

Abstract
Low-density lipoprotein cholesterol (LDL-C) is a most important risk factor for developing coronary artery disease (CAD) and other forms of atherosclerotic cardiovascular disease (CVD) and a major focus of CVD risk reduction with lifestyle and statins. Unfortunately residual risk of CVD remains in patients with familial hypercholesterolaemia and/or statin intolerance in whom adequate LDL-C lowering is not accomplished with lifestyle and statins. PCSK9 is a serine protease that binds the LDL receptor (LDL-R) and acts as a chaparone for endocytosis and shuttling the PCSK9-LDLR complex to lysosomes for degradation. In the absence of PCSK9 the LDLR-LDL-C complex dissociates and LDL-R is recycled back to the cell surface. Humanised monoclonal antibodies (evolocumab, alirocumab, bocolicumab) have been developed that increase LDL-R by ~2-fold and lower LDL-C by up to 75 percent. This effect is synergistic to that of statins with the only common adverse effect is a local injection site reaction. At present, ongoing Phase III CVD outcome trials with PCSK9 inhibitors offer promise that patients with LDL-C levels that remain elevated can decrease CVD events and related mortality.
Keywords: PCSK9, CHD, CVD, statins, LDL-C, LDL receptor, prevention
Elevated levels of low-density lipoprotein cholesterol (LDL-C) have long been established as one of the most important risk factors for developing coronary artery disease (CAD) and other forms of atherosclerotic cardiovascular disease (CVD).[1,2] Targeting LDL-C reduction has been effective in lowering cardiovascular risk.[3,4] The use of HMG-CoA reductase inhibitors (statins) for both primary and secondary cardiovascular disease prevention has become first-line therapy and has resulted in reduction of cardiovascular (CV) events and overall mortality.[3] More recent guidelines from the American College of Cardiology (ACC) and American Heart Association (AHA) have recommended intensification of statin therapy based on calculated atherosclerotic CVD risk and not based on LDL-C levels alone.[5] However, despite reduction in LDL-C by 18–41 % (moderate doses) and 40–60 % (higher doses or more potent statins), there remains significant residual cardiovascular risk. In addition, there are many otherindividuals that have been unable to achieve sufficient LDL-C lowering or have been intolerant to the drug completely.[6] Other cholesterol lowering agents (bile acid sequestrants, ezetimibe, niacin, fibrates) have been used in conjunction with statins to achieve historic LDL and/or non-HDL-C goals, however at this time their addition has not been proven to have added value in clinical cardiovascular outcomes.[7–10] Due to continued inability to achieve adequate clinical benefit and ongoing incidence of statin intolerance, research for alternative therapies is of utmost priority. The discovery of proprotein convertase subtilisin/kexin Type 9 (PCSK9) has opened the possibility for effective and adjunctive therapy for those who are not optimised with statins while providing therapy in lowering cardiovascular risk in subjects who are intolerant and have little alternatives.
LDL Cholesterol Cellular Metabolism
Most LDL is cleared from the plasma by LDL receptors on hepatocytes. At neutral pH, the LDL-receptor (LDL-R) binds to apolipoprotein B-100 (apoB100) on LDL and LDL endocytosis ensues.[11] In the acidic environment of the endosome, the LDL-R folds into a closed formation state, thereby releasing LDL.[12] LDL-R is then recycled to the cell surface while LDL is transferred to a proteasome targeting it for further processing.[13] The inability of LDLR-LDLP complex to dissociate routes the complex for degradation, thereby reducing LDL-R surface concentration. Regulation of the LDL-R is also driven by the intracellular concentration of cholesterol. In the setting of high intracellular cholesterol levels, LDL-R levels decrease and activity of HMG CoA synthase and reductase decrease by more than 90 %. Thus, in the setting of drugs that lower intracellular cholesterol, such as statins, LDL-R levels are increased and serum LDL-C levels are decreased.
Cellular mechanism regulating cholesterol homeostasis is complex and involves balance between intracellular biosynthesis and exogenous uptake of cholesterol. Cholesterol uptake and intracellular biosynthesis is regulated by sterol regulatory element-binding protein (SREBP), a transcription factor located in the endoplasmic reticulum. When cholesterol levels on the endoplasmic reticulum membrane are low, a sterol sensor binds to SREBP and signals its movement from the endoplasmic reticulum to the Golgi apparatus where SREBP is further activated and transported to the nucleus.[14] In the nucleus, SREBP acts as a transcription factor, increasing HMG coA reductase and LDLR gene expression.[13]
Genetics of PCSK9
PCSK9 was first discovered as a ninth member of the subtilase subfamily,[15] Its function was initially largely unknown until its role in hypercholesterolaemia was realised in a group of French families with autosomal dominant familial hyperlipidaemia (FH). These individuals did not exhibit the usual genetic mutations in the LDLR (LDL receptor) or APOB (apolipoprotein B) genes.[16] Instead, the genotype of these FH cohorts revealed multiple missense mutations in the PCSK9 gene resulting in defective intracellular PCSK9 processing (see Table 1). A gain of function mutation results from the defective pro-PCSK9 processing and causes either an over expression of PCSK9 or an increased affinity for the LDL receptor.[17] These gain of function mutations have shown to significantly decrease LDL receptors by as much as 35 % and consequently increase serum LDL-C levels.[18] Targeting PCSK9 as a therapeutic strategy was conceptualised after loss of function mutations were discovered in both African American and Caucasian populations (see Table 1).[19,20] Subjects with low LDL-C in the Dallas Heart Study had PCSK9 sequencing that identified two non-sense mutations in 2 % of the African American cohort resulting in low levels of PCSK9 and a marked reduction of plasma LDL-C of 40 %.[21] Similarly, missense mutations were also discovered in the Caucasian population that was associated with 30 % decrease in LDL-C.[19] Subjects with loss of function mutations and its association with ischaemic heart disease have been evaluated in a meta-analysis. In multiple groups of subjects with loss of function mutations in the PCSK9 gene, there was a 13 % decrease of LDL-C that was associated with 30 % risk reduction in incidence of ischemic heart disease.[20] The higher than expected CVD risk reduction is thought to be from life-long exposure to very low LDL-C levels.
Table 1: Genetic Mutations of the PCSK9 Gene.
| Type of Mutation | Examples | Effects | |
|---|---|---|---|
| Gain of function mutation | Missense | S127R F216L D374Y C161T I474V |
Increase levels of PCSK9 or affinity for LDL receptor |
| Loss of function mutation | Nonsense Missense |
Y142X C679X R46L |
Introduce stop codon |
Premature termination signal and truncated protein. LDL = low-density lipoprotein.
Molecular Mechanism of PCSK9
PCSK9 is a serine protease that belongs to a subfamily of subtilisin proteases responsible for activation or inactivation of other proteins, such as hormones, growth factors and other enzymes. It was initially identified as a seceretory proprotein convertase neural apoptosis-regulated convertase-1 (NARC-1) prior to the discovery of its role in cholesterol metabolism.[15] PCSK9 proprotein is processed in the endoplasmic reticulum where it undergoes autocatalytic cleavage producing a cleaved, but bound, prodomain and catalytic subunit. The bound prodomain makes the catalytic domain enzymatically inactive, unlike other serine proteases, acting instead as a binding protein when secreted. In the extracellular space, PCSK9 binds to LDL receptor via its catalytic subunit while its C-terminal subunit acts as a chaparone for endocytosis and shuttling the PCSK9-LDLR-LDL complex to lysosomes for degradation.[22] In the absence of PCSK9 and in the acidic environment of the clathrin coated endosome, LDLR-LDL complex dissociates and LDLR is recycled back to the cell surface (see Figure 1).[23]
Figure 1: PCSK9, LDL-P and LDL-R Cellular Metabolism.
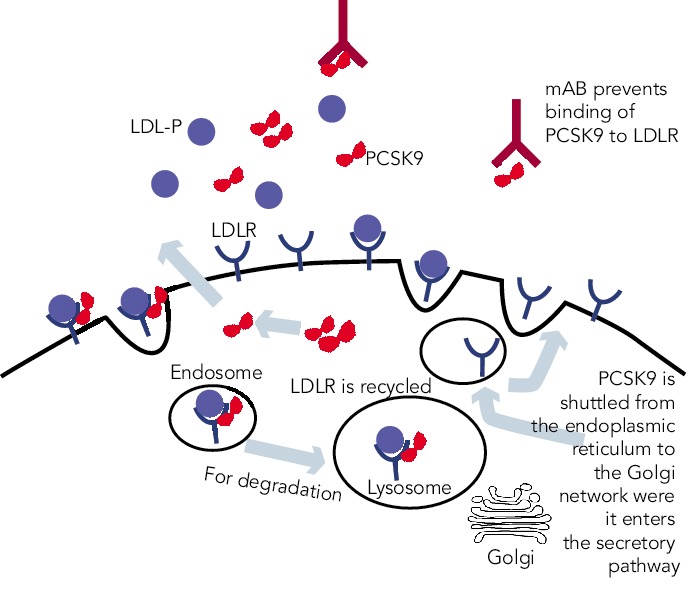
PCSK 9 enters the secretory pathway through the Golgi network. Circulating PCSK9 binds to LDL-R on cellular surface of hepatocytes. Bound LDL-P to LDL-R is internalised along with PCSK9. In the endosome, the bound PCSK9 targets LDL-R in the endosome for degradation, subsequently lowering LDL-R levels and increasing circulating LDL-P and LDL-C. LDL-R = LDL receptor; LDL-P = LDL particle.
The PCSK9 gene is located on chromosome 1p32 and its expression is also regulated by intracellular cholesterol, via SREBP-2.[24] Synthesis of PCSK9 occurs mostly in the liver, small intestine and kidney and is secreted as a soluble enzyme.[25] After secretion, 30–40 % of PCSK9 binds to LDL, a process that may inhibit its uptake of LDL by LDLR.[26] Low intracellular levels of cholesterol not only stimulate the synthesis of LDL-R but also PCSK9, serving as a counter-regulatory mechanism to maintain intracellular delivery of cholesterol. In the setting of statin, fibrate and ezetimibe use, PCSK9 expression is up-regulated due to low intracellular cholesterol levels.[27] The degree of PCSK9 expression correlates with degree of LDL-C levels in subjects treated with statin, attenuating the cholesterol-lowering effects of statin.[28] Thus, PCSK9 inhibition is additive to statin therapy and play a synergistic role in its lipid-lowering effects.
Inhibition of PCSK9
PCSK9 inhibition is currently achieved by either preventing PCSK9 synthesis or inhibiting the binding of PCSK9 to LDL-R. The most clinically advanced mechanism by which inhibition of PCSK9 has been achieved is through the development of monoclonal antibodies. Other mechanisms, such as mimetic peptides, antisense RNA inhibition and natural binding inhibitors are mostly in preclinical development (see Table 2).
Table 2: Approaches and Mechanism of PCSK9 Inhibition.
| Approaches to PCSK9 Inhibition | Mechanism | Example | Phase |
|---|---|---|---|
| Gene Silencing | Antisense oligonucleotide Small interfering RNA (siRNA) |
ISIS pharmaceutical[1a] Santaris pharma[2a] Alnylam Pharmaceuticals[3a] |
Preclinical Phase I |
| Mimetic Peptides | Peptide that mimics EGFA domain of LDLR and binds PCSK9[4a,5a] Peptide that mimics PCSK9 and binds LDLR[6a] |
Genentech, Inc |
Preclinical Preclinical |
| Small molecule inhibitors | Target binds of PCSK9 | Natural inhibitor Annexin[7a,8a] Berberine[9a,10a] BMS-962476[11a] |
Preclinical |
| Monoclonal antibodies | Targeting catalytic domain of PCSK9 | Amgen AMG145 (evolocumab) Regeneron SAR236553/REGN7270 Pfizer RN316 (bococizumab) Novartis LGT209 Genentech MPSK3169A |
Phase I - III |
LDLR = LDL receptor
Monoclonal Antibodies
In 2009, the first successful development of monoclonal antibody against PCSK9 was developed by Chan et al.[29] The fully human monoclonal antibody (mAB) binds to both the catalytic and prodomain sites preventing PCSK9 binding to LDL-R.[29] Monoclonal AB was shown to increase LDL receptor levels by 1.7-2.2-fold and had synergistic effects when administered concomitantly with statin.[29] Phase I studies showed significant efficacy in reducing LDL-C levels with minimal adverse events.[30,31] Almost simultaneously, multiple monoclonal antibodies have been developed with rapid completion of phase II trials and now ongoing phase III clinical trials.[32–34]
Alirocumab (REGN727/SAR236553)
Initial phase I clinical trials of alirocumab showed a dose-dependent LDL-C lowering effects. In one phase I clinical trial, escalating subcutaneous doses of 50, 100, and 150 mg doses were compared to placebo, lowering LDL-C by 39, 54 and 61 %, respectively (see Table 3).[31] Phase II clinical trials have evaluated alirocumab’s use in heterozygous FH and in subjects with hyperlipidaemia on either stable doses of statin or escalating doses of statin (low and high dose atorvastatin). In the phase II clinical trial evaluating the use of alirocumab in the background of stable doses of atorvastatin (low or high dose) in subjects with LDL-C ≥ 100 mg, LDL-C decreased by as much as 72 % with 150 mg administered every two weeks.[35,36] This study also showed significant decreases in apolipoprotein B (apoB), non-high density lipoprotein cholesterol (non-HDL-C) and lipoprotein (a) [Lp(a)].[35] The mechanism by which Lp(a) is lowered is unknown, but maybe related to the decrease in the availability of LDL-apoB [known to decrease Lp(a)] and the increased clearance of apolipoprotein(a).[37]
Table 3: Monoclonal Antibodies Dose and LDL-C Reduction.
| Monoclonal Antibodies | Reduction of LDL-C from Baseline |
|---|---|
| Alirocumab | |
| 150 mg every two weeks 300 mg every four weeks |
66–72 % 43–48 % |
| Evolocumab | |
| 140 mg every two weeks 420 mg every four weeks |
51–76 % 48–71 % |
| Bococizumab | |
| 150 mg every two weeks 300 mg every four weeks |
33 % (0.5 mg/kg) – 85 % (18 mg/kg) Mean reduction 53 mg/dL Mean reduction 45 mg/dL |
low-density lipoprotein cholesterol
In the phase II clinical trial evaluating the safety and efficacy of escalating dose of alirocumab in subjects with heterozygous FH, alirocumab was given either every two (Q2 week) or four (Q4 week) weeks in the background statin use with or without ezetimibe.[38] Alirocumab was well-tolerated and effective in adequately treating subjects with FH (68 % reduction in LDL-C with 150 mg SC dose Q2 weeks compared with 11 % in the placebo group).[38] The ODYSSEY program is currently underway and is a collection of 14 clinical phase III trials evaluating the efficacy and safety of alirocumab in various patient populations and clinical settings. All trials in the program are designed to treat to goal levels of LDL-C and evaluates efficacy in patient population that are not optimised with current standard therapies. This program involves over 2,000 international sites with over 23,500 subjects.[39] The largest study will be evaluating clinical cardiovascular outcomes (ODYSSEY-OUTCOMES), which is enrolling subjects with prior history of acute coronary syndrome already on optimised-guideline based therapy.[40] In the recent European Society of Cardiology (ESC) scientific meeting, preliminary results from the ODYSSEY program were presented. Particularly intriguing was a post-hoc analysis of the ODYSSEY LONG TERM trial evaluating LDL-C lowering effects of alirocumab in patients with heterozygous FH or patients with high CV risk. Not surprisingly, alirocumab was shown to decrease LDL-C levels by 61 % compared to placebo. However, after only one year of the intervention post-hoc analysis showed a lower risk for CV events (HR 0.46, CI 0.26-0.82, p = <0.01) (ESC Barcelona Spain 2014).
Evolocumab (AMG 145)
Evolocumab, a full human monoclonal antibody, is administered subcutaneously either as every two weeks or every four weeks dosing regimen. Koren at al. showed that escalating doses of evolocumab as a monotherapy in both Q2 week or Q4 week dosing administered subcutaneously to subjects with history of hypercholesterolaemia and low cardiovascular risk (Framingham risk score < 10 %) decreased LDL-C from baseline by up to 48 %.[41,42] Stein et al. in the phase II Goal achievement after utilising an anti-PCSK9 antibody in statin intolerant subjects (GAUSS) evaluated the efficacy and safety of evolocumab in patients with statin intolerance.[43] Patients receiving escalating doses experienced a significantly reduced LDL-C compared to placebo. At 280 mg dose, LDL-C was reduced by 41 %.[43] When escalated to a dose of 420 mg, LDL-C was reduced by 51 %.[43] This study was expanded to a phase III trial in subjects with a history of hypercholesterolemia not at previously established National Cholesterol Education Program Adult Treatment Program III (NCEP-ATP III) risk-based goals and intolerant to statins.[44] As in the phase II results, any regimen administered significantly reduced LDL-C compared to ezetimibe, with the highest reduction (61 %) seen with evolocumab 420 mg + ezetimibe 10 mg. When administered to subjects who were already taking statin, administration of evolocumab further decreased LDL-C by 63–75 % compared to placebo.[45] The use of evolocumab was also studied in subjects with heterozygous FH already on maximized tolerated lipid therapy (stable dose of statin with or without ezetimibe).[46,47] As in other studies, the largest additional decrease in LDL-C was seen with dose of 420 mg Q4 week (55 % additional LDL-C reduction).[44] In subjects with known CAD or CAD risk equivalent and not at NCEP-ATP III goal of LDL-C ≤ 70 mg/dL, evolocumab was studied as an adjunct to stable doses of statin with or without ezetimibe.[48] When administered in escalating doses either as a Q2 week or Q4 week regimen significant reductions in LDL-C occurred. The Q2 week regimen at 140 mg, achieved the highest LDL-C reduction at 12 weeks (64 %). Each dosing group also significantly achieved NCEP-ATP III treatment goal when compared to placebo (90 % of those in the Q2 week 140 mg group achieved LDL-C ≤ 70 mg/dL).[48] An analysis from this study also showed therapy with evolocumab decreased Lp(a) by 23 % compared to placebo.[49] As previously discussed, mechanisms by which this effect occurs is unknown.
The use of evolocumab was also studied in homozygous FH, a rare but severely elevated LDL-C, wherein very high CVD risk with possible death in childhood occurs. In the small study by Stein et al., the effect of evolocumab was studied in both LDL-R negative subjects and LDL-R defective patients. Evolocumab significantly reduced LDL-C by 26 % in only the LDL-R defective subjects. In a larger, randomised controlled trial that enrolled homozygous FH, evolocumab consistently decreased LDL-C by 23 %.[50]
There are currently phase III clinical trials that have shown safety and efficacy in lowering LDL-C with evolocumab. In a 12-week phase III clinical trial, evolocumab use in combination with moderate or high-intensity statin showed significant reduction in LDL-C (up to 75 % reduction when administered every two or four weeks).[45] Another phase III study evaluating evolocumab use as a monotherapy also showed significant decrease in LDL-C (57 % more reduction compared to placebo and 40 % more reduction than ezetimibe).[42] In a 12-week phase III study evaluating evolocumab use in subjects with intolerance to statin, those treated with evolocumab had significant reduction of LDL-C compared to ezetimibe (53–56 % vs 37–39 % p< 0.001).[44] Patients that were previously enrolled in prior phase II studies (GAUSS, RUTHERFORD, LAPLACE-TIMI 57, and MENDEL) were evaluated in the Open-Label Study of Long-Term Evaluation Against LDL-C (OSLER) trial. This study showed that subjects who continued to take evolocumab for the duration of the year on a monthly dosing regimen, maintained decreased LDL-C levels, whereas those that discontinued the study drug resumed their baseline levels.[51] The Further cardiovascular outcomes research with PCSK9 inhibition in Subjects with Elevated CV Risk (FOURNIER) is currently enrolling to evaluate CV outcomes with evolocumab use in subjects already on guideline-based lipid therapy.
Bococizumab (RN316/PF04950615)
The monoclonal antibody bococizumab has undergone phase I clinical trial and shown that single and escalating intravenous and subcutaneous dose significantly lowered LDL-C by as much as 54 % with 150 mg Q2 week regimen without significant adverse effects.[52] Currently, two phase III trials are underway studying bococizumab effects on cardiovascular outcomes in high risk subjects. In one of these outcomes trials, bococizumab use will be evaluated in subjects who are at high risk and have an LDL-C level of 70-100 mg/dL (SPIRE1). In another CV outcomes study, safety and efficacy of bobocizumab will be evaluated in high risk subjects who have higher than LDL 100 mg/ dL, despite high-dose statin or at the highest tolerated dose (SPIRE2) (see Table 4).
Table 4: Phase III Clinical Trials in Subjects with Hypercholesterolaemia Treated with Monoclonal Antibodies.
| Monoclonal Antibody | Name of Phase III | Population |
|---|---|---|
| Evolocumab | MENDEL-2 GAUSS-2 LAPLACE-2 FOURIER |
Subjects with hypercholesterolaemia, Framingham Risk score < 10%, monotherapy Statin-intolerant subjects, compared to ezetimibe Subjects with hypercholesterolaemia, statin intolerance, ezetimibe controlled Subjects treated with evolocumab on high or low dose statin Evaluating cardiovascular outcomes in subjects with hypercholesterolaemia and elevated risk cardiovascular risk |
| Alirocumab | ODYSSEY COMBO-I CHOICE I CHOICE II LONG TERM OUTCOMES |
Global Phase III program subjects treated with maximally tolerated statin therapy Alirocumab administered every four weeks compared with placebo Alirocumab as monotherapy compared to other non-statin lipid lowering therapies Alirocumab use in the background of lipid lowering therapies and long term safety and efficacy Alirocumab effects in cardiovascular outcomes in subjects with acute coronary syndrome |
| Bocolicumab | SPIRE-1 SPIRE-2 SPIRE-IS SPIRE-HR SPIRE-LL |
CV outcomes in subjects with high risk and LDL-C ≤ 70 but ≤ 100 mg dL CV outcomes in subjects with high risk and LDL-C ≥ 100 mg dL Subjects who are intolerant to statin Subjects with high or very high risk for CV events Subjects with primary hyperlipidaemia at high or very high risk |
Other Targets for PCSK9 Inhibition
Other pharmacologic approaches to PCSK9 inhibition have been proposed; many are still in preclinical development. Direct inhibition of PCSK9 can be attained using small mimetic peptides called adnectins. Mimetic peptides of the PCSK9 binding domain for LDL have been shown to decrease LDL-R degradation.[53,54] Mimetic peptides of the binding domain of LDL-R can also bind to PCSK9 and prevent its interaction with LDL-R.[53,55] Another approach in PCSK9 inhibition is gene silencing techniques. Antisense oligonucleotides (ASO) are in preclinical development. ASO administration in mice has been shown to increase hepatic LDL-R receptor concentration.[56] Antisense RNAs (siRNA) are also being developed to target PCSK9 mRNA, shown in non-human primates to reduce LDL-C by up to 50–70 percent.[57] Natural inhibitors, such as annexin A2, a protein expressed in many tissues, inhibit PCSK9 and increase LDL-R.[58,59] Berberine, a natural occurring plant alkaloid has been shown to also decrease PCSK9 mRNA expression and increase LDL-R in vitro and animal studies.[60,61]
Adverse Events
Serious adverse events from monoclonal antibodies targeting PCSK9 are rare. The most common adverse reactions are local injection site reactions. In the GAUSS phase II trial evaluating subjects that have intolerance to statin, myalgias were the most common adverse event but had low incidence overall.[43] Alirocumab had similar adverse reactions between placebo and treatment groups in its phase II trials. In a dose escalating study of alirocumab, one of 152 subjects receiving a dose of alirocumab developed cutaneous leukocytoclastic vasculitis that was successfully treated with prednisone.[35] Most common adverse reactions that caused discontinuation of medication in these trials were mild injection site reactions (erythema, pruritis, discoloration, haematoma, swelling). For evolocumab, the most common treatment related adverse reaction was not only injection site reaction (pain), but also headache and skin burning sensation.[46]
Conclusion
PCSK9 Inhibition for Use in Cardiovascular Disease Prevention
Although statins have revolutionised lipid therapy and secondary cardiovascular disease prevention, there remains a significant residual risk that can be further targeted. Certain populations may benefit from additive therapies: those with persistent elevation of LDL-C or residual risk despite optimal guideline-based therapy. Subjects with familial hypercholesterolaemia, for example, have been difficult to treat; despite maximum doses of lipid therapies, many subjects may not achieve sufficient reduction of LDL-C. Intolerance of statin, due to moderate to severe myalgias and/or a 5-fold increase in creatinine kinase have also limited the beneficial use of optimised lipid therapy, and this statin-induced myopathy may represent up to 10 % of treated patients in a primary care setting.[62] At present, PCSK9 inhibitors have successfully shown to significantly reduce LDL-C, non-HDL-C and Lp(a) in different patient population and clinical settings. As we look to continued phase III clinical trials, particularly those measuring CV outcomes, PCSK9 inhibitors offer promise to patients with significant residual risk that treatment with PCSK9 inhibitors can further lower cardiovascular outcome events and related mortality.
Table 2 references
- 1a.Shan L, Pang L, Zhang R et al. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Commun. 2008;375:69–73. doi: 10.1016/j.bbrc.2008.07.106. [DOI] [PubMed] [Google Scholar]
- 2a.McNutt MC, Kwon HJ, Chen C et al. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J Biol Chem. 2009;284:10561–70. doi: 10.1074/jbc.M808802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3a.Du F, Hui Y, Zhang M et al. Novel domain interaction regulates secretion of proprotein convertase subtilisin/kexin type 9 (PCSK9) protein. J Biol Chem. 2011;286:43054–61. doi: 10.1074/jbc.M111.273474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a.Graham MJ, Lemonidis KM, Whipple CP et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. 2007;48:763–7. doi: 10.1194/jlr.C600025-JLR200. [DOI] [PubMed] [Google Scholar]
- 5a.Gupta N, Fisker N, Asselin MC et al. A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression in vitro and in vivo. PloS One. 2010;5:e10682. doi: 10.1371/journal.pone.0010682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6a.Frank-Kamenetsky M, Grefhorst A, Anderson NN et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11915–20. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7a.Mayer G, Poirier S, Seidah NG. Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J Biol Chem. 2008;283:31791–801. doi: 10.1074/jbc.M805971200. [DOI] [PubMed] [Google Scholar]
- 8a.Seidah NG, Poirier S, Denis M et al. Annexin A2 is a natural extrahepatic inhibitor of the PCSK9-induced LDL receptor degradation. PloS one. 2012;7:e41865. doi: 10.1371/journal.pone.0041865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Cameron J, Ranheim T, Kulseth MA et al. Berberine decreases PCSK9 expression in HepG2 cells. Atherosclerosis. 2008;201:266–73. doi: 10.1016/j.atherosclerosis.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 10a.Kong W, Wei J, Abidi P et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. 2004;10:1344–51. doi: 10.1038/nm1135. [DOI] [PubMed] [Google Scholar]
- 11a.Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein engineering, design & selection: PEDS. 2011;24:3–9. doi: 10.1093/protein/gzq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 1.Gordon T, Kannel WB, Castelli WP, Dawber TR. Lipoproteins, cardiovascular disease, and death. The Framingham study. Arch Intern Med. 1981;141:1128–31. [PubMed] [Google Scholar]
- 2.Sharrett AR, Ballantyne CM, Coady SA et al. Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: The Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2001;104:1108–13. doi: 10.1161/hc3501.095214. [DOI] [PubMed] [Google Scholar]
- 3.Baigent C, Keech A, Kearney PM et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 4.LaRosa JC, He J, Vupputuri S. Effect of statins on risk of coronary disease: a meta-analysis of randomized controlled trials. JAMA. 1999;282:2340–6. doi: 10.1001/jama.282.24.2340. [DOI] [PubMed] [Google Scholar]
- 5.Stone NJ, Robinson JG, Lichtenstein AH et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2889–934. doi: 10.1016/j.jacc.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Sampson UK, Fazio S, Linton MF. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep. 2012;14:1–10. doi: 10.1007/s11883-011-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Investigators A-H, Boden WE, Probstfield JL et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 8.Januszkiewicz L. [The ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus]. Kardiologia polska. 2010;68:853, 4, 5. discussion. [PubMed] [Google Scholar]
- 9.Villines TC, Stanek EJ, Devine PJ et al. The ARBITER 6-HALTS Trial (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol 6-HDL and LDL Treatment Strategies in Atherosclerosis): final results and the impact of medication adherence, dose, and treatment duration. J Am Coll Cardiol. 2010;55:2721–6. doi: 10.1016/j.jacc.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 10.Group HTC, Landray MJ, Haynes R et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–12. doi: 10.1056/NEJMoa1300955. [DOI] [PubMed] [Google Scholar]
- 11.Gent J, Braakman I. Low-density lipoprotein receptor structure and folding. CMLS. 2004;61:2461–70. doi: 10.1007/s00018-004-4090-3. [DOI] [PubMed] [Google Scholar]
- 12.Huang S, Henry L, Ho YK et al. Mechanism of LDL binding and release probed by structure-based mutagenesis of the LDL receptor. J Lipid Res. 2010;51:297–308. doi: 10.1194/jlr.M000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer’s bottle to Scap’s MELADL. J Lipid Res. 2009;50(Suppl):S15–27. doi: 10.1194/jlr.R800054-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–40. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 15.Seidah NG, Benjannet S, Wickham L et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:928–33. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abifadel M, Varret M, Rabes JP et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nature genetics. 2003;34:154–6. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 17.Allard D, Amsellem S, Abifadel M et al. Novel mutations of the PCSK9 gene cause variable phenotype of autosomal dominant hypercholesterolemia. Human Mutation. 2005;26:497. doi: 10.1002/humu.9383. [DOI] [PubMed] [Google Scholar]
- 18.Benjannet S, Rhainds D, Essalmani R et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279:48865–75. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- 19.Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 20.Benn M, Nordestgaard BG, Grande P et al. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta-analyses. J Am Coll Cardiol. 2010;55:2833–42. doi: 10.1016/j.jacc.2010.02.044. [DOI] [PubMed] [Google Scholar]
- 21.Cohen J, Pertsemlidis A, Kotowski IK et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nature Genetics. 2005;37:161–5. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto T, Lu C, Ryan RO. A two-step binding model of PCSK9 interaction with the low density lipoprotein receptor. J Biol Chem. 2011;286:5464–70. doi: 10.1074/jbc.M110.199042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo Surdo P, Bottomley MJ, Calzetta A et al. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO reports. 2011;12:1300–5. doi: 10.1038/embor.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maxwell KN, Soccio RE, Duncan EM et al. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res. 2003;44:2109–19. doi: 10.1194/jlr.M300203-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Seidah NG, Prat A. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov. 2012;11:367–83. doi: 10.1038/nrd3699. [DOI] [PubMed] [Google Scholar]
- 26.Kosenko T, Golder M, Leblond G et al. Low density lipoprotein binds to proprotein convertase subtilisin/kexin type-9 (PCSK9) in human plasma and inhibits PCSK9-mediated low density lipoprotein receptor degradation. J Biol Chem. 2013;288:8279–88. doi: 10.1074/jbc.M112.421370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Careskey HE, Davis RA, Alborn WE et al. Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res. 2008;49:394, 8. doi: 10.1194/jlr.M700437-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Dubuc G, Chamberland A, Wassef H et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–9. doi: 10.1161/01.ATV.0000134621.14315.43. [DOI] [PubMed] [Google Scholar]
- 29.Chan JC, Piper DE, Cao Q et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9820–5. doi: 10.1073/pnas.0903849106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dias CS, Shaywitz AJ, Wasserman SM et al. Effects of AMG 145 on low-density lipoprotein cholesterol levels: results from 2 randomized, double-blind, placebo-controlled, ascending-dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol. 2012;60:1888–98. doi: 10.1016/j.jacc.2012.08.986. [DOI] [PubMed] [Google Scholar]
- 31.Stein EA, Mellis S, Yancopoulos GD et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–18. doi: 10.1056/NEJMoa1105803. [DOI] [PubMed] [Google Scholar]
- 32.Blom DJ, Hala T, Bolognese M et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370:1809–19. doi: 10.1056/NEJMoa1316222. [DOI] [PubMed] [Google Scholar]
- 33.Giugliano RP, Desai NR, Kohli P et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet. 2012;380:2007–17. doi: 10.1016/S0140-6736(12)61770-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roth EM, Taskinen MR, Ginsberg HN et al. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: results of a 24 week, double-blind, randomized Phase 3 trial. Int J Cardiol. 2014;176:55–61. doi: 10.1016/j.ijcard.2014.06.049. [DOI] [PubMed] [Google Scholar]
- 35.McKenney JM, Koren MJ, Kereiakes DJ et al. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59:2344–53. doi: 10.1016/j.jacc.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 36.Roth EM, McKenney JM, Hanotin C et al. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891–900. doi: 10.1056/NEJMoa1201832. [DOI] [PubMed] [Google Scholar]
- 37.Raal FJ, Giugliano RP, Sabatine MS et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol. 2014;63:1278–88. doi: 10.1016/j.jacc.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 38.Stein EA, Gipe D, Bergeron J et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36. doi: 10.1016/S0140-6736(12)60771-5. [DOI] [PubMed] [Google Scholar]
- 39.Kastelein JJ, Robinson JG, Farnier M et al. Efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia not adequately controlled with current lipid-lowering therapy: design and rationale of the ODYSSEY FH studies. Cardiovascular drugs and therapy/sponsored by the International Society of Cardiovascular Pharmacotherapy. 2014;28:281–9. doi: 10.1007/s10557-014-6523-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roth EM, Diller P. Alirocumab for hyperlipidemia: physiology of PCSK9 inhibition, pharmacodynamics and Phase I and II clinical trial results of a PCSK9 monoclonal antibody. Future Cardiology. 2014;10:183–99. doi: 10.2217/fca.13.107. [DOI] [PubMed] [Google Scholar]
- 41.Koren MJ, Scott R, Kim JB et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomised, double-blind, placebo-controlled, phase 2 study. Lancet. 2012;380:1995–2006. doi: 10.1016/S0140-6736(12)61771-1. [DOI] [PubMed] [Google Scholar]
- 42.Koren MJ, Lundqvist P, Bolognese M et al. Anti-PCSK9 Monotherapy for Hypercholesterolemia: The MENDEL-2 Randomized, Controlled Phase III Clinical Trial of Evolocumab. J Am Coll Cardiol. 2014;63:2531–40. doi: 10.1016/j.jacc.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 43.Sullivan D, Olsson AG, Scott R et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA. 2012;308:2497–506. doi: 10.1001/jama.2012.25790. [DOI] [PubMed] [Google Scholar]
- 44.Stroes E, Colquhoun D, Sullivan D et al. Anti-PCSK9 Antibody Effectively Lowers Cholesterol in Patients With Statin Intolerance: The GAUSS-2 Randomized, Placebo-Controlled Phase 3 Clinical Trial of Evolocumab. J Am Coll Cardiol. 2014;63:2541–8. doi: 10.1016/j.jacc.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 45.Robinson JG, Nedergaard BS, Rogers WJ et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA. 2014;311:1870–82. doi: 10.1001/jama.2014.4030. [DOI] [PubMed] [Google Scholar]
- 46.Raal F, Scott R, Somaratne R et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation. 2012;126:2408–17. doi: 10.1161/CIRCULATIONAHA.112.144055. [DOI] [PubMed] [Google Scholar]
- 47.Raal FJ, Stein EA, Dufour R PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet. 2014. [DOI] [PubMed]
- 48.Desai NR, Giugliano RP, Zhou J et al. AMG 145, a monoclonal antibody against PCSK9, facilitates achievement of national cholesterol education program-adult treatment panel III low-density lipoprotein cholesterol goals among high-risk patients: an analysis from the LAPLACE-TIMI 57 trial (LDL-C assessment with PCSK9 monoclonal antibody inhibition combined with statin thErapy-thrombolysis in myocardial infarction 57). J Am Coll Cardiol. 2014;63:430–3. doi: 10.1016/j.jacc.2013.09.048. [DOI] [PubMed] [Google Scholar]
- 49.Desai NR, Kohli P, Giugliano RP et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL-C Assessment with Proprotein Convertase Subtilisin Kexin Type 9 Monoclonal Antibody Inhibition Combined with Statin Therapy (LAPLACE)-Thrombolysis in Myocardial Infarction (TIMI) 57 trial. Circulation. 2013;128:962–9. doi: 10.1161/CIRCULATIONAHA.113.001969. [DOI] [PubMed] [Google Scholar]
- 50.Raal FJ, Honarpour N, Blom DJ Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2014. [DOI] [PubMed]
- 51.Koren MJ, Giugliano RP, Raal FJ et al. Efficacy and safety of longer-term administration of evolocumab (AMG 145) in patients with hypercholesterolemia: 52-week results from the Open-Label Study of Long-Term Evaluation Against LDL-C (OSLER) randomized trial. Circulation. 2014;129:234–43. doi: 10.1161/CIRCULATIONAHA.113.007012. [DOI] [PubMed] [Google Scholar]
- 52.Ling H, Burns TL, Hilleman DE. An update on the clinical development of proprotein convertase subtilisin kexin 9 inhibitors, novel therapeutic agents for lowering low-density lipoprotein cholesterol. Cardiovascular Therapeutics. 2014;32:82–8. doi: 10.1111/1755-5922.12056. [DOI] [PubMed] [Google Scholar]
- 53.Shan L, Pang L, Zhang R et al. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Commun. 2008;375:69–73. doi: 10.1016/j.bbrc.2008.07.106. [DOI] [PubMed] [Google Scholar]
- 54.Du F, Hui Y, Zhang M et al. Novel domain interaction regulates secretion of proprotein convertase subtilisin/kexin type 9 (PCSK9) protein. J Biol Chem. 2011;286:43054–61. doi: 10.1074/jbc.M111.273474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McNutt MC, Kwon HJ, Chen C et al. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J Biol Chem. 2009;284:10561–70. doi: 10.1074/jbc.M808802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Graham MJ, Lemonidis KM, Whipple CP et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. 2007;48:763–7. doi: 10.1194/jlr.C600025-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.Frank-Kamenetsky M, Grefhorst A, Anderson NN et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11915–20. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seidah NG, Poirier S, Denis M et al. Annexin A2 is a natural extrahepatic inhibitor of the PCSK9-induced LDL receptor degradation. PloS one. 2012;7:e41865. doi: 10.1371/journal.pone.0041865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mayer G, Poirier S, Seidah NG. Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J Biol Chem. 2008;283:31791–801. doi: 10.1074/jbc.M805971200. [DOI] [PubMed] [Google Scholar]
- 60.Cameron J, Ranheim T, Kulseth MA et al. Berberine decreases PCSK9 expression in HepG2 cells. Atherosclerosis. 2008;201:266–73. doi: 10.1016/j.atherosclerosis.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 61.Kong W, Wei J, Abidi P et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nature Medicine. 2004;10:1344–51. doi: 10.1038/nm1135. [DOI] [PubMed] [Google Scholar]
- 62.Eckel RH. Approach to the patient who is intolerant of statin therapy. J Clin Endocrinol Metab. 2010;95:2015–22. doi: 10.1210/jc.2009-2689. [DOI] [PubMed] [Google Scholar]