

Abstract
Primordial dwarfism (PD) is a group of rare genetically heterogeneous disorders consisted of disorders with intrauterine growth retardation continued through the life. SOFT syndrome with characteristics of short stature, onychodysplasia, facial dysmorphism, and hypotrichosis has been presented as a subtype of PD. Only 20 cases of SOFT syndrome have been reported in world to date, but none of them were not in Iran. Our case was 6.5-year-old girl with a complaint of growth retardation including height of 97 cm (Z = −4.6 standard deviation [SD]) and weight of 14 kg (Z = −4 SD) referred to growth clinic. She had a prominent forehead, triangular face, short limbs, malformed nails, and crowded teeth and her psychomotor function was normal. Laboratory and karyotype tests were normal while she was homozygous for c.G491A mutation of POC1A gene thus SOFT syndrome diagnosis was confirmed for her and recombinant growth hormone therapy was discontinued.
Keywords: Growth retardation, primordial dwarfism, short stature, SOFT syndrome
Introduction
Short stature as a common complaint of pediatrics health clinics is in association with various underlying etiologies. This condition may be detected in a well-child during routine child visits or following a chronic condition. In a patient with short stature, a complete history of patients' medical, social, and familial state should be evaluated. This medical history can reflect underlying etiology of this condition as it can reveal an intrauterine or after birth growth retardation, an acute or chronic condition and renal, gastrointestinal, or hormonal impairments.[1]
Primordial dwarfism (PD) is one of the rare genetically heterogeneous disorders consisted of a group of disorders with intrauterine growth retardation that would continue after the birth as well, although there is no chromosomal or hormonal disorder.[2]
Variety of clinical features for a description of PD has been recommended previously, while the common one is head circumference. Microcephaly is the most common finding of these patients that causes small size of the brain. Some subtypes of PD with microcephaly are Seckel syndrome, microcephalic osteodysplastic PD type I and type II. On the other hand, there are few PD syndromes with normal head circumference that have relatively normal intelligence. These syndromes include Silver–Russell syndrome, Mulibrey nanism, and 3M syndrome.[3]
A new disorder known as SOFT syndrome with characteristics of short stature, onychodysplasia, facial dysmorphism, and hypotrichosis has been presented as a new subtype of PD. This disorder is incompatible with usual features of other PD subtypes as these children have macrocephaly relatively, but due to low speed of head circumference growth, they have significant microcephaly in adulthood.[4,5] Mutation of POC1A has been presented previously as the responsible gene for SOFT incidence.[3]
In following, a case of SOFT syndrome from Iran is presented.
Case Report
A 6.5-year-old girl with a chief complaint of growth retarda tion referred to growth center affiliated to Isfahan University of Medical Sciences.
Physical examination
Her height at referral time was 97 cm (Z = −4.6 standard deviation [SD]), her weight was 14 kilograms (Z = −4 SD), and her head circumference was 50.5 cm (25 percentile). The patient was treated with growth hormone since 2.5-year-old, but no significant response was found.
In physical examination, her motor function and social network were normal. In addition, her intelligence quotient (IQ based on Raven test) was 113. In her appearance, she had dolichocephaly, prominent forehead, triangular face, pointed chin, and brief scattered hair at the upper parts of the head. She had short fingers and limbs with malformed nails. Her teeth were crowded. She did not have organomegaly and genitalia were feminine and normal [Figure 1].
Figure 1.
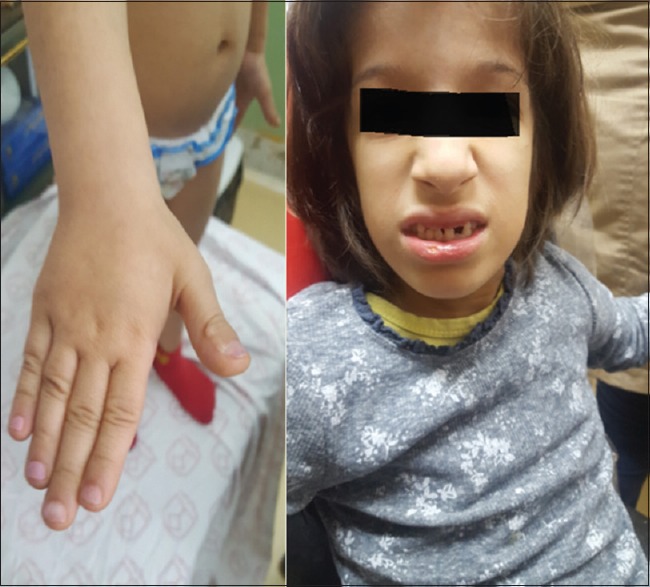
Patients appearance. This photograph presents facial dysmorphism including; a triangular face, pointed chin, sparse hair, nail hypoplasia, brachydactyly, crowded teeth and frontal bossing
Past medical history
In the patient's medical history, a sonography of 29th gestational week was presented with manifestations of lower normal limitation of amniotic fluid with fetus weight under three percentile. The fetus was at the risk of intrauterine growth retardation.
She was born in 39 gestational weeks through cesarean section because of meconium passage; with birth weight of 2100 g (Z = −3.1 SD), birth length of 46 cm (3 percentile), and head circumference of 32 cm (15 percentile). Due to meconium aspiration, she was admitted to Neonatal Intensive Care Unit (NICU), and due to hypotonia and metabolic acidosis, brain magnetic resonance imaging, and metabolic screening tests were done for her that were normal. In obtained echocardiography, she had atrial septal defect (ASD), ventricular septal defect (VSD), and patent ductus arteriosus (PDA). She was discharged from NICU after 24 days.
In the 6th month, she underwent another echocardiography again. This one presented that ASD and VSD have repaired spontaneously, but PDA had remained. Thus, she underwent surgical repair for her PDA when she was 8 months. As patients with SOFT syndrome would not have cardiac presentations; this abnormality may be irrelevant with her presentations of SOFT syndrome.
In the 13th month, she referred to endocrine clinic for her short stature as her weight, height, and head circumference was 4.8 Kg (Z = −4.6 SD), 62 cm (Z = −5.1 SD), and 44 cm (25 percentile), respectively.
In 2.5-year-old, growth hormone treatment was initiated for her due to the height of 73 cm (Z = −4.7 SD).
Family History
She was the only child of the Iranian relative parents with mother's height of 166 cm and father's height of 181 cm. The mother did not have any history of miscarriage.
In her family history, parents have a common cousin with SOFT syndrome, but he is living in Denmark. He is a 1-year-old boy with the normal developmental state that studies in usual schools. Now, he has 128 cm height and 37 kg weight. He was treated with growth hormone for 3 years, and because of lack of response, the remedy was discontinued.
Laboratory and Karyotyping Test
In this step, thyroid function tests, complete blood count, urine analysis, Vitamin D level, growth hormone and IGFBP3, sweat test, celiac test, Sudan black test, and urinary tract sonography were requested. All of them were normal.
When she was 5.5-year-old, bone surveys showed hypoplastic iliac bones, shallow acetabuli, short femoral necks, metaphyseal irregularity, flat epiphysis of knees, short phalanges, short and broad metacarpal and matatarsal bones, cupping of metaphysis, and cone-shaped epiphysis of phalanges [Figure 2].
Figure 2.
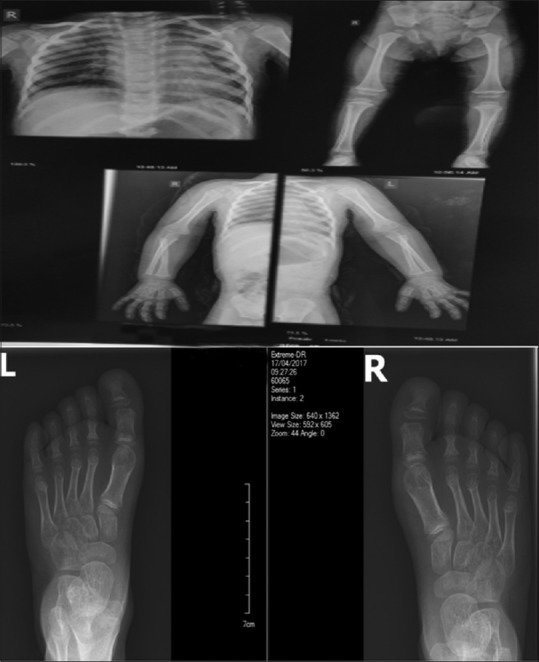
Minimal cupping at the phalangeal basis, short phalanges, short and broad metacarpal and metatarsal bones, cone-shaped epiphysis and metaphyseal cupping at middle and distal phalanges. Narrow and vertical appearance of the iliac bone with shallow acetabuli and short femoral necks. Knee metaphyseal flaring with mild epiphyseal dysplasia and slender diaphysis of long bones. Short fibula and tall vertebrae are seen
Therefore, skeletal dysplasia diagnosis was suspected for her and FGFR3 genetic test was done to rule out of achondroplasia/hypochondroplasia. However, the result of this test was negative. In addition, her chromosomal karyotyping was 46XX.
Genetic study
By all mentioned facts, genetic test of POC1A was requested for all family members. Her father and mother were heterozygous for c.G491A gene and the patient was homozygous.
Treatment
Although the patient was under recombinant growth hormone therapy with the proof of SOFT syndrome diagnosis, the growth hormone therapy was discontinued.
Discussion
In the current study, we present a case of SOFT syndrome that was the first reported case of this syndrome in Iran. A patient with short stature (Z = −4.6 SD) and severe underweight (Z = −4) referred to University Growth Clinic. The patient was normocephalic with normal IQ. She had skeletal abnormalities including; prominent forehead, triangular face, short fingers and limbs, malformed nails, and irregular teeth. In addition, brief scattered hair at upper parts of her head was seen. Various laboratory and karyotype tests were normal. Thus, genetic test of POC1A was assessed for her and by positive result of this genetic test, diagnosis of SOFT syndrome was confirmed for her.
Normocephalic PD is attributed to following syndromes including SOFT, 3M syndrome, Mulibrey nanism, and Russell–Silver syndrome. These syndromes have overlapping clinical findings that complicate their differentiation.[3]
Characteristics of patients with SOFT disorder have been presented as extremely reduced head circumference of adulthood while it would be normal at early childhood. The patients usually have normal social and motor development. Dolichocephaly, frontal bossing, triangular face, pointed chin, and sparse hair have been reported as facial dysmorphism features. In addition, high-pitched voice can be detected in these patients. They have hypoplastic fingernails and brachydactyly too. Metaphysical irregularity of long bones, hypoplasticity of sacrum and pelvic, delayed ossification of bones and cone-shaped epiphyses are major skeletal findings of SOFT-affected patients. In general, mentioned skeletal and facial dysmorphism features and growth retardation are common in all case of SOFT syndrome.[2] Some patients have hypogonadism and secondary sex characteristic delay and oligo-azospermia;[5] while rarely precocious puberty has been presented in SOFT-affected patients.[3]
Growth retardation findings during pregnancy can be found using limb size through ultrasonography. Unusually, shortened limbs have been presented during pregnancy in their second trimester while all others had a history of growth retardation in their third trimester of pregnancy.[6] In the current patient, growth retardation was found in her third-trimester ultrasonography.
Shalev et al. presented cases of two Arab Muslim families who had inherited following features in autosomal recessive manner. These nine patients had intrauterine growth retardation found in the second trimester through ultrasonography. They had normal psychomotor function while their normal head circumference in their childhood had been turned to markedly microcephaly through their adulthood. Their final height was consistent with a normal individual with 6–8 years of age. Short long bones, hair paucity in face and nail abnormalities were detected in these individuals. In addition, they had facial dysmorphism of triangular face and prominent nose.[5,6]
The other point about SOFT patients similar to all other PD patients is lack of expectable response to recombinant human growth hormone therapy, as the use of this remedy in these patients may cause metabolic syndrome developing.[7] This nonresponse to growth hormone therapy in our patient made us abandon recombinant human growth hormone remedy.
Several gene mutations have been presented for combined bone and skin developmental defects. Most of these genes are responsible for extracellular matrix protein formation.[8] On the other hand, studies are increasingly presenting new genetic defects in association with skeletal and ectodermal defects.[9] Recently, variety of genetic tests has been provided to play role in the diagnosis of PD. As mentioned above, normocephalic PDs have clinical features in common that necessitates detecting of particular gene sequences responsible for the incidence of that specific phenotype. Carrying a mutation of POC1A gene has been attributed to SOFT syndrome by other studies.[10] Poc1 gene in human consists of two subtypes of POC1A and POC1B. This gene contains WD40 domain which is responsible for the centriole/basal body centriole contact. Centrioles play a significant role in cell mitosis. In addition, centrosomal functions are important for Golgi assembly. Previously nonsense mutations of this gene was presented as underlying etiology of hypomorphism in SOFT patients[4] while there are studies that have presented rare missense mutations responsible for this phenotype.[3,5] Moreover, Chen et al. have reported that exon 10 part of mRNA mutation detected in POC1A was responsible for the incidence of SOFT syndrome, as this mRNA caused dysfunction of WD40 domain which poses centriole dysfunction.[11] In general, variety of mutations has been found in POC1A gene while all were attributed to SOFT syndrome incidence. Table 1 compares skeletal manifestations of POC1A-positive patients presented in various studies.
Table 1.
Skletal manifestation of proved POC1A mutation positive patients
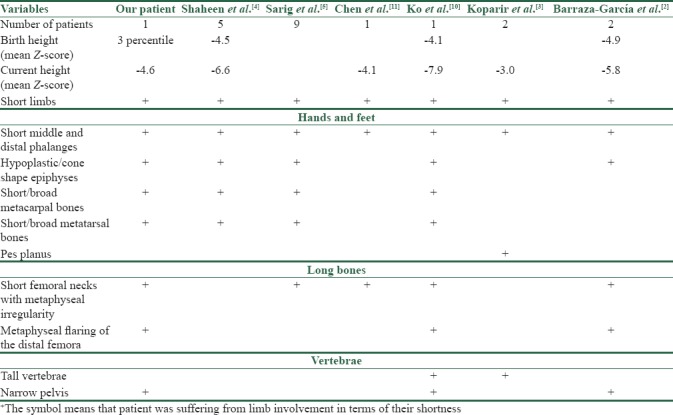
This finding was similar to the patient of our study that was homozygous for POC1A gene presents autosomal recessive role of this gene for this phenotype incidence.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Rogol AD, Hayden GF. Etiologies and early diagnosis of short stature and growth failure in children and adolescents. J Pediatr. 2014;164:S1–14.e6. doi: 10.1016/j.jpeds.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 2.Barraza-García J, Iván Rivera-Pedroza C, Salamanca L, Belinchón A, López-González V, Sentchordi-Montané L, et al. Two novel POC1A mutations in the primordial dwarfism, SOFT syndrome: Clinical homogeneity but also unreported malformations. Am J Med Genet A. 2016;170A:210–6. doi: 10.1002/ajmg.a.37393. [DOI] [PubMed] [Google Scholar]
- 3.Koparir A, Karatas OF, Yuceturk B, Yuksel B, Bayrak AO, Gerdan OF, et al. Novel POC1A mutation in primordial dwarfism reveals new insights for centriole biogenesis. Hum Mol Genet. 2015;24:5378–87. doi: 10.1093/hmg/ddv261. [DOI] [PubMed] [Google Scholar]
- 4.Shaheen R, Faqeih E, Shamseldin HE, Noche RR, Sunker A, Alshammari MJ, et al. POC1A truncation mutation causes a ciliopathy in humans characterized by primordial dwarfism. Am J Hum Genet. 2012;91:330–6. doi: 10.1016/j.ajhg.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarig O, Nahum S, Rapaport D, Ishida-Yamamoto A, Fuchs-Telem D, Qiaoli L, et al. Short stature, onychodysplasia, facial dysmorphism, and hypotrichosis syndrome is caused by a POC1A mutation. Am J Hum Genet. 2012;91:337–42. doi: 10.1016/j.ajhg.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shalev SA, Spiegel R, Borochowitz ZU. A distinctive autosomal recessive syndrome of severe disproportionate short stature with short long bones, brachydactyly, and hypotrichosis in two consanguineous arab families. Eur J Med Genet. 2012;55:256–64. doi: 10.1016/j.ejmg.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 7.Bober MB, Niiler T, Duker AL, Murray JE, Ketterer T, Harley ME, et al. Growth in individuals with majewski osteodysplastic primordial dwarfism type II caused by pericentrin mutations. Am J Med Genet A. 2012;158A:2719–25. doi: 10.1002/ajmg.a.35447. [DOI] [PubMed] [Google Scholar]
- 8.Bateman JF, Boot-Handford RP, Lamandé SR. Genetic diseases of connective tissues: Cellular and extracellular effects of ECM mutations. Nat Rev Genet. 2009;10:173–83. doi: 10.1038/nrg2520. [DOI] [PubMed] [Google Scholar]
- 9.Uppal S, Diggle CP, Carr IM, Fishwick CW, Ahmed M, Ibrahim GH, et al. Mutations in 15-hydroxyprostaglandin dehydrogenase cause primary hypertrophic osteoarthropathy. Nat Genet. 2008;40:789–93. doi: 10.1038/ng.153. [DOI] [PubMed] [Google Scholar]
- 10.Ko JM, Jung S, Seo J, Shin CH, Cheong HI, Choi M, et al. SOFT syndrome caused by compound heterozygous mutations of POC1A and its skeletal manifestation. J Hum Genet. 2016;61:561–4. doi: 10.1038/jhg.2015.174. [DOI] [PubMed] [Google Scholar]
- 11.Chen JH, Segni M, Payne F, Huang-Doran I, Sleigh A, Adams C, et al. Truncation of POC1A associated with short stature and extreme insulin resistance. J Mol Endocrinol. 2015;55:147–58. doi: 10.1530/JME-15-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]