent visits for hydroxyurea monitoring (11%, N=4). Thirty-three percent (N=12) reported multiple reasons for previously refusing hydroxyurea. Among those concerned about treatment side effects (N=18), 2 families specifically cited fertility concerns, but families most commonly reported non-specific side effect concerns.
Forty-five percent of participants (N=29) reported no previous hydroxyurea offer. Fifty-nine percent (N=17/29) of these participants had documentation in their medical record that a previous hydroxyurea offer had been declined. Participants who reported no previous hydroxyurea offer were less likely to have had a regular hematology clinic visit in the last year compared with participants who reported a previous offer (69% versus 92%, p=0.019).
Hydroxyurea initiation after Q-SHIP
Fifty-five percent of participants (N=36) started hydroxyurea after Q-SHIP. There were no significant differences in demographics, intervention-related variables, or markers of SCD clinical severity between participants who started hydroxyurea and those who did not (TABLE 3). Hydroxyurea initiation was not significantly influenced by the participants’ previous reason for refusing hydroxyurea (p=0.41). Most patients who had wanted more information about hydroxyurea (N=8/10), had side effect concerns (N=12/18) or had multiple concerns (N=7/12) started hydroxyurea; and 45% (N=5/11) of those who had reported that their child’s disease was not severe enough to warrant treatment started hydroxyurea. No patients in this cohort permanently discontinued hydroxyurea because of side effects or toxicity. After a median of 49 weeks (IQR 28, 62 weeks), 83% (N=30) of those who started hydroxyurea after Q-SHIP were still taking hydroxyurea at a recent follow-up visit. Two patients stopped hydroxyurea because their families decided it was unnecessary. Four patients were lost to follow-up (defined as no medical encounter from 4/1/17 to 7/1/17).
TABLE 3. Comparison of Q-SHIP Participants Who Did and Did Not Start Hydroxyurea.
There were no differences between those who did and did not start hydroxyurea according to previous hydroxyurea offer, participation in a group session or while hospitalized, or clinical variables including baseline hemoglobin and %HbF, ED visits, or intensive care unit admission.
| Started HU after Q-SHIP N=36 |
Did not start HU after Q-SHIP N=30 |
p-value | |
|---|---|---|---|
| Age, median years (IQR) | 7.5 (5.0, 15.3) | 9.3 (3.1, 16.3) | 0.94 |
| Reported previous HU offera | 22/35 (63%) | 14/30 (47%) | 0.19 |
| Group Q-SHIP session with other patient families | 20 (56%) | 15 (50%) | 0.66 |
| Attended Q-SHIP… …after only an ED visit …during a hospitalization …after a hospitalization |
13 (36%) 8 (22%) 15 (42%) |
10 (33%) 6 (20%) 14 (47%) |
0.81 0.83 0.68 |
| Days from ED or hospital discharge to Q-SHIP participation, median (IQR) | 5 (2, 20) | 8 (3, 28) | 0.59 |
| Baseline hemoglobin, mean g/dl (SD) | 8.3 (1.2) | 8.6 (1.0) | 0.29 |
| Baseline %HbF, mean % (SD)b | 15.0 (9.5) | 16.9 (9.3) | 0.43 |
| Number of ED visits or hospitalizations for pain or ACS in prior 2 years, median (IQR) | 3 (2, 5) | 2 (2, 4) | 0.29 |
| Prior PICU admission or transfer | 12 (33%) | 5 (17%) | 0.12 |
| Number of prior transfusions, median (IQR) | 2 (1, 5) | 1 (0, 4) | 0.12 |
n=65, one patient did not compete pre-session questionnaire;
n=63, three patients did not have baseline %HbF.
Participants who started and continued hydroxyurea after Q-SHIP (N=30) had a significant increase in their MCV and %HbF. Median increase in MCV was 8.6 (IQR 5.0, 17.7, p<0.0001) with median follow-up of 38 weeks (IQR 21, 53 weeks). Median increase in %HbF was 5.7% (IQR 2.5, 9.8, p=0.0001) with median follow-up of 29 weeks (IQR 18, 45 weeks). Six participants were excluded in the %HbF comparison because they did not have a pre- or post- %HbF value. During the follow-up time period, three additional Q-SHIP participants started hydroxyurea more than 3 months after participating in Q-SHIP.
At 12 weeks after discharge from their ED encounter, Q-SHIP participants had started hydroxyurea more often than non-participants (53% vs 20%, p=0.0004). At the most recent follow-up visit, Q-SHIP participants were significantly more likely to be taking hydroxyurea than non-participants (50% vs. 20% p=0.001) after a median follow-up of one year (FIGURE 2).
FIGURE 2. Interval Hydroxyurea Use in Q-SHIP Participants and Non-Participants During Follow-Up.
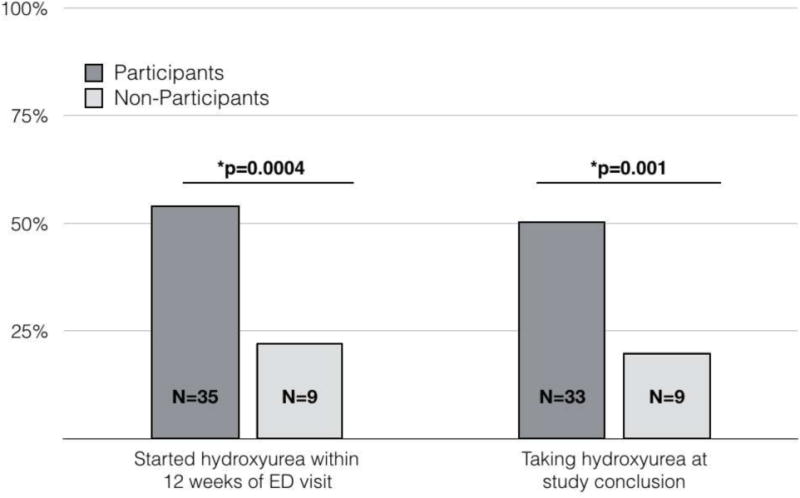
Hydroxyurea initiation in Q-SHIP participants and non-participants over time. Twelve weeks from ED presentation, Q-SHIP participants were significantly more likely to start hydroxyurea than non-participants (35/66 participants versus 9/46 non-participants, p=0.0004). At the study’s conclusion, significantly more Q-SHIP participants were taking hydroxyurea than non-participants (33/66 participants versus 9/46 non-participants, p=0.001).
Hydroxyurea Use Among ED Patients Over Time
The proportion of patients with SCA presenting to the ED who were taking hydroxyurea (assessed annually in the month of February) has steadily increased since the implementation of Q-SHIP in 2016 (FIGURE 3). Among patients otherwise eligible for Q-SHIP, the proportion on hydroxyurea increased from 32/57 (56%) in February 2016 to 44/55 (80%) in February 2018, p=0.0069.
FIGURE 3. Steady Increase in the Proportion of Patients in the ED taking Hydroxyurea Since Start of Q-SHIP.
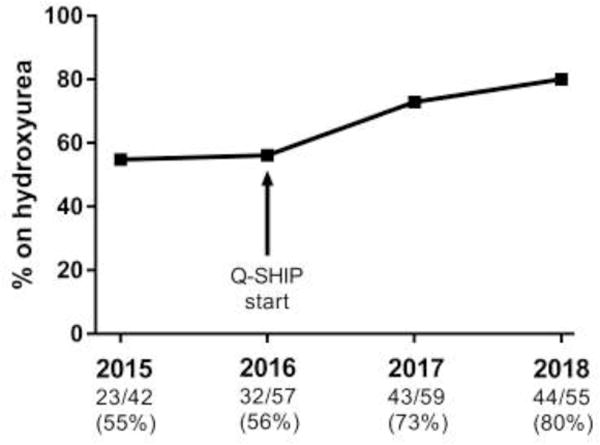
Among patients with SCA followed at CNHS, aged >9 months, not on chronic transfusion who presented to the ED for pain and/or ACS during the month of February, the number of patients on hydroxyurea is shown for 2015, 2016, 2017, and 2018. There was no significant increase in this proportion when comparing the year before Q-SHIP was started (February 2015) to the baseline proportion at the start of Q-SHIP (February 2016): 55% vs. 56%, p=0.89. In contrast, there was a significant increase in this proportion when comparing this baseline proportion (February 2016) to two years after the start of Q-SHIP (February 2018): 56% vs. 80%, p=0.0069.
DISCUSSION
After an acute SCA complication requiring ED care, follow-up in a clinic devoted to hydroxyurea education in untreated children led over half of participating families to begin and continue hydroxyurea. Most patients who started hydroxyurea shortly after a pain or ACS episode demonstrated evidence of treatment adherence. These results suggest that a clinic focused on hydroxyurea that incorporates patient and parent perspectives, and also includes time for discussion of concerns, has a positive influence on families’ treatment choices.
In this study, we were surprised that 45% of families reported no prior offer of hydroxyurea, given that providers at CNHS typically offer hydroxyurea to all children with SCA as per current NHLBI guidelines. Indeed, for most families who reported no previous hydroxyurea offer there actually was documentation that they had been offered hydroxyurea. However, these families were also less likely to have had a hematology clinic visit in the last year. Inaccurate parent and patient recall of medical information is a recognized, but perhaps underappreciated, phenomenon. Our finding has two implications. First, regular hematology follow-up may help families retain essential information about SCD treatment. Second, if families do not necessarily remember discussing hydroxyurea, then revisiting indications for treatment on consecutive visits may not be futile.
How information about hydroxyurea is presented affects its use. For example, a center that “strongly recommends” hydroxyurea for all children with SCA 5 years or older recently reported that over 90% of their patients were on hydroxyurea.(13) The NHLBI guidelines state that families should be “offered” hydroxyurea; this may lead to ambiguous language from providers who offer, rather than recommend, treatment. Providers may also be communicating misgivings about hydroxyurea including concerns about carcinogenicity, teratogenicity, and adverse effects on fertility.(21,31) At some European institutions, hematologists do not universally offer hydroxyurea to patients with SCA due to concerns about late and unknown effects.(12, 24) A recent qualitative study identified two distinct patterns of communication among American pediatric hematologists when discussing hydroxyurea with patients with SCA and their families. Bakshi et al describe a “collaborative approach” involving discussion of all treatment options with families versus a “proponent approach” defined by advocating for a pre-established treatment plan.(32) In the Q-SHIP model, we adopted a “proponent approach,” advocating for hydroxyurea initiation with participants.
Q-SHIP creates time dedicated to hydroxyurea education. Some pediatric hematologists report that they lack the time to explain the risks and benefits of hydroxyurea.(21) Like general pediatricians,(33) pediatric hematologists have more topics to cover during a routine SCA clinic visit than time allows. Due to these other priorities and time constraints, implementing a shared decision making model for initiating hydroxyurea therapy in children with SCA is difficult.(27) Q-SHIP’s focus on hydroxyurea allows time for this discussion, and signifies to families that our clinical team believes this treatment is worth additional effort to discuss.(34)
The video shown during Q-SHIP includes portions of videos that were produced by outside academic centers and patient advocacy groups. This helps incorporate patient perspectives and also reinforces that other centers make similar recommendations about hydroxyurea. At the University of Florida, a video featuring patients with SCA speaking positively about hydroxyurea inspired viewers’ interest in starting therapy.(35) Collaboration and resource sharing among centers may provide reinforcement for a single center’s practices and help providers at centers nationally and internationally meet evidence-based care for children with SCD. Videos may especially benefit patients with limited health literacy, a measure of a person’s ability to understand health information and make health decisions. Written materials about SCD for patients and families often exceed the average literacy level of adults in the United States and may not be appropriate for all parents of children with SCA.(36,37) Finally, videos on the internet may be helpful to families as parents can review these videos with other decision-makers outside of clinic.
Similar hydroxyurea initiation rates were observed regardless of whether Q-SHIP was conducted with groups of different families together or when a single family participated. The benefits of group clinic visits, especially for underserved populations, include opportunities for increased provider time and intensive education.(38,39) A randomized trial comparing group medical appointments to individual medical appointments for patients with SCD is ongoing,(40) and novel SCD clinic structures designed to improve care and distribute knowledge are being explored in both resource-rich and resource-poor settings.(41) This kind of research may help centers with limited resources lead thorough and thoughtful hydroxyurea initiation decision making sessions for families.
Although the majority of families who participated in Q-SHIP started hydroxyurea, a significant number did not initiate treatment even with this intensive education. This failure to start hydroxyurea was not associated with any particular prior rationale for refusing hydroxyurea. We had hypothesized that patients who had experienced fewer acute SCD complications would be less likely to accept hydroxyurea, but indicators of disease severity were not associated with hydroxyurea initiation after Q-SHIP. This observation is consistent with other studies that found patients’ and parents’ desire to pursue hematopoietic stem cell transplant for SCD is not associated with the patient’s disease severity.(42–44) Research is needed to better understand whether these families’ rejection of therapy rests on surmountable objections to treatment. At the end of this study, Q-SHIP participants were significantly more likely to be on hydroxyurea compared with non-participants. The non-participants in this study are an imperfect control group because, although they did not differ in demographic or clinical characteristics from participants, their lack of participation was non-random. Compared with participating families, this population may have distinct barriers to hydroxyurea initiation that were not explored by this study. These patients may require more resources or be especially reluctant to start treatment.
Our finding that the proportion of patients presenting to the ED with complications of SCA who were taking hydroxyurea increased after the implementation of Q-SHIP provides additional evidence that this initiative helped increase overall hydroxyurea use at our institution. Although it is possible this increased hydroxyurea use over time was unrelated to Q-SHIP, no increase in hydroxyurea use was observed in the year immediately before the start of Q-SHIP (FIGURE 3). This data also suggests that Q-SHIP’s benefits are sustainable. The program did require considerable effort (~5 hours/week) to identify all eligible patients and refer them to the education session during the study period, but currently the program continues without this intensive screening of all ED patients. Although we have observed a decreased number of referrals to the weekly Q-SHIP session without this screening, this trend also reflects the fact that, due to increased hydroxyurea use, fewer Q-SHIP eligible patients are presenting to the ED. Q-SHIP has likely also helped change the culture of our institution such that hydroxyurea is more likely to be offered to eligible patients at every opportunity.
The current study has several limitations. We cannot evaluate if the success of Q-SHIP was more influenced by the proximity to a recent acute SCA complication, the dedicated clinic time to hydroxyurea education, or an inclination of participants towards initiating hydroxyurea even before attending Q-SHIP. We now accept referrals to Q-SHIP for any patient who is eligible for hydroxyurea but not receiving therapy; a recent hospitalization or ED encounter is no longer a pre-requisite for participation. This project was not a randomized clinical trial and we thus lack an ideal control group to measure the effect of Q-SHIP on hydroxyurea initiation. Nonetheless, our study design reflects real-world implementation of an intervention. This is a single center study where specific clinic time is reserved for a weekly Q-SHIP session. This kind of protected clinic time may not be feasible at centers with fewer patients with SCA, but Q-SHIP can be adapted to the context of the implementing site. For example, the interval between ED or hospital discharge and Q-SHIP session was not significantly associated with hydroxyurea initiation, suggesting that timing Q-SHIP within one week of the acute SCA complication is not critical. A center might choose to offer monthly rather than weekly sessions. We did not measure parental education, household income, or family size, variables that may have influenced participation in Q-SHIP and initiation of hydroxyurea. Finally, Q-SHIP sessions were led by a single physician and participants may have been uniquely influenced by this person.
This study demonstrated that Q-SHIP helps overcome barriers to hydroxyurea prescription and acceptance. By providing adequate time and mixed-media resources for a thoughtful exchange of information between a clinician and family at a time when the family may be more receptive to this information, questions and concerns about hydroxyurea can be more fully explored and addressed. This study also identified a gap in provider-patient communication about hydroxyurea, and provides support for a solution: targeted education focused on hydroxyurea after an SCA complication. Modifications to this intervention and other novel interventions should be studied in order to continue to improve hydroxyurea treatment use among patients with SCA.
Supplementary Material
Acknowledgments
We thank the patients and families who make this work possible, the CNHS SCD team, hematology-oncology fellows, and ED providers for their essential support of this work. We also thank the Children’s Medical Center Dallas; the Monroe Carrell Jr Children’s Hospital at Vanderbilt; the Philadelphia/Delaware Valley Chapter of the Sickle Cell Disease Association of America; the University of Florida; the National, Heart, Lung and Blood Institute; and the Central-Northern New Jersey Sickle Cell Network, Newark Beth Israel Medical Center and the Children’s Hospital of New Jersey for making their videos publicly available so they could be used as part of Q-SHIP.
L.P. was supported by Award Number T32HL110841-04 at the conception of this study. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Advancing Translational Sciences of the National Institutes of Health.
Abbreviations
- ACS
Acute chest syndrome
- CNHS
Children’s National Health System
- ED
Emergency Department
- Hb
Hemoglobin
- IQR
Interquartile Range
- Lung and Blood Institute, NHLBI
National Heart
- PICU
Pediatric Intensive Care Unit
- Q-SHIP
Quick-Start Hydroxyurea Initiation Project
- SCA
Sickle cell anemia
- SCD
Sickle cell disease
APPENDIX 1
Q-SHIP Questionnaire
Survey administered to parents or patient (if >18 years) prior to Q-SHIP participation.

APPENDIX 2
Hydroxyurea for Sickle Cell Disease: A Guide for Starting Treatment
A handbook for families developed by the interdisciplinary SCD team at CNHS. The topics of this handbook are covered during a Q-SHIP session.

APPENDIX 3
Children’s National Hydroxyurea Treatment Guidelines
Guidelines created by the sickle cell team at CNHS that outline standard of care clinical practices regarding hydroxyurea for SCD.


Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflicts of interest.
Portions of this study were presented as oral abstracts at the American Society of Pediatric Hematology/Oncology annual meeting, ≪ ≫, ≪ ≫, and the American Society of Hematology annual meeting, ≪ ≫, ≪ ≫.
References
- 1.Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010;115:5300–11. doi: 10.1182/blood-2009-04-146852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quarmyne M-O, Dong W, Theodore R, Anand S, Barry V, Adisa O, Buchanan ID, Bost J, Brown RC, Joiner CH, Lane PA. Hydroxyurea effectiveness in children and adolescents with sickle cell anemia: A large retrospective, population-based cohort. Am J Hematol. 2017;92:77–81. doi: 10.1002/ajh.24587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thornburg CD, Files BA, Luo Z, Miller ST, Kalpatthi R, Iyer R, Seaman P, Lebensburger J, Alvarez O, Thompson B, Ware RE, Wang WC, BABY HUG Investigators Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood. 2012;120:4304–10. doi: 10.1182/blood-2012-03-419879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Estepp JH, Smeltzer MP, Kang G, Li C, Wang WC, Abrams C, Aygun B, Ware RE, Nottage K, Hankins JS. A clinically meaningful fetal hemoglobin threshold for children with sickle cell anemia during hydroxyurea therapy. Am J Hematol. 2017;330:1639. doi: 10.1002/ajh.24906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang WC, Oyeku SO, Luo Z, Boulet SL, Miller ST, Casella JF, Fish B, Thompson BW, Gross SD, BABY HUG Investigators Hydroxyurea Is Associated with Lower Costs of Care of Young Children with Sickle Cell Anemia. Pediatrics. 2013;132:677–83. doi: 10.1542/peds.2013-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nevitt SJ, Jones AP, Howard J. Hydroxyurea (hydroxycarbamide) for sickle cell disease. In: Nevitt SJ, editor. Cochrane Database Syst Rev. 4. 2017. p. CD002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hankins JS, McCarville MB, Rankine-Mullings A, Reid ME, Lobo CLC, Moura PG, Ali S, Soares DP, Aldred K, Jay DW, Aygun B, Bennett J, Kang G, Goldsmith JC, Smeltzer MP, Boyett JM, Ware RE. Prevention of conversion to abnormal transcranial Doppler with hydroxyurea in sickle cell anemia: A Phase III international randomized clinical trial. Am J Hematol. 2015;90:1099–105. doi: 10.1002/ajh.24198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, Sarnaik S, Odame I, Fuh B, George A, Owen W, Luchtman-Jones L, Rogers ZR, Hilliard L, Gauger C, Piccone C, Lee MT, Kwiatkowski JL, Jackson S, Miller ST, Roberts C, Heeney MM, Kalfa TA, Nelson S, Imran H, Nottage K, Alvarez O, Rhodes M, Thompson AA, Rothman JA, Helton KJ, Roberts D, Coleman J, Bonner MJ, Kutlar A, Patel N, Wood J, Piller L, Wei P, Luden J, Mortier NA, Stuber SE, Luban NL, Cohen AR, Pressel S, Adams RJ. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet. 2016;387:661–70. doi: 10.1016/S0140-6736(15)01041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lê P-Q, Gulbis B, Dedeken L, Dupont S, Vanderfaeillie A, Heijmans C, Huybrechts S, Devalck C, Efira A, Dresse MF, Rozen L, Benghiat FS, Ferster A. Survival among children and adults with sickle cell disease in Belgium: Benefit from hydroxyurea treatment. Pediatr Blood Cancer. 2015;62:1956–61. doi: 10.1002/pbc.25608. [DOI] [PubMed] [Google Scholar]
- 10.Fitzhugh CD, Hsieh MM, Allen D, Coles WA, Seamon C, Ring M, Zhao X, Minniti CP, Rodgers GP, Schechter AN, Tisdale JF, Taylor JG., 6th Hydroxyurea-Increased Fetal Hemoglobin Is Associated with Less Organ Damage and Longer Survival in Adults with Sickle Cell Anemia. PLOS ONE. 2015;10:e0141706–13. doi: 10.1371/journal.pone.0141706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nottage KA, Ware RE, Aygun B, Smeltzer M, Kang G, Moen J, Wang WC, Hankins JS, Helton KJ. Hydroxycarbamide treatment and brain MRI/MRA findings in children with sickle cell anaemia. Br J Haematol. 2016;175:331–8. doi: 10.1111/bjh.14235. [DOI] [PubMed] [Google Scholar]
- 12.Phillips K, Healy L, Smith L, Keenan R. Hydroxyurea therapy in UK children with sickle cell anaemia: A single-centre experience. Pediatr Blood Cancer. 2017;19:e26833. doi: 10.1002/pbc.26833. [DOI] [PubMed] [Google Scholar]
- 13.Ghafuri DL, Chaturvedi S, Rodeghier M, Stimpson S-J, McClain B, Byrd J, DeBaun MR. Secondary benefit of maintaining normal transcranial Doppler velocities when using hydroxyurea for prevention of severe sickle cell anemia. Pediatr Blood Cancer. 2017;64:e26401. doi: 10.1002/pbc.26401. [DOI] [PubMed] [Google Scholar]
- 14.Thornburg CD, Calatroni A, Panepinto JA. Differences in health-related quality of life in children with sickle cell disease receiving hydroxyurea. J Pediatr Hematol Oncol. 2011;33:251–4. doi: 10.1097/MPH.0b013e3182114c54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, Orringer E, Bellevue R, Olivieri N, Eckman J, Varma M, Ramirez G, Adler B, Smith W, Carlos T, Ataga K, DeCastro L, Bigelow C, Saunthararajah Y, Telfer M, Vichinsky E, Claster S, Shurin S, Bridges K, Waclawiw M, Bonds D, Terrin M. Effect of Hydroxyurea on Mortality and Morbidity in Adult Sickle Cell Anemia. JAMA. 2003;289:1645–51. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- 16.NHLBI. The Management of Sickle Cell Disease. 2002:1–206. [Google Scholar]
- 17.NHLBI. Evidence-Based Management of Sickle Cell Disease: Expert Panel. 2014:1–161. [Google Scholar]
- 18.Creary SE, Chisolm DJ, Koch TL, Zigmont VA, Lu B, O’Brien SH. Hydroxyurea use in Children with Sickle Cell Disease: Do Severely Affected Patients Use It and Does It Impact Hospitalization Outcomes? Pediatr Blood Cancer. 2016;63:844–7. doi: 10.1002/pbc.25894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stettler N, McKiernan CM, Melin CQ, Adejoro OO, Walczak NB. Proportion of adults with sickle cell anemia and pain crises receiving hydroxyurea. JAMA. 2015;313:1671–2. doi: 10.1001/jama.2015.3075. [DOI] [PubMed] [Google Scholar]
- 20.Lanzkron S, Haywood C, Segal JB, Dover GJ. Hospitalization rates and costs of care of patients with sickle-cell anemia in the state of Maryland in the era of hydroxyurea. Am J Hematol. 2006;81:927–32. doi: 10.1002/ajh.20703. [DOI] [PubMed] [Google Scholar]
- 21.Brandow AM, Jirovec DL, Panepinto JA. Hydroxyurea in children with sickle cell disease: practice patterns and barriers to utilization. Am J Hematol. 2010;85:611–3. doi: 10.1002/ajh.21749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zumberg MS, Reddy S, Boyette RL, Schwartz RJ, Konrad TR, Lottenberg R. Hydroxyurea therapy for sickle cell disease in community-based practices: a survey of Florida and North Carolina hematologists/oncologists. Am J Hematol. 2005;79:107–13. doi: 10.1002/ajh.20353. [DOI] [PubMed] [Google Scholar]
- 23.Lanzkron S, Haywood C, Hassell KL, Rand C. Provider barriers to hydroxyurea use in adults with sickle cell disease: a survey of the Sickle Cell Disease Adult Provider Network. J Natl Med Assoc. 2008;100:968–73. [PubMed] [Google Scholar]
- 24.de Montalembert M, Roberts I. Sickle cell disease: primum non nocere (first do no harm) Haematologica. 2010;95:4–5. doi: 10.3324/haematol.2009.020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oyeku SO, Driscoll MC, Cohen HW, Trachtman R, Pashankar F, Mullen C, Giardina PJ, Velazco N, Racine AD, Green NS. Parental and other factors associated with hydroxyurea use for pediatric sickle cell disease. Pediatr Blood Cancer. 2013;60:653–8. doi: 10.1002/pbc.24381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haywood C, Beach MC, Lanzkron S, Strouse JJ, Wilson R, Park H, Witkop C, Bass EB, Segal JB. A systematic review of barriers and interventions to improve appropriate use of therapies for sickle cell disease. J Natl Med Assoc. 2009;101:1022–33. doi: 10.1016/s0027-9684(15)31069-5. [DOI] [PubMed] [Google Scholar]
- 27.Crosby LE, Shook LM, Ware RE, Brinkman WB. Shared decision making for hydroxyurea treatment initiation in children with sickle cell anemia. Pediatr Blood Cancer. 2015;62:184–5. doi: 10.1002/pbc.25124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elander J, Beach MC, Haywood C. Respect, trust, and the management of sickle cell disease pain in hospital: comparative analysis of concern-raising behaviors, preliminary model, and agenda for international collaborative research to inform practice. Ethn Health. 2011;16:405–21. doi: 10.1080/13557858.2011.555520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thornburg CD, Calatroni A, Telen M, Kemper AR. Adherence to hydroxyurea therapy in children with sickle cell anemia. J Pediatr. 2010;156:415–9. doi: 10.1016/j.jpeds.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hankins J, Hinds P, Day S, Carroll Y, Li C-S, Garvie P, Wang W. Therapy preference and decision-making among patients with severe sickle cell anemia and their families. Pediatr Blood Cancer. 2007;48:705–10. doi: 10.1002/pbc.20903. [DOI] [PubMed] [Google Scholar]
- 31.Berthaut I, Bachir D, Kotti S, Chalas C, Stankovic K, Eustache F, Ravel C, Habibi A, Brailly-Tabard S, Lévy-Dutel L, Bleibtreu A, Simon T, Galactéros F, Lionnet F, Mandelbaum J. Adverse effect of hydroxyurea on spermatogenesis in patients with sickle cell anemia after six months of treatment. Blood. 2017;130:2354–6. doi: 10.1182/blood-2017-03-771857. [DOI] [PubMed] [Google Scholar]
- 32.Bakshi N, Sinha CB, Ross D, Khemani K, Loewenstein G, Krishnamurti L. Proponent or collaborative: Physician perspectives and approaches to disease modifying therapies in sickle cell disease. In: Young B, editor. PLOS ONE. Vol. 12. 2017. p. e0178413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belamarich PF, Gandica R, Stein REK, Racine AD. Drowning in a sea of advice: pediatricians and American Academy of Pediatrics policy statements. Pediatrics. 2006;118:e964–78. doi: 10.1542/peds.2006-0652. [DOI] [PubMed] [Google Scholar]
- 34.Henry Louis Gates J. The Signifying Monkey. Oxford University Press; 2014. [Google Scholar]
- 35.LaVista JM, Treise DM, Dunbar LN, Ritho J, Hartzema AG, Lottenberg R. Development and evaluation of a patient empowerment video to promote hydroxyurea adoption in sickle cell disease. J Natl Med Assoc. 2009;101:251–7. doi: 10.1016/s0027-9684(15)30853-1. [DOI] [PubMed] [Google Scholar]
- 36.McClure E, Ng J, Vitzthum K, Rudd R. A Mismatch Between Patient Education Materials About Sickle Cell Disease and the Literacy Level of Their Intended Audience. Prev Chronic Dis. 2016;13:E64. doi: 10.5888/pcd13.150478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carden MA, Newlin J, Smith W, Sisler I. Health literacy and disease-specific knowledge of caregivers for children with sickle cell disease. Pediatr Hematol Oncol. 2016;33:121. doi: 10.3109/08880018.2016.1147108. [DOI] [PubMed] [Google Scholar]
- 38.Machuca H, Arevalo S, Hackley B, Applebaum J, Mishkin A, Heo M, Shapiro A. Well Baby Group Care: Evaluation of a Promising Intervention for Primary Obesity Prevention in Toddlers. Child Obes. 2016;12:171–8. doi: 10.1089/chi.2015.0212. [DOI] [PubMed] [Google Scholar]
- 39.Coker TR, Windon A, Moreno C, Schuster MA, Chung PJ. Well-Child Care Clinical Practice Redesign for Young Children: A Systematic Review of Strategies and Tools. Pediatrics. 2013;131:S5–S25. doi: 10.1542/peds.2012-1427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Madderom MJ, Heijdra J, Utens EMWJ, Polinder S, Rijneveld AW, Cnossen MH. A randomized controlled trial studying the effectiveness of group medical appointments on self-efficacy and adherence in sickle cell disease (TEAM study): study protocol. BMC Hematology. 2016;16:21. doi: 10.1186/s12878-016-0058-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shook LM, Farrell CB, Kalinyak KA, Nelson SC, Hardesty BM, Rampersad AG, Saving KL, Whitten-Shurney WJ, Panepinto JA, Ware RE, Crosby LE. Translating sickle cell guidelines into practice for primary care providers with Project ECHO. Med Educ Online. 2016;21:33616. doi: 10.3402/meo.v21.33616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kodish E, Lantos J, Stocking C, Singer PA, Siegler M, Johnson FL. Bone marrow transplantation for sickle cell disease. A study of parents’ decisions. N Engl J Med. 1991;325:1349–53. doi: 10.1056/NEJM199111073251905. [DOI] [PubMed] [Google Scholar]
- 43.Van Besien K, Koshy M, Anderson-Shaw L, Talishy N, Dorn L, Devine S, Yassine M, Kodish E. Allogeneic stem cell transplantation for sickle cell disease. A study of patients’ decisions. Bone Marrow Transplant. 2001;28:545–9. doi: 10.1038/sj.bmt.1703208. [DOI] [PubMed] [Google Scholar]
- 44.Meier ER, Dioguardi JV, Kamani N. Current attitudes of parents and patients toward hematopoietic stem cell transplantation for sickle cell anemia. Pediatr Blood Cancer. 2015;62:1277–84. doi: 10.1002/pbc.25446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.