

Abstract
A long-standing question concerning X-chromosome inactivation (XCI) has been how some genes avoid the otherwise stable chromosome-wide heterochromatinization of the inactive X chromosome. As 20% or more of human X-linked genes escape from inactivation, such genes are an important contributor to sex differences in gene expression. Although both human and mouse have genes that escape from XCI, more genes escape in humans than mice, with human escape genes often clustering in larger domains than the single escape genes of mouse. Mouse models offer a well-characterized and readily manipulated system in which to study XCI, but given the differences in genes that escape it is unclear whether the mechanism of escape gene regulation is conserved. To address conservation of the process and the potential to identify elements by modelling human escape gene regulation using mouse, we integrated a human and a mouse BAC each containing an escape gene and flanking subject genes at the mouse X-linked Hprt gene. Escape-level expression and corresponding low promoter DNA methylation of human genes RPS4X and CITED1 demonstrated that the mouse system is capable of recognizing human elements and therefore can be used as a model for further refinement of critical elements necessary for escape from XCI in humans.
Introduction
X-chromosome inactivation (XCI) epigenetically silences one X chromosome in every cell in female mammals, yet a significant number of genes on the human inactive X chromosome (Xi) are able to escape silencing and continue to be expressed, albeit at lower levels than their active X chromosome (Xa) copy (1). As the majority of human 45, X conceptuses do not survive to term, with the ones that do suspected to be mosaic (2), there is strong evidence that the second sex chromosome is crucial for viability. Escape genes also contribute to male and female susceptibility to disease, including a protective role in females against cancer (3), and as a risk factor in females for developing autoimmune disorders (reviewed in 4). In addition to the importance to disease, escape genes offer a means of exploring epigenetic gene regulation, yet little is known about the mechanisms that allow them to resist the chromosome-wide silencing of XCI.
Genes that escape from XCI have been identified using a variety of approaches; both directly on the basis of Xi gene expression, or indirectly by using features such as histone marks or DNA methylation (DNAm) (reviewed in 5). Escape genes share active histone marks commonly associated with actively transcribed genes on the Xa and autosomes, as well as hypomethylation of CpG islands in promoters and hypermethylation of gene bodies. At the level of expression, a gene is generally called as escaping if the proportion of transcription from the Xi relative to the Xa is greater than 10% (1), although variations to this threshold have been used (6). Ideally, female cells with known expressed polymorphisms are used to obtain this allelic ratio between Xs; however, in cases where there is no allelic information available, the differences between females (Xa/Xi) and males (who have a single Xa) have been used to predict the X-inactivation status of the gene. Studies using DNAm to predict XCI status also take advantage of this sex difference, as genes escaping from XCI tend to have low levels of promoter DNAm similar to the Xa copy in males, while genes subject to inactivation typically have significantly greater DNAm at promoters (7,8).
A recent integration of escape status calls from multiple investigations suggests that 12–20% of genes escape from inactivation and are expressed from both the Xa and the Xi in human (9). In comparison, only 3–7% of X-linked genes escape in mouse (6), with the lower number of escape genes likely playing a role in the less severe phenotype seen in a 45, X mice (reviewed in 10). The range in percentage of genes escaping XCI is largely accounted for by a subset of escape genes that show individual, strain (in mouse) or tissue-specific expression. Additionally, escape status calls for some genes are discordant between studies. Much of the difference in number of escapees between human and mouse results from clusters of up to 15 genes within an escape domain in humans, while in mice escape genes are predominantly singletons or associated with a non-coding RNA gene (11), although several newly identified variable genes contribute to small regions of escape (12). While the mechanisms of escape from XCI are still unknown, the fact that approximately one-half of the mouse escape genes also escape in humans suggests some conservation of the elements and mechanisms involved. Local regulatory elements driving escape have been proposed in mouse (13,14). Either these differ in human, or there is a larger bystander effect in humans whereby a single gene may be under strict regulatory control, but a lack of stable adjacent boundaries allows spread of open chromatin and neighbouring genes to also escape.
Multiple bioinformatic studies have sought to identify genomic components involved in allowing a gene to escape from XCI, but have yet to find a robust identifier of escapees, suggesting that the mechanism involves multiple components. We have proposed three broad categories of DNA elements that play a role in the ability of a gene to escape from XCI: a depletion in waystations, often suggested to be repetitive sequences, which are thought to propagate the silencing signal along the X chromosome; enrichment of boundary elements, for example CTCF, which act as insulators between active and inactive domains; and lastly sequences termed escape elements that allow the gene to avoid complete silencing (15). Location on the X chromosome, as well as overall chromosomal ultrastructure and interactions between loci, have also been hypothesized to influence regulation of genes on the Xi (reviewed in 5). The strongest evidence for an intrinsic escape element and boundary sequence comes from several studies of the mouse escape gene Kdm5c (previously Jarid1c, Smcx). Random integration of two separate BACs each containing Kdm5c, as well as subject flanking genes, at four different locations on the mouse X chromosome in female embryonic stem cells (ESCs) demonstrated ongoing escape from inactivation of Kdm5c after differentiation, while the flanking genes maintained their expected inactive state (16). A follow-up to this study analysed partially deleted integrations of the transgene and showed that deletion of the 3′ end of Kdm5c extended the escape domain by disrupting proper silencing of three endogenous genes adjacent to Kdm5c, indicating a likely location of a boundary sequence (17). Kdm5c has a human homolog that also escapes XCI, but unlike the mouse, which is a lone escaper, human KDM5C is in a domain of several genes that escape from inactivation.
Given the differences in escape frequency and distribution between mice and humans, it is likely that there may be differences between the regulatory processes; however, human ESCs, as well as induced pluripotent stem cells, remain a limited model for studying XCI since most already have an Xi or an eroded Xi with partial gene reactivation and epigenetic instability that is maintained through differentiation (18,19). Mouse models have been much better characterized as a consistent developmental model for XCI, therefore in this study we explore the conservation of mechanisms of escape from XCI between species in order to determine whether the processes are sufficiently conserved to allow modelling of human escape from XCI in mouse.
We have previously assessed over 1.5 Mb of primarily autosomal human transgenic DNA integrated at the Hprt locus on the mouse X chromosome for evidence of escape from XCI (20), anticipating that escape might be frequent given that waystations have been suggested to be depleted on autosomes (21). However, most transgenes were subject to XCI, with only one truncated autosomal gene, PHB, identified as being expressed from the Xi. While finding that one transgene escapes from XCI demonstrates that expression is possible from this location on the Xi, the rarity of escape observed for BACs integrated at Hprt may reflect a resilience of the docking site to Xi expression. Alternatively, the BAC carrying PHB could contain some combination of escape elements and boundary factors to block silencing, which otherwise is capable of spreading across constructs over 195 kb even if they lack additional waystations. As the majority of the BACs tested were of autosomal origin, we now revisit this system to determine if the Hprt site recognizes intrinsic escape elements of X-linked genes. We integrated one of the BACs carrying Kdm5c previously shown to escape XCI (16) into the locus, and knock-in mice carrying the BAC on the Xi generally showed an increase in Kdm5c expression as well as hypomethylation at the gene’s promoter, validating the Hprt docking site as permissive to X-linked escape. To address the conservation of escape status when human escape genes are integrated into mouse, we further generated transgenic mice containing a human BAC with the escape gene RPS4X, discordant gene CITED1 and subject gene ERCC6L. Expression and promoter DNAm analyses demonstrated that mouse was able to correctly recapitulate the escape and subject statuses of the human genes, therefore suggesting that intrinsic escape elements may share recognizable properties between mouse and human.
Results
Generation of transgenic mice with BAC knock-ins on the inactive X chromosome
To ensure that the Hprt locus is permissive for escape, we chose to use BAC RP23–391D18 which contains the mouse gene Kdm5c, previously shown to escape from XCI at four integration sites in a mouse cell line (16). This BAC also contains subject (in mouse) genes Tspyl2, Kantr and Gpr173. Our choice of human BAC was driven by several criteria as outlined in Figure 1. The vector used to retrofit the BACs with homology arms for integration into Hprt was designed to work with BACs from the RPCI-11 library (22). In order to ensure that putative elements were present in the construct, the BAC should contain an escape gene flanked by subject genes, ideally all with broad expression levels and/or CpG island promoters to monitor DNAm. Our restriction to small domains biases our assessment of escape elements to regions that more closely resemble the single escape genes in mice, as capturing larger human escape domains in a single BAC is not feasible.
Figure 1.
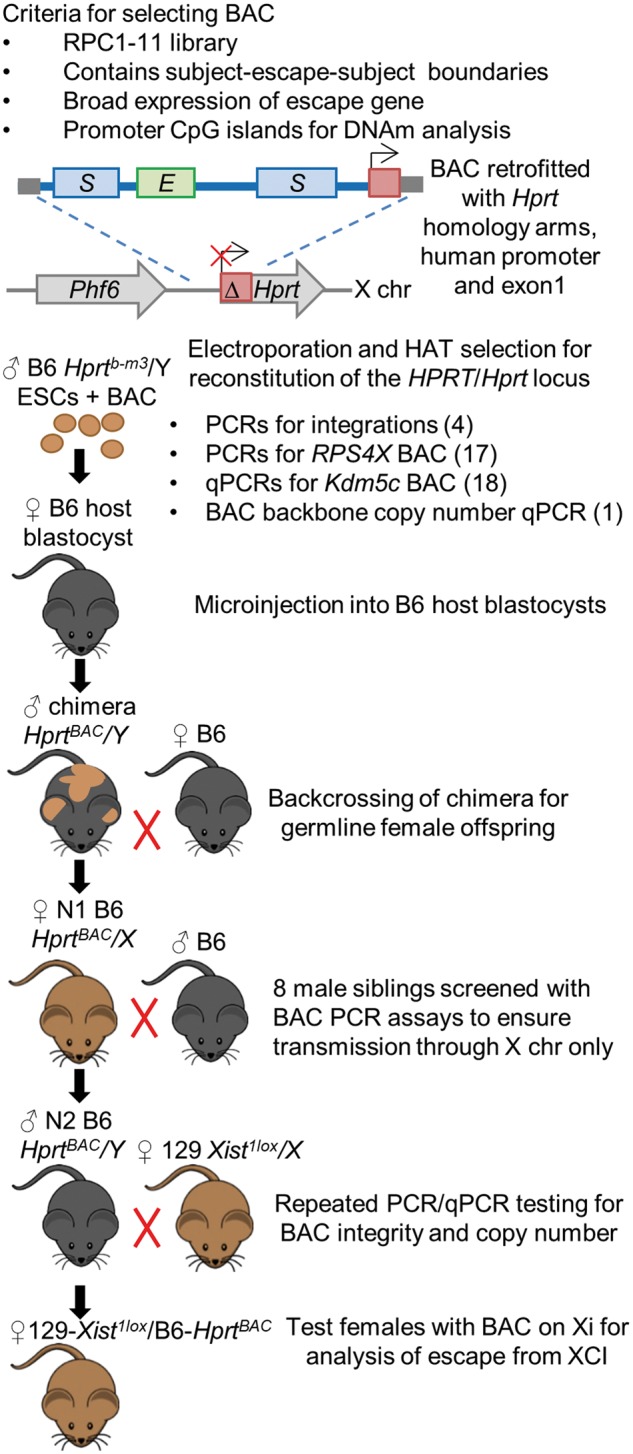
Generation of transgenic mice with BAC knock-ins on the Xi. Flow diagram showing criteria for selecting BACs containing genes that escape from XCI, followed by the breeding scheme after electroporation of the BAC into male mouse ESCs. Clones with successful integration at the Hprt locus are selected for with HAT media, followed by PCR and qPCR screening. Positive clones are microinjected into host blastocysts. ESCs have the Aw–Jallele to allow us to follow coat colour and choose appropriate chimera offspring where the ESCs containing the BAC have gone germline. Only N1 females carry the BAC as it is transmitted on the X chromosome. We screened N1 male siblings by PCR assays for the BAC to ensure there was no autosomal transmission and did not detect any bands. BACs were again tested for intactness and copy number at the N1 and N2 stages, before breeding N2 male mice with the BAC on their X to 129Xist1lox females. Females from this cross could inherit either X from their mother, generating our test females with the BAC always on the Xi as well as knock-in females with random XCI.
We selected human BAC RP11–1145H7 as it contains multiple human genes with different XCI status calls. The primate-specific escape gene RPS4X and discordant gene CITED1 are in the centre bounded by subject genes ERCC6L and HDAC8 (truncated), giving us confidence that human escape and boundary elements for regulating expression from the Xi were present within the BAC, and we could test if they were recognized by mouse.
Selected BACs were targeted by homologous recombination to the Hprtb-m3 deletion on the mouse X chromosome in C57BL/6 (B6) male ESCs (Fig. 1). Proper integration was selected for with hypoxanthine aminopterin thymidine (HAT) media, as the BAC constructs contained a complementary sequence that rescues HPRT activity through creation of a chimeric locus consisting of the human HPRT promoter and exon 1 and mouse Hprt exons 2–9 (23).
Validation of the intactness of the knock-ins utilized assays approximately every 10 kb along the BACs (Supplementary Material, Table S2). As the mouse Kdm5c BAC is from B6 and was integrated into a B6 background, qPCR assays of genomic DNA were performed and showed single copy integration with no major deletions, both in ESCs prior to blastocyst microinjection, as well as in N2 males (Supplementary Material, Fig. S1A). Human-specific PCR assays approximately every 10 kb along the BAC RP11–1145H7 integration confirmed that the BAC was intact with no major deletions, both in ESCs prior to blastocyst microinjection as well as in N1 female offspring of chimeras. Copy number qPCR of the BAC backbone vector suggested that two copies of the human BAC had integrated (Supplementary Material, Fig. S1B). Negative PCR assays for the BAC and backbone vector in eight male offspring of chimeras indicated that there was no autosomal transmission of the BAC, and thus both copies were likely linked on the X chromosome.
To examine expression of the transgenes from the Xi only, B6 N2 male mice carrying the BAC at Hprt were crossed with 129S1/SvImJ (129) females carrying a deletion at the Xist gene responsible for initiation of XCI, resulting in experimental female offspring that always carried the BAC on their Xi (129-Xist1lox/B6-HprtBAC).
An X-linked mouse gene escapes inactivation at Hprt
To determine if an X-linked gene can escape from XCI when integrated at Hprt, we analysed our female mice with BAC RP23–391D18, carrying mouse escape gene Kdm5c (129-Xist1lox/B6-HprtKdm5c) on the Xi (Fig. 2A). Knock-in male mice carrying a copy of the BAC on their single Xa (B6-HprtKdm5c/129-Y), as well as knock-in females with random XCI (129-XistWT/B6-HprtKdm5c) were also generated and assessed. As the genes on the BAC have endogenous copies, the use of a cross with informative polymorphisms would have improved our sensitivity to detect expression from the integrated allele; however, we wanted to test the mouse lines designed for integration of human BACs (22). We therefore included wild-type 129/B6 male and female controls as a baseline for expression and DNAm (Fig. 2B). Genes on BAC RP23–391D18 were examined by RT-qPCR to assess expression in brain, liver and spleen for six mice of each genotype (Fig. 2C), as escape has been described to vary between individuals and tissues.
Figure 2.

Analysis of BAC RP23–391D18 shows Kdm5c escapes XCI at Hprt. (A) Integration of Kdm5c BAC at Hprt; genes on the BAC expected to escape from XCI in green, subject in blue and genes at integration site (both known to be subject to XCI) in grey. Genes with RT-qPCR and DNAm assays are indicated. (B) Description of genotypes, six mice for each. (C) Normalized to Pgk1, RT-qPCR of both Kdm5c and Tspyl2 expression in knock-in males (blue) shows significantly more expression than wild-type males (green) in brain and spleen demonstrating that our transgene is expressed on an Xa. Kdm5c expression from the non-random Xi in knock-in females (yellow) is higher than wild-type females (purple) suggesting escape of the transgene, but does not reach significance in brain (unpaired t-test). Expression of knock-in females with random XCI (pink) is generally higher than non-random females (yellow). Tspyl2 expression from the Xi was significant in spleen only but not supported by DNAm. (D) Average DNAm of Kdm5c shows promoter hypomethylation in knock-in females (yellow), which supports the expression trend of escape from the Xi. Tspyl2, HPRT and Phf6 show hypermethylated promoters in knock-in females suggesting they are subject to XCI. Knock-in females are compared to wild-type females for all assays except HPRT as wild-type females do not carry the human gene (Mann–Whitney t-test, significance denoted by asterisks; P-value <0.001***, 0.001–0.01**, 0.01–0.05*, >0.05 ns).
Increased expression in knock-in males carrying an additional copy of both Kdm5c and Tspyl2, compared to wild-type males, was significant in two tissues and provides evidence that our integrated BAC has retained all necessary elements for functional transcription from the X when it is active. To examine escape from XCI, expression from the Xi was analysed in 129-Xist1lox/B6-HprtKdm5c females with two endogenous copies of Kdm5c (one Xa and one Xi) plus our transgenic copy on the Xi, relative to wild-type females (one endogenous Xa and Xi copy). Expression of Kdm5c from skewed transgenic females was significantly higher than wild-type females in liver and spleen only, with a trend toward higher expression in brain. Tspyl2 has been previously reported to be subject upon random integration into the X (16,24) consistent with our results in brain and liver; however, we did see a significant increase in expression in spleen.
Measuring DNAm of genes with CpG islands has been a robust indirect approach to examine XCI status as backup to, or in lieu of, expression analysis, therefore we established assays for the CpG-island promoters of Kdm5c and Tspyl2. We also examined DNAm at the promoters of the integrated human HPRT and the closest mouse endogenous gene to the integration site, Phf6, to see if there were any upstream or downstream regulatory effects of our integration. DNAm is shown as an average of at least three CpGs for both endogenous and transgenic alleles where applicable (Fig. 2D). Our transgenic males mirrored the wild-type males in all assays typically showing hypomethylation of the analysed gene, as expected given they have a single active X. As with expression, we compared our transgenic females with the knock-in on the Xi to wild-type females to observe if the additional Xi copy raised (indicating transgene is silenced) or lowered (indicating transgene is expressed) DNAm levels. The Kdm5c promoter DNAm in the transgenic females remained low like the wild-type females and both males, reinforcing that all three copies of the gene are capable of expression. High promoter DNAm of neighbouring BAC gene Tspyl2 in the skewed knock-in females suggests that the gene is subject to inactivation. It also generally shows a slight increase in DNAm from wild-type females, which is expected if there is now a second inactive copy raising methylation. HPRT and Phf6 both showed hypermethylation in females similar to gene averages previously seen at these sites (20) suggesting that the elements permitting escape from XCI at the transgenic Kdm5c locus are not affecting the surrounding environment. Kantr and Gpr173 were not assessed as they do not have promoter CpG islands and neither has been previously suggested to escape from XCI.
The integration of extra genes could be detrimental to the mouse, and selection against them being expressed on the Xa could lead to non-random XCI in females. Thus, we used expression of an X-linked SNP to assess knock-in females without the Xist deletion for deviations from random XCI. There was no consistent skewing of inactivation toward one X being silenced more often than the other (Supplementary Material, Fig. S2A). This is in agreement with both expression and DNAm of non-escaping BAC genes Tspyl2 and HPRT where they are now on an Xa approximately half of the time, thereby contributing to generally higher expression levels and lower DNAm compared to the experimentally skewed females. Lack of a negative influence of the transgene was also supported by normal breeding and genotype ratios of the experimental mice (Supplementary Material, Table S1). As the expression differences between transgenic and wild-type females were not dramatic, an additional set of four 129-Xist1lox/B6-HprtKdm5c females were tested separately; however, these were not significantly different in Kdm5c expression from wild-type females, although they continued to show hypomethylation at the promoter (Supplementary Material, Fig. S3), supporting that Kdm5c escapes XCI. Tspyl2 remains hypermethylated in additional females, and loses significance of expression in spleen.
Human X-linked genes escape inactivation at Hprt
We next analysed our transgenic mice carrying human BAC RP11–1145H7 (Fig. 3A) to determine if the BAC carried the necessary elements for escape, and if mouse could recognize them. Male mice carrying the knock-in on their Xa (B6-HprtRPS4X/129-Y), knock-in females with random XCI (129-XistWT/B6-HprtRPS4X) and females carrying the knock-in on their Xi (129-Xist1lox/B6-HprtRPS4X) were assessed for expression and DNAm in brain, liver and spleen for six mice of each genotype (Fig. 3B). In all tissues, knock-in males expressed RPS4X, demonstrating that the transgene is capable of expression from the Xa. CITED1 expression was detected from the male Xa in brain, but was not detected at significant levels in liver or spleen. Significant expression of ERCC6L was not detected in any of the three tissues we initially examined for any genotype, however, expression was detected in mouse embryonic fibroblasts (MEFs) derived from two knock-in males (Supplementary Material, Fig. S4). These expression patterns are consistent with data in the corresponding tissues in human (Supplementary Material, Fig. S5).
Figure 3.

Analysis of BAC RP11–1145H7 shows RPS4X and CITED1 escape XCI at Hprt. (A) Integration of the RPS4X BAC at Hprt; genes on the BAC expected to escape from XCI in green, subject in blue, variable and discordant in yellow, and genes at integration site (both known to be subject to XCI) in grey. Genes with RT-qPCR and DNAm assays are indicated. (B) Description of genotypes, six mice for each. (C) Normalized to Pgk1, RT-qPCR of RPS4X expression in brain shows that the transgene is active on a male X, and escapes inactivation at ∼50% when on the Xi in brain and liver, with a slightly lower level in spleen. An adjacent discordant gene CITED1 also escapes from the Xi in females at similar levels in brain. CITED1 expression was not detected in liver and spleen. Expression from female Xi is shown as percentage of the male X (red text). (D) Average DNAm of skewed knock-in females shows a hypomethylated RPS4X promoter in all tissues, CITED1 is low but different between tissues, and ERCC6L, HPRT and Phf6 are significantly hypermethylated compared to knock-in males in all tissues (Mann–Whitney t-test, significance from hypomethylated males denoted by asterisks; P-value <0.001***, 0.001–0.01**, 0.01–0.05*, >0.05 ns).
In knock-in females where expression is measured only from the Xi, RPS4X and CITED1 both appear to be expressed at approximately half the level of the male Xa in brain, with RPS4X also showing similar escape levels in liver and spleen. Knock-in female mice with random XCI displayed higher expression levels than the skewed female mice (Fig. 3C). No ERCC6L expression was detected from MEFs derived from two skewed knock-in females (Supplementary Material, Fig. S4).
Promoter CpG islands associated with BAC genes RPS4X, CITED1 and ERCC6L were examined, with all three appearing hypomethylated in males. Xi knock-in females have ongoing hypomethylation of the RPS4X promoter in all tissues, which supports that the gene escapes from XCI. CITED1 DNAm is low in brain where the gene is expressed from the Xi; however, is slightly increased in other tissues. The ERCC6L promoter is hypomethylated in males suggesting the transgene is capable of expression as seen in the male MEFs, despite expression not being detectable in the other tissues we examined. ERCC6L is hypermethylated in Xi knock-in females implying that it is subject to inactivation, concordant with the lack of expression seen in knock-in female MEFs. All mice show Phf6 and HPRT DNAm averages similar to those previously seen at these sites (20), demonstrating that escape of RPS4X and CITED1 was not spreading into neighbouring genes on the Xi (Fig. 3D). The lack of DNAm at RPS4X, and consistency in expression levels between skewed female knock-in mice after multiple generations of breeding, further demonstrates that both copies of the human BAC are on the X chromosome, and both copies of RPS4X are escaping inactivation.
Expressed X-linked SNP analysis of knock-in females with random XCI showed no consistent skewing of inactivation toward one X being silenced more often than the other (Supplementary Material, Fig. S2B). This is in agreement with both expression and DNAm of non-escaping BAC genes ERCC6L and HPRT where contribution of an Xa in addition to an Xi in these females presents itself in generally higher expression levels and lower DNAm compared to the experimentally skewed females. Lack of a detrimental influence of the transgene was also supported by normal breeding and genotype ratios of the experimental mice (Supplementary Material, Table S1).
Elements regulating RPS4X and CITED1 escape from XCI
A broad array of DNA elements is proposed to play a role in the ability of a gene to escape from XCI (depletion of waystations, enrichment of boundary elements between active and inactive domains, and presence of escape elements), as well as factors involved in chromatin ultrastructure. Considering this, we used available datasets try and demarcate regions that could support the escape and silenced profiles on our human BAC by profiling repetitive content of the transgenes, patterns of transcription-factor binding and contact domain boundaries (Fig. 4A). Co-localization of boundary factor CTCF, cohesion components SMC3 and RAD21, as well as YY1 potentially mark a boundary between subject gene ERCC6L and escape gene RPS4X, however, similar sites exist in the corresponding region in mouse (Fig. 4B), which does not escape from XCI. Additionally, kilobase-resolution Hi-C data of the BAC in its endogenous location in GM12878 reveals a contact domain boundary between ERCC6L and RPS4X, although in several other female cell lines examined at 5 kb resolution the end of the domain sometimes shifts from downstream of RPS4X to end between CITED1 and RPS4X so that the two genes are not always in the same domain (25). It is possible that such a shift in boundaries is responsible for the variability we see in CITED1 DNAm and previously recorded discordance in escape status between studies.
Figure 4.

The RPS4X BAC contains elements that may contribute to escape from XCI. (A) Using available datasets, we examined potential elements that may aid in setting up an escape domain around human RPS4X and CITED1 (shaded for escape) displayed in the UCSC browser (hg19 assembly). Factors associated with escape from XCI as well as components of structural complexes to form boundaries line up between silenced gene ERCC6L and escape gene RPS4X in female cell line GM12878. In GM12878 as well as several other female cell lines, a contact domain boundary is located within the region contained on the BAC, although the end shifts depending on cell line and resolution to either include RPS4X and CITED1 in the same domain, or end between the two (grey to black) (25). (B) The corresponding region in mouse is subject to inactivation yet retains similar elements to the human region. Transcription factor binding sites as well as contact domain (black) (25) are for CH12 cells in the UCSC browser (mm9 assembly), with the exception of YY1 binding which was done in embryonic stem cells (49). Truncated genes HDAC8 and PIN4 not labelled.
Discussion
The use of sex-reversed mice (the four core genotype model) has demonstrated that the sex chromosomes are important contributors to many phenotypic differences between males and females (reviewed in 26), likely partially due to the genes that escape from XCI. As more genes escape from XCI in humans, their phenotypic contribution is likely even larger, yet given the differences in number of escape genes between human and mice, it is unknown how different the mechanism of expression from an otherwise heterochromatic chromosome may be between the species. Previous studies have demonstrated that Kdm5c harbours an intrinsic escape element (16,17). As those studies examined random integrations on the Xi, they could not be recapitulated for our assessment of DNA elements. We therefore turned to the Hprt docking site, previously suggested to support expression from the Xi (20). Using the Kdm5c BAC examined by others (16,17), and a combination of direct expression and indirect DNAm assays, we established that Hprt can support escape gene expression. Our Kdm5c transgene showed expression from the Xi in the range of 20–80% of the Xa across our three examined tissues, which is similar to what has been previously reported. Levels have been shown to vary widely between mouse strains, tissue type, cell type and developmental stage, with Xi expression relative to the Xa ranging from 20–70% [in vivo: (6,27,28); in vitro: reviewed in 12]. As KDM5C has been linked to intellectual disability disorders in humans (29) there may be tighter regulation of a third copy of the gene in a developing female brain, which is where we saw the lowest (and non-significant) level of Xi expression.
Having confirmed the utility of the docking site with the Kdm5c BAC, we further used the Hprt site to examine a human escape domain containing RPS4X and CITED1. RPS4X is a broadly expressed (Supplementary Material, Fig. S5) primate-specific escape gene with Y homology retained in human but not mouse (30), which may have led to the loss of a drive for extra gene dosage in mouse and subsequent lack of conservation of the DNA elements necessary for escape (31). However, the mouse is able to recognize the intrinsic element present at the human gene, demonstrating likely conservation of the elements, or at a minimum conservation of the machinery recognizing the element(s). Recent examination of neural progenitor cell clones suggests that Rps4x, as well as neighbouring genes Ercc6l and Cited1, are occasionally capable of variable escape in some mouse cell types suggesting that some sensitivity to escape may remain (12,14).
ERCC6L has been well-characterized as subject in humans (1,7), in agreement with our lack of expression in female MEFs and hypermethylation across all tissues in skewed knock-in females. It is not well expressed in brain, liver or spleen in humans which may be why were unable to detect expression in our core dataset (Supplementary Material, Fig. S5).
CITED1 has previously been called escape by DNAm analysis (7) but subject by expression in Xi somatic cell hybrids (1). CITED1 shows tissue-specific expression in humans in testis, hypothalamus and pituitary (Supplementary Material, Fig. S5), which agrees with our ability to detect expression in brain only. Expression of CITED1 from the Xi along with a hypomethylated promoter in brain gives us confidence that it escapes in this tissue. Interestingly, CITED1 DNAm increases in tissues where it is not expressed; however, it is still in the range where we call escape, although there is not a clear threshold of DNAm at which genes become subject to XCI. Our previous X-chromosome-wide DNAm study generally called genes as escape if, in females with random XCI, they had 0–20% DNAm and a difference of less than 10% from males, yet left an uncallable zone between these and the subject genes that generally had >30% methylation (7). By that metric we would call CITED1 an escape gene in liver and uncallable in spleen. It is, however, important to note that we have observed silencing of HPRT with DNAm at its promoter in female mice with random XCI as low as ∼13% (20).
The integration and recapitulation of escape gene expression from our human BAC in mouse demonstrates that escape elements have a conserved recognition and mechanism across species, and putative escape elements must lie within ∼112 kb for RPS4X from the subject ERCC6L promoter to the end of the BAC (Fig. 4). Both BACs must also carry boundaries to contain the open state, as spread of hypomethylation was not seen into Phf6 or HPRT, previously detected when an autosomal transgene escaped from XCI (20). CTCF binding has been hypothesized to be responsible for setting boundaries around escapees, with differential binding responsible for creating the larger domains of escape genes in humans than in mouse, which tend to be solo (32). Additionally, it has been suggested that shifting the binding site of CTCF in different cell lines can adjust which genes in a region are escaping (6) and may be part of the explanation for genes that variably escape. On a larger scale, allelic ultrastructural studies of the X chromosome have revealed that in both humans and mice the Xi forms a distinctive superdomain structure rather than the topologically associated domains (TADs) that are observed across the Xa and the autosomes (14,33), and that Xi-specific CTCF sites correspond to the escaping long non-coding RNA loci involved in the superloop formation. Yet evidence also suggests that continued transcription of genes escaping from XCI along with binding of factors such as CTCF may enable the maintenance or re-creation of TAD structures around regions containing escape genes (14). Given the plethora of CTCF sites available on the X, and that CTCF sites could not insulate a transgenic reporter gene from X-inactivation (34), it is unlikely that CTCF acts alone as a complete escape element, although it is a likely contributor to form an escape domain on the BAC.
The clustering of human escape genes is suggestive of domain-based regulation and so RPS4X and CITED1 may be under control of the same element; however, there is growing evidence for promoter-proximal elements being involved in escape (13,14,35). Indeed, some escape genes have alternative promoters that can differ in status (36). In silico analysis of the transcription start sites (TSS) of escape genes in humans has found significant over-representations of YY1 transcription factor binding motif and ChIP-seq peaks, and similar to CTCF, YY1 occupancy is significantly biased toward the Xi at loci that are frequent contacts of Xi-specific superloops (37). Additionally, components of the cohesin complex (RAD21, SMC3) have been found to co-localize with CTCF at these sites in both human and mouse female cells (38), again highlighting that there is likely a structural component to escape.
Previous studies on the X and X; autosome translocations have found correlations between repetitive content and whether genes are subject to or escaping from XCI. Using large windows (± 50–100 kb) around the TSS or full transcribed regions of genes (39,40) as well as more promoter-centric methods (41), LINEs are seen to be significantly enriched in regions surrounding genes that are subject to inactivation, while Alu repetitive elements and short motifs were significantly enriched in those that escape inactivation. Given that the escape genes on the BAC are small and located in close proximity to subject genes, we undertook a promoter-centric approach (±5 kb around TSS and 15 kb upstream of the TSS) to see if the Alu-enrichment, LINE-depletion pattern holds for the escape genes RPS4X and CITED1 compared to subject gene ERCC6L. We found little difference in content between the three genes that would support using these sequences for predicting escape on our BAC. Overall, it is possible that all genes are capable of escape, but need the correct environmental set-up to do so which we suspect is a combination of multiple factors.
In an attempt to harness the potential of the second allele to protect from X-linked disease in females, recent studies have explored shRNA and pharmacological reactivation of the Xi, but have determined that such approaches tend to reactivate a substantial portion of silenced genes (42). Therefore, understanding of how expression from the Xi is limited to only a subset of genes on the X might provide insights into utilization of such therapeutic approaches. Importantly, despite a lack of conservation in number and distribution of genes that escape from XCI, we have demonstrated the ability of mouse to recognize human elements regulating escape from XCI, at least for this subject-escape-subject region, thereby providing a model system for the exploration of these elements to see how various deletions affect silencing and escape of nearby genes. Integration of multiple BACs from larger escape domains such as that including KDM5C, or the pseudoautosomal region 1, would start to address the commonality of such escape elements across the human X.
Materials and Methods
Construct generation
BACs RP23–391D18 and RP11–1145H7 (CHORI, BACPAC Resources Center) were each retrofitted using the lambda recombination system (43) allowing the addition of Hprt homologous recombination targeting arms to integrate constructs into the Hprt gene on the mouse X chromosome (23,44). PCRs spanning retrofit junctions as well as pulsed-field gel electrophoresis confirmed proper retrofit of construct (Supplementary Material, Table S2 for primer information).
Generation of mouse strains
BAC DNA was purified using the Nucleobond XTRA BAC kit (Macherey-Nagel) and linearized with I-SceI. The BAC constructs were electroporated into male C57BL/6NTac (Taconic, Hudson, NY) ESCs (mEMS6131) carrying the Hprtb-m3 deletion (N11 backcrossing from C57BL/6J [The Jackson Laboratory [JAX], Bar Harbor, ME, Stock 002171]) and homozygous for the Aw-j agouti allele (N10 backcrossing from B6.129 [JAX, Stock 00051]); with a BTX ECM 630 Electro cell manipulator (BTX, USA). ESC clones were selected in HAT media for reconstitution of the HPRT/Hprt locus, isolated and DNA purified. qPCR and PCR with primers spanning approximately every 10 kb along the construct to test intactness were performed for RP23–391D18 and RP11–1145H7 BACs, respectively. Number of integrations was tested using copy-number qPCR assays of BAC backbone regions common to both libraries (Supplementary Material, Table S2 for primer information). Approximately 100 ng of DNA was added to a master mix containing 0.16 μl Maxima Hot Start Taq (Fermentas, USA) with 2 μl 10× buffer and 2 μl 25 mm MgCl2, 1 μl EvaGreen dye (Biotium, USA), 0.16 μl 25 mm dNTPs, 0.2 μl of each 25 μm forward and reverse primers and sterile dH2O to 20 μl. qPCR was performed in triplicate for each sample using a StepOnePlus Real-Time PCR System (Applied Biosystems, Germany) with conditions as follows for all primer sets: 95°C for 5 min; followed by 40 cycles of 95°C for 15 s, 60°C for 30 s, and 72°C for 1 min; and a melt curve stage of 95°C for 15 s, 60°C for 1 min and an increase of 0.3°C until 95°C. Testing for multiple Tm peaks for primer specificity, as well as removal of sample outliers that significantly deviated from their associated group were performed using the StepOne software v2.1. Intactness and copy number assays were performed in ESCs prior to microinjection, as well as in N1 and N2 generations. Copy number was analysed using comparative CT method, normalized to Hbb-bs control assay and then to male wild-type controls. ESC derivation and culture was conducted as we have described previously (44). Targeted ESC clones were microinjected into C57BL/6J (JAX, Stock 000664) blastocysts to generate chimeras that were subsequently bred to C57BL/6J females to obtain female germline offspring carrying the BAC insert. The female germline offspring were then bred to C57BL/6J males and backcrossing to C57BL/6J (B6) continued such that mice used in this study were N3 or higher.
The floxed Xist strain 129-Xisttm2Jae [Mutant Mouse Regional Resource Centre, Chapel Hill, NC, Stock 029172-UNC (45)] was crossed to the cre-deleter strain 129-ACTBCre (N7 backcrossing from C57BL/6J [JAX, Stock 003376]) to generate females carrying the Xist deletion (129-Xist1lox/X). The 129-Xist1lox strain was maintained by backcrossing to strain 129S1/SvImJ (129) (JAX, Stock 002448) (46). Females with the Xist deletion were then crossed to males with the BAC construct integrated at the Hprt locus (B6-HprtBAC/Y) to generate F1 129-Xist1lox/B6-HprtBAC and F1 129-XistWT/B6-HprtBAC females. This Xist knockout has been shown to render the X chromosome carrying it unable to inactivate (20,47), thereby resulting in the knock-in X chromosome with an intact Xist becoming the Xi. As controls, females with the BAC construct (B6-HprtBAC/X) were crossed to 129 males to produce F1 B6-HprtBAC/129-Y males. Chi-square tests were performed to assess breeding outcomes of the experimental mice. Approval for the generation and breeding of mice carrying the BAC constructs and Xist alleles was obtained from the University of British Columbia Committee on Animal Care (A14–0294).
Tissue collection and DNA and RNA extraction
Adult mouse (over 8 weeks) livers, spleens and brains were macrodissected and flash frozen with liquid nitrogen, then stored at −80°C for no more than 6 months before processing. For MEFs, 13.5 days post-coital embryos were isolated, the head and red organs removed, the remaining embryo individually minced with suction and expulsion using an 18-gauge needle in feeder medium (10% fetal bovine serum in D-MEM) and plated into a T75 flask. Two days following collection, cultures were rinsed with PBS, trypinized and re-plated in their original flasks to achieve maximal cell dispersal and to rid the cultures of debris. Confluency was typically achieved two days after re-plating and at this point cells from individual embryos were frozen for future expansion.
DNA and RNA extraction was performed using TRIzol Reagent (Invitrogen, USA), according to the manufacturer’s protocol. A total of 50–100 mg samples of each liver and spleen were used, while an entire sagittal half of brain was homogenized to control for cellular heterogeneity in this tissue. Nucleic acids were quantified by UV spectrophotometry (Ultraspec 2000, Pharmacia Biotech). RNA extractions were diluted to concentrations of 1 μg/μl and treated with 1 μl DNase I with 10× buffer (Roche) and 1 μl Ribolock (Thermo Fisher Scientific) in a volume of 50 μl at 37°C for 1 h followed by heat inactivation at 75°C for 10 min.
Expression analysis
For analysis of transcription, 2 μg of DNased RNA extracted from tissues was converted to cDNA using standard reverse transcription conditions with Random Hexamer Primers (Thermo Fisher Scientific) and 200U M-MLV Reverse Transcriptase (Invitrogen, USA). Reactions were carried out at 42°C for 2 h followed by a 5-min incubation at 95°C. qPCR was used to determine relative transcription levels of transgenes compared to stable housekeeping gene Pgk1 (48) in mice carrying the BAC constructs (Supplementary Material, Table S2 for primer information). Samples were run in triplicate using the same reaction set-up, run conditions, analysis of Tm peaks and outliers as described previously for the copy number qPCR. Negative controls of RNA without reverse transcriptase were also run to ensure that the samples contained no DNA contamination. Expression levels were quantified using the comparative CT method and tested for significant differences between groups using the unpaired t-test with Welch’s correction in GraphPad Prism 5.
DNA methylation and SNP analyses
Using the EZ DNA Methylation-Gold Kit (Zymo Research, USA), 500 ng of DNA was bisulphite converted following the manufacturer’s instructions. Internal bisulphite conversion controls were included in the pyrosequencing assays to monitor complete conversion of DNA. Each 25 μl pyrosequencing PCR was performed with 10× PCR buffer (Qiagen, Germany), 0.2 mm dNTPs, 0.125 units Hot Start Taq DNA polymerase (Qiagen, Germany), 0.25 mm forward primer, 0.25 mm reverse primer and 12–35 ng bisulfite-converted DNA. Conditions for PCR were 95°C for 15 min, 50 cycles of 94°C for 30 s, 55°C for 30 s, 72°C for 1 min and finally 72°C for 10 min. One forward or reverse primer was biotinylated, depending on which strand contained the target region to be sequenced, to subsequently isolate the strand of interest for pyrosequencing. Template preparation for pyrosequencing was done according to the manufacturer’s protocol, using 10–15 μl of PCR products. CDT tips were used to dispense the nucleotides for pyrosequencing, using the PyroMark MD machine (Qiagen, Germany). Each human promoter assay was tested in at least one mouse sample without the target transgene to ensure the specificity of the human primers. At least three CpGs in an island were evaluated and averaged per assay. Significance was tested using the Mann–Whitney t-test in GraphPad Prism 5.
Pyrosequencing was performed as above using primers that amplify a single-nucleotide polymorphism of the Fln locus from cDNA of knock-in females without the Xist deletion to determine level of skewing by relative expression of the B6 and 129 alleles (Supplementary Material, Table S2 for primer information).
Supplementary Material
Supplementary Material is available at HMG online. Our supplementary material contains data from the Genotype-Tissue Expression (GTEx) Project, supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH and NINDS. The data used for the analyses described in this manuscript were obtained from: the GTEx Portal on 12/01/17.
Supplementary Material
Acknowledgements
We thank past and present members of the Brown and Wasserman laboratory, specifically Bradley Balaton, for helpful discussion. We also thank Tess C. Lengyell of the Mouse Animal Production Services (MAPS) team at CMMT for microinjection and breeding.
Conflict of Interest statement. None declared.
Funding
This work was supported by the Canadian Institutes of Health Research (MOP-119586 to C.J.B. and E.M.S.). S.B.P was supported by a Natural Sciences and Engineering Research Council of Canada doctoral award. Funding to pay the Open Access publication charges for this article was provided by Canadian Institutes of Health Research (MOP-119586 to C.J.B. and E.M.S.).
References
- 1. Carrel L., Willard H.F. (2005) X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature, 434, 400–404. [DOI] [PubMed] [Google Scholar]
- 2. Hook E.B., Warburton D. (2014) Turner syndrome revisited: review of new data supports the hypothesis that all viable 45, X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum. Genet., 133, 417–424. [DOI] [PubMed] [Google Scholar]
- 3. Dunford A., Weinstock D.M., Savova V., Schumacher S.E., Cleary J.P., Yoda A., Sullivan T.J., Hess J.M., Gimelbrant A.A., Beroukhim R.. et al. (2017) Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat. Genet., 49, 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carrel L., Brown C.J. (2017) When the Lyon(ized chromosome) roars: ongoing expression from an inactive X chromosome. Philos. Trans. R. Soc. Lond. B Biol. Sci., 372, 20160355.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balaton B.P., Brown C.J. (2016) Escape artists of the X chromosome. Trends Genet., 32, 348–359. [DOI] [PubMed] [Google Scholar]
- 6. Berletch J.B., Ma W., Yang F., Shendure J., Noble W.S., Disteche C.M., Deng X. (2015) Escape from X inactivation varies in mouse tissues. PLoS Genet., 11, e1005079.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cotton A.M., Price E.M., Jones M.J., Balaton B.P., Kobor M.S., Brown C.J. (2015) Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum. Mol. Genet., 24, 1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cotton A.M., Lam L., Affleck J.G., Wilson I.M., Peñaherrera M.S., McFadden D.E., Kobor M.S., Lam W.L., Robinson W.P., Brown C.J. (2011) Chromosome-wide DNA methylation analysis predicts human tissue-specific X inactivation. Hum. Genet., 130, 187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balaton B.P., Cotton A.M., Brown C.J. (2015) Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol. Sex. Dif., 6, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Disteche C.M., Berletch J.B. (2015) X-chromosome inactivation and escape. J. Genet., 94, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reinius B., Shi C., Hengshuo L., Sandhu K., Radomska K.J., Rosen G.D., Lu L., Kullander K., Williams R.W., Jazin E. (2010) Female-biased expression of long non-coding RNAs in domains that escape X-inactivation in mouse. BMC Genomics, 11, 614.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marks H., Kerstens H.H.D., Barakat T.S., Splinter E., Dirks R.A.M., van Mierlo G., Joshi O., Wang S.-Y., Babak T., Albers C.A.. et al. (2015) Dynamics of gene silencing during X inactivation using allele-specific RNA-seq. Genome Biol., 16, 149.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Calabrese J.M., Sun W., Song L., Mugford J.W., Williams L., Yee D., Starmer J., Mieczkowski P., Crawford G.E., Magnuson T. (2012) Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell, 151, 951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giorgetti L., Lajoie B.R., Carter A.C., Attia M., Zhan Y., Xu J., Chen C.J., Kaplan N., Chang H.Y., Heard E., Dekker J. (2016) Structural organization of the inactive X chromosome in the mouse. Nature, 535, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang C., Chapman A.G., Kelsey A.D., Minks J., Cotton A.M., Brown C.J. (2011) X-chromosome inactivation: molecular mechanisms from the human perspective. Hum. Genet., 130, 175–185. [DOI] [PubMed] [Google Scholar]
- 16. Li N., Carrel L. (2008) Escape from X chromosome inactivation is an intrinsic property of the Jarid1c locus. Proc. Natl. Acad. Sci. USA, 105, 17055–17060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Horvath L.M., Li N., Carrel L. (2013) Deletion of an X-inactivation boundary disrupts adjacent gene silencing. PLoS Genet., 9, e1003952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patel S., Bonora G., Sahakyan A., Kim R., Chronis C., Langerman J., Fitz-Gibbon S., Rubbi L., Skelton R.J.P., Ardehali R.. et al. (2017) Human embryonic stem cells do not change their X inactivation status during differentiation. Cell Rep., 18, 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sahakyan A., Kim R., Chronis C., Sabri S., Bonora G., Theunissen T.W., Kuoy E., Langerman J., Clark A.T., Jaenisch R., Plath K. (2017) Human naive pluripotent stem cells model X chromosome dampening and X inactivation. Cell Stem Cell, 20, 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang C., McLeod A.J., Cotton A.M., de Leeuw C.N., Laprise S., Banks K.G., Simpson E.M., Brown C.J. (2012) Targeting of >1.5 Mb of human DNA into the mouse X chromosome reveals presence of cis-acting regulators of epigenetic silencing. Genetics, 192, 1281–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gartler S.M., Riggs A.D. (1983) Mammalian X-Chromosome inactivation. Annu. Rev. Genet., 17, 155–190. [DOI] [PubMed] [Google Scholar]
- 22. Schmouth J.-F., Bonaguro R.J., Corso-Diaz X., Simpson E.M. (2012) Modelling human regulatory variation in mouse: finding the function in genome-wide association studies and whole-genome sequencing. PLoS Genet., 8, e1002544.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bronson S.K., Plaehn E.G., Kluckman K.D., Hagaman J.R., Maeda N., Smithies O. (1996) Single-copy transgenic mice with chosen-site integration. Proc. Natl. Acad. Sci. USA, 93, 9067–9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsuchiya K.D., Greally J.M., Yi Y., Noel K.P., Truong J.-P., Disteche C.M. (2004) Comparative sequence and X-inactivation analyses of a domain of escape in human xp11.2 and the conserved segment in mouse. Genome Res., 14, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rao S.S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S., Aiden E.L. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arnold A.P., Reue K., Eghbali M., Vilain E., Chen X., Ghahramani N., Itoh Y., Li J., Link J.C., Ngun T., Williams-Burris S.M. (2016) The importance of having two X chromosomes. Philos. Trans. R. Soc. Lond. B Biol. Sci., 371, 20150113.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sheardown S., Norris D., Fisher A., Brockdorff N. (1996) The mouse Smcx gene exhibits developmental and tissue specific variation in degree of escape from X inactivation. Hum. Mol. Genet., 5, 1355–1360. [DOI] [PubMed] [Google Scholar]
- 28. Carrel L., Hunt P.A., Willard H.F. (1996) Tissue and lineage-specific variation in inactive X chromosome expression of the murine Smcx gene. Hum. Mol. Genet., 5, 1361–1366. [DOI] [PubMed] [Google Scholar]
- 29. Jensen L.R., Amende M., Gurok U., Moser B., Gimmel V., Tzschach A., Janecke A.R., Tariverdian G., Chelly J., Fryns J.-P.. et al. (2005) Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet., 76, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bellott D.W., Hughes J.F., Skaletsky H., Brown L.G., Pyntikova T., Cho T.-J., Koutseva N., Zaghlul S., Graves T., Rock S.. et al. (2014) Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature, 508, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park C., Carrel L., Makova K.D. (2010) Strong purifying selection at genes escaping X chromosome inactivation. Mol. Biol. Evol., 27, 2446–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Filippova G.N., Cheng M.K., Moore J.M., Truong J.-P., Hu Y.J., Nguyen D.K., Tsuchiya K.D., Disteche C.M. (2005) Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development. Dev. Cell, 8, 31–42. [DOI] [PubMed] [Google Scholar]
- 33. Deng X., Ma W., Ramani V., Hill A., Yang F., Ay F., Berletch J.B., Blau C.A., Shendure J., Duan Z., Noble W.S., Disteche C.M. (2015) Bipartite structure of the inactive mouse X chromosome. Genome Biol., 16, 152.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ciavatta D., Kalantry S., Magnuson T., Smithies O. (2006) A DNA insulator prevents repression of a targeted X-linked transgene but not its random or imprinted X inactivation. Proc. Natl. Acad. Sci. USA, 103, 9958–9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mugford J.W., Starmer J., Williams R.L., Calabrese J.M., Mieczkowski P., Yee D., Magnuson T. (2014) Evidence for local regulatory control of escape from imprinted X chromosome inactivation. Genetics, 197, 715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goto Y., Kimura H. (2009) Inactive X chromosome-specific histone H3 modifications and CpG hypomethylation flank a chromatin boundary between an X-inactivated and an escape gene. Nucl. Acids Res., 37, 7416–7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen C.-Y., Shi W., Balaton B.P., Matthews A.M., Li Y., Arenillas D.J., Mathelier A., Itoh M., Kawaji H., Lassmann T.. et al. (2016) YY1 binding association with sex-biased transcription revealed through X-linked transcript levels and allelic binding analyses. Sci. Rep., 6, 37324.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang F., Deng X., Ma W., Berletch J.B., Rabaia N., Wei G., Moore J.M., Filippova G.N., Xu J., Liu Y.. et al. (2015) The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol., 16, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bala Tannan N., Brahmachary M., Garg P., Borel C., Alnefaie R., Watson C.T., Thomas N.S., Sharp A.J. (2014) DNA methylation profiling in X; autosome translocations supports a role for L1 repeats in the spread of X chromosome inactivation. Hum. Mol. Genet., 23, 1224–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Z., Willard H.F., Mukherjee S., Furey T.S. (2006) Evidence of influence of genomic DNA sequence on human X chromosome inactivation. PLoS Comp. Biol., 2, e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cotton A.M., Chen C.-Y., Lam L.L., Wasserman W.W., Kobor M.S., Brown C.J. (2014) Spread of X-chromosome inactivation into autosomal sequences: role for DNA elements, chromatin features and chromosomal domains. Hum. Mol. Genet., 23, 1211–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lessing D., Dial T.O., Wei C., Payer B., Carrette L.L.G., Kesner B., Szanto A., Jadhav A., Maloney D.J., Simeonov A.. et al. (2016) A high-throughput small molecule screen identifies synergism between DNA methylation and Aurora kinase pathways for X reactivation. Proc. Natl. Acad. Sci. USA, 113, 14366–14371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu D., Ellis H.M., Lee E.C., Jenkins N.A., Copeland N.G., Court D.L. (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA, 97, 5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang G.S., Banks K.G., Bonaguro R.J., Wilson G., Dreolini L., de Leeuw C.N., Liu L., Swanson D.J., Goldowitz D., Holt R.A., Simpson E.M. (2009) Next generation tools for high-throughput promoter and expression analysis employing single-copy knock-ins at the Hprt1 locus. Genomics, 93, 196–204. [DOI] [PubMed] [Google Scholar]
- 45. Csankovszki G., Panning B., Bates B., Pehrson J.R., Jaenisch R. (1999) Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat. Genet., 22, 323–324. [DOI] [PubMed] [Google Scholar]
- 46. Simpson E.M., Linder C.C., Sargent E.E., Davisson M.T., Mobraaten L.E., Sharp J.J. (1997) Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat. Genet., 16, 19–27. [DOI] [PubMed] [Google Scholar]
- 47. Gribnau J., Luikenhuis S., Hochedlinger K., Monkhorst K., Jaenisch R. (2005) X chromosome choice occurs independently of asynchronous replication timing. J. Cell. Biol., 168, 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boda E., Pini A., Hoxha E., Parolisi R., Tempia F. (2009) Selection of reference genes for quantitative real-time RT-PCR studies in mouse brain. J. Mol. Neurosci., 37, 238–253. [DOI] [PubMed] [Google Scholar]
- 49. Sigova A.A., Abraham B.J., Ji X., Molinie B., Hannett N.M., Guo Y.E., Jangi M., Giallourakis C.C., Sharp P.A., Young R.A. (2015) Transcription factor trapping by RNA in gene regulatory elements. Science, 350, 978–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.