

Abstract
Increased airway smooth muscle (ASM) mass is believed to underlie the relatively fixed airway hyperresponsiveness (AHR) in asthma. Developments of therapeutic approaches to reverse airway remodeling are impeded by our lack of insight on the mechanisms behind the increase in mass of contractile ASM cells. Increased expression of laminin, an extracellular matrix protein, is associated with asthma. Our studies investigate the role of laminin-induced ASM survival signals in the development of increased ASM and AHR. Antagonizing laminin integrin binding using the laminin-selective competing peptide, YIGSR, and mimicking laminin with exogenous α2-chain laminin, we show that laminin is both necessary and sufficient to induce ASM cell survival, concomitant with the induction of ASM contractile phenotype. Using siRNA, we show that the laminin-binding integrin α7β1 mediates this process. Moreover, in laminin-211-deficient mice, allergen-induced AHR was not observed. Notably, ASM cells from asthmatic airways express a higher abundance of intracellular cell survival proteins, consistent with a role for reduced rates of cell apoptosis in development of ASM hyperplasia. Targeting the laminin-integrin α7β1 signaling pathway may offer new avenues for the development of therapies to reduce the increase in mass of contractile phenotype ASM cells that underlie AHR in asthma.—Tran, T., Teoh, C. M., Tam, J. K. C., Qiao, Y., Chin, C. Y., Chong, O. K., Stewart, A. G., Harris, T., Wong, W. S. F., Guan, S. P., Leung, B. P., Gerthoffer, W. T., Unruh, H., and Halayko, A. J. Laminin drives survival signals to promote a contractile smooth muscle phenotype and airway hyperreactivity.
Keywords: apoptosis, phenotype plasticity, integrin
Airway wall remodeling (AWR) and airway hyperresponsiveness (AHR) are two prominent features of asthma that contribute to chronic symptoms. AWR is defined by a number of structural changes, including increased mass of contractile airway smooth muscle (ASM) cells, and fibrosis resulting from the accumulation of extracellular matrix (ECM) proteins that include laminin (1). Increased ASM mass is a principal factor underlying excessive and fixed AHR in asthma (2).
There has been considerable investigation into the molecular signals that regulate ASM proliferation in vitro to explain ASM tissue hypertrophy in patients with asthma. However, despite studies spanning more than a decade, there remains only limited evidence for increased proliferation of ASM cells as the mechanism for ASM mass accumulation in situ (1, 3, 4); this suggests that other cellular mechanisms play a prominent role. For example, ASM cell hypertrophy, which parallels phenotype maturation of ASM cells, has been described using in vitro models (5, 6); and reduced physiological apoptosis of ASM cells may lead to ASM hyperplasia over time. These two issues and their possible relationship were investigated in this study. Moreover, we explored whether capacity for ASM cell maturation (hypertrophy) induced by laminin is linked with concomitant inhibition of apoptosis, both in cell culture and in a murine model of allergen-induced asthma.
ASM cells are key contributors to fibrosis through their capacity to secrete ECM proteins, including laminin. Notably, increased airway wall immunoreactivity for laminin correlates with asthma severity (7). We were the first to show that contractile phenotype ASM cells express laminin-211, and, with the use of laminin-competing peptide tyrosine-isoleucine-glycine-serine-arginine (YIGSR), that endogenous laminin-211 expression is essential for ASM cells to acquire and maintain an enlarged contractile phenotype (8). Moreover, the laminin-binding integrin α7β1 mediates and is required for the effects of laminin-211 on ASM phenotype (9), suggesting that changes in the endogenous expression of laminin and integrin α7β1 are determinants of ASM phenotype and function during disease pathogenesis.
The potential for interaction between laminin and integrins on ASM cells to regulate ASM survival has not previously been investigated, but, herein, we propose that laminin-211, via integrin α7β1, activates antiapoptotic pathways and suppresses proapoptotic signals in ASM cells to promote maturation (hypertrophy) of contractile phenotype in ASM cells. Thus, our study investigates a new mechanistic role for laminin matrix changes in asthma that supports disease-relevant remodeling of ASM to influence AHR.
MATERIALS AND METHODS
Cell culture
Human ASM cell lines were generated using MMLV retroviral transfection to facilitate stable integration of the human telomerase reverse transcriptase (hTERT) gene (8). Primary human ASM cell cultures were obtained from macroscopically healthy segments of the second to fourth generation main bronchus from patients with (n=4; 50% male, 50% female; mean±sd age: 35±17 yr, range: 14–54 yr) or without asthma (n=4; 50% male, 50% female; age: 58±15 yr, range: 38–70 yr), who underwent lung resection surgery (8). All procedures were approved by the Institutional Review Board of the National University of Singapore (NUS) and of the University of Melbourne. To induce ASM cells toward a contractile phenotype, confluent cultures were switched to DMEM supplemented with 1% ITS for up to 7 d (8). LY294002 and wortmannin (Cell Signaling Technology, Danvers, MA, USA) were added at the time of serum deprivation and again either every 2 d each time serum-free medium was replaced (LY294002) or 3×/d (wortmannin) for 7 d. Laminin peptide (YIGSR; Sigma, St. Louis, MO, USA) was prepared in distilled water. For laminin-coating experiments, laminin preparations from Engelbreth-Holm-Swarm murine sarcoma (Sigma) and human placenta tissues (Millipore, Singapore) were used (8).
Western blot analysis
Human ASM cells from d 0 (basal) and following 7 d serum deprivation (d 7) were lysed using ice-cold lysis buffer (100 mM NaCl; 10 mM Tris HCl, pH 7.5; 2 mM EDTA; 0.5% deoxycholate; 1% Triton X-100; 1 mM phenylmethylsulfonylfluoride; 10 mM MgCl2; 5 μg/ml aprotinin; and 100 μM sodium orthovanadate) and centrifuged at 9600 g, as described previously (8). Protein content was determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA). Total proteins (10–12 μg/well) were separated on either 10 or 12% SDS-polyacrylamide gels, then electrotransferred to nitrocellulose membranes for Western blot analysis. Membranes were blocked with 5% skim milk containing Tris-buffered saline (10 mM Tris HCl, pH 8, and 150 mM NaCl) and 0.1% Tween-20 followed by overnight incubation with primary antibodies sm-α-actin, β-actin, and α2-chain laminin (Sigma); Bcl-2, Bax, poly-(ADP-ribose) polymerase (PARP), phospho-Akt (Ser473), and Akt total (Cell Signaling); integrin α7 (Abcam, Cambridge, UK) and integrin α7 (Guido Tarone, University of Torino, Turin, Italy). The membranes were developed by subsequent incubation with secondary horseradish peroxidase-conjugated antibody, and immunoreactive bands were visualized by enhanced chemiluminescence (Amersham; GE Healthcare, Singapore). Band intensities were scanned and quantified using Epson Perfection V200 Photo (Seiko Epson, Nagano, Japan) and TotalLab software (Nonlinear Dynamics, Newcastle, UK). β-Actin (Sigma) was used to normalize for equal loading of all samples. Protein results are expressed as fold increment over basal (d 0) relative to β-actin. The above procedure was also performed for mouse lung tissue.
Caspase 3 activity assay
Caspase 3 activity was measured using a fluorometric assay, according to manufacturer's instructions (Alexis Biochemical, San Diego, CA, USA). Briefly, cell lysates that were prepared for protein analysis were also used to measure the level of caspase 3 activity. Each cell lysate (25 μl) was transferred to 96-well plates in duplicates, and an equal volume of reaction master mix was added. The master mix consisted of 2× reaction buffer (20 mM HEPES, 4 mM EDTA, 20 mM KCl, and 3 mM MgCl2), 0.25 μl of DTT (10 mM), and 2.5 μl of the DEVD-AFC caspase 3 substrates (20 mM). Caspase 3 inhibitor (25 μl of 10 mM DEVD-CHO) was added to the reaction mixture (without the substrate), and subsequently, 2.5 μl of the substrate was added directly into the well and then incubated at 37°C for 4 h. In pilot studies, we determined that an incubation of 4 h was the optimal duration for increased levels of caspase 3 activity. Subsequently, plates were examined using a spectrofluorometer (Tecan, Männedorf, Switzerland) with excitation and emission wavelength of 400 and 505 nm, respectively, to measure the caspase 3 enzyme activity based on the cleavage of fluorogenic substrate DEVD-AFC. Blanks containing 25 μl of DMSO were prepared, and mixture samples were then incubated at 37°C for 4 h as mentioned above. After subtraction of the blanks, results in each sample were divided with the protein concentration for the respective protein lysate sample and then expressed as percentage relative to control (d 0).
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) staining
Human ASM cells were grown to confluence on 4-well chamber slides. Cells were fixed in neutral buffered formalin and permeabilized in 0.2% Triton X-100 before staining with TUNEL reagent (Roche Diagnostics, Indianapolis, IN, USA) for 1 h in a dark humidified 37°C incubator. After washing, nuclei were counterstained with diamidino-2-phenylindole (DAPI) and mounted using Prolong Gold Antifade reagent (Life Technologies, Carlsbad, CA, USA). Apoptotic cells were detected under a fluorescence microscope (Zeiss AxioImager Z1; Carl Zeiss, Oberkochen, Germany) using excitation wavelength of 450–500 nm, and emission wavelength of 515–565 nm (green). Relevant positive control (cells treated with staurosporine, 0.1 μm, 7 h) and negative (without TdT) controls were included in the experiment.
Apoptosis in mouse lung sections were detected using a TUNEL assay kit (GenScript Corp., Piscataway, NJ, USA), according to the manufacturer's instructions. Briefly, tissue sections were deparaffinized, rehydrated, and treated with proteinase K. After 2 washes in PBS, sections were incubated with labeling buffer consisting of biotin-11-dUTP and TdT buffer for 60 min in a humidified chamber at 37°C. The labeling buffer was discarded, replaced by fresh buffer (50 μl) containing streptavidin-HRP, and allowed to incubate for 30 min at 37°C in a humidified chamber. This was followed by development with DAB working solution. The end-labeling reaction was stopped by washing the sections with PBS and counterstained with Mayer hematoxylin (Dako, Glostrup, Denmark) and finally mounted with Histomount (National Diagnostics, Atlanta, GA, USA). Sections without TdT enzyme added to the TUNEL mixture were used as negative control, while sections treated with DNase I (25 U/μl for 10 min) to induce DNA strand degradation before labeling step were used as a positive control.
Immunocytochemistry
Human ASM cells that were grown to confluence on 25-mm2 glass coverslips were fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 in PBS for 5 min, blocked with 1% BSA in PBS for 1 h, and labeled with mouse monoclonal anti-sm-α-actin monoclonal antibody (1:500; Sigma) for 2 h. The cells were then incubated with Alexa Fluor (AF)-488 goat anti-mouse immunoglobulin G (IgG; 1:200; Invitrogen, Singapore) for 2 h to detect primary antibody bound to labeled cells. To monitor nuclear morphology, nuclei were labeled with Hoechst 33258 (10 μg/ml; Invitrogen) before the coverslips were mounted using fluorescent mounting medium (Dako). Thereafter, fluorescence micrographs were obtained using a Nikon Eclipse TE2000-U microscope (Nikon, Tokyo, Japan). Cells with condensed or fragmented nuclei were considered apoptotic.
Small interfering RNA (siRNA) preparation
The siRNA Generation Kit (Gene Therapy Systems; AMS Biotechnology, Abingdon, UK) was used to prepare siRNA from human ASM cDNA with primers that amplified integrin α7 cDNA, according to our previous work (9). Briefly, the day before siRNA transfection, confluent human ASM cells were reseeded into 6-well plates in DMEM and 10% FBS, so that 50–60% confluence was achieved 16 h later. Transfection of siRNA (1 μg/ml) was performed with Genesilencer reagent (Gene Therapy Systems), according to manufacturer's instructions, and cells were maintained in serum-free DMEM and ITS thereafter. A second transfection was performed 3 d later at the same time that culture medium was refreshed. Experiments were terminated following 6 d serum deprivation based on preliminary experiments, which showed effective silencing of integrin α7 mRNA and protein expression for ≥6 d for siRNA experiments. Transfection efficiency was >95%, as we have previously reported (9). For negative control studies, the transfection protocol was performed in the absence of siRNA, as well as in the presence of a nonspecific siRNA [green fluorescent protein (GFP) siRNA].
For α2-chain laminin siRNA experiments, LAMA-2-specific ON-TARGETplus SMARTpool siRNA and ON-TARGETplus nontargeting siRNA (scramble siRNA) were purchased from Dharmacon (Thermo Fisher, Rockford, IL, USA). LAMA-2-specific ON-TARGETplus SMARTpool siRNA contains a mixture of 4 SMARTselection-designed siRNAs targeting one LAMA-2 gene. The 4 SMARTselection-targeted LAMA-2 sequences were GCAAUGACAUACUCGAUGA, GAAAGGAAUUUAUGACAGU, GCUCCUGUCUGAUAUGUAA, and GCACUGGGCCACCGUCAUA. ON-TARGETplus nontargeting siRNA was used as a negative control siRNA (NT siRNA) with ≥4 mismatches to any human, mouse, or rat gene. Human ASM cells were serum starved and transfected with 50 nM of siRNA using Dharmafect 2 transfection agent (TA; Thermo Fisher). A second transfection was performed 3 d later at the same time that culture medium was refreshed, based on preliminary experiments that showed effective silencing of α2-chain laminin protein expression (Sigma; 1:500) for ≥3 d for siRNA experiments. Experiments were terminated following 7 d serum deprivation.
Sensitization and allergen challenge protocol
Female mice, aged 6 to 8 wk, were purchased from The Jackson Laboratory (Lama2−/−; Jackson Laboratory, Bar Harbor, ME, USA) and the Animal Resource Centre (wild-type, C57BL/6J; NUS). All procedures were approved by the Institutional Guidelines for Animal Care and Use Committee of NUS. Mice were sensitized by intraperitoneal (i.p.) injections of 20 μg of ovalbumin (OVA) and 4 mg aluminum hydroxide constituted in 0.1 ml of saline on d 0 and 14. On d 22, 23, and 24, mice were challenged with 1% OVA aerosol for 30 min. Aerosols were generated using an ultrasonic nebulizer (DeVilbiss 2000; DeVilbiss Healthcare, Castle Hill, NSW, Australia) on the indicated days. Control mice received saline aerosols using an identical protocol. At 24 h after the last challenge, mouse airway reactivity was measured. Mice were anesthetized by i.p. injection of 100 μl/10 g body weight anesthetic mixture (ketamine: 7.5 mg/ml; medetomidine: 0.1 mg/ml; NUS), and then a tracheotomy was performed by insertion of a blunted 19-gauge polyethylene catheter into the trachea, according to methods of Bao et al. (10). Mice were then intubated with a cannula that was connected to a multipurpose tube that leads the pneumotach, ventilator, and nebulizer within the FinePointe Series RC Sites (Buxco Research Systems, Wilmington, NC, USA). The system was calibrated for air flow and air pressure. Mice were ventilated at a fixed breathing rate of 140 breaths/min and the airway resistance (RI) in response to nebulized saline (PBS) followed by increasing concentrations of nebulized methacholine (0.5–8 mg/ml) were recorded using the Biosystem XA data acquisition and analysis software (Buxco Research Systems).
Immunohistochemistry for assessment of sm-α-actin and hematoxylin and eosin (H&E)
Lung tissue sections were deparaffinized with xylene and rehydrated with graded alcohol. Antigen retrieval was performed by heating the slides in the antigen unmasking solution (Vector Laboratories, Burlingame, CA, USA). Immunoreactivity staining was carried out using the Vector Mouse on Mouse (M.O.M.) immunodetection kit (Vector Laboratories), according to manufacturer's instructions. Briefly, slides were incubated with the Dako peroxidase blocking solution (DakoCytomation, Carpenteria, CA, USA) and M.O.M. mouse Ig blocking reagent. Slides were then incubated with a primary antibody directed at sm-α-actin (1:500; DakoCytomation), followed by subsequent incubations with the biotinylated anti-mouse IgG reagent and the Vectastain Elite ABC reagent (Vector Laboratories). Counterstaining was performed with hematoxylin. The area of sm-α-actin-positive cells was calculated on the basis of a modification of a previously described method (11), and the cellSens dimension software (Olympus, Hamburg, Germany) was used for analysis. Digital photographs of lung sections were analyzed at ×20. Sm-α-actin-positive areas were measured by a single observer in a blinded fashion.
Measurement of Ig levels
Cardiac puncture was performed to collect blood from mice 24 h after the last OVA or saline challenge. Serum OVA-specific IgG1, IgG2a, and IgE (all BD Biosciences, San Diego, CA, USA) titers were measured by ELISA as described previously (10), with modification of the dilution of sera (1:400); plates were read at 570 nm.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 5 (GraphPad, San Diego, CA, USA) using 1-way ANOVA with repeated measures followed by Bonferroni's post hoc t test. A probability value of P < 0.05 was considered significant.
RESULTS
Laminin is required for ASM cell survival and phenotype maturation
Following prolonged serum deprivation, the contractile phenotype marker protein sm-α-actin was markedly elevated in cells that acquire an enlarged, elongate morphology characteristic of contractile ASM cells. In the presence of a selective laminin-competing peptide, YIGSR, the spontaneous increase in sm-α-actin was prevented, a finding that confirms that endogenous laminin is required for ASM cell maturation (Fig. 1A). YIGSR is a selective inhibitor of laminin binding to integrins that blocks cell adhesion and migration on laminin, as well as angiogenesis, tumor growth, and metastasis (12). We have previously shown that the inhibitory effect of YIGSR on ASM contractile phenotype acquisition is not the result of peptide-induced cell toxicity or cell detachment (8).
Figure 1.
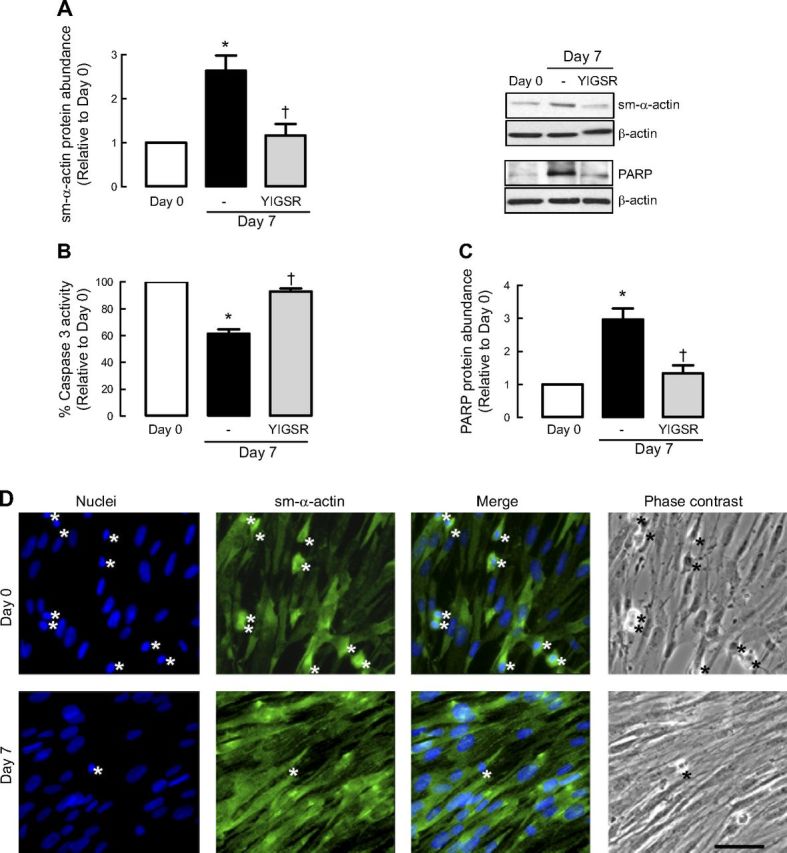

Laminin is required for ASM phenotype maturation and cell survival. A–C) Human ASM cells were treated in the absence or presence of selective laminin-competing peptide YIGSR and then serum deprived for 7 d before protein abundance for sm-α-actin (A), caspase 3 activity (B), and protein abundance for PARP (C) were determined. Results are representative of 3 independent experiments. Data are expressed as fold increment over basal (d 0) relative to β-actin protein abundance. *P < 0.05 vs. d 0; †P < 0.05 vs. d 7 response in absence of peptide. D) Representative immunofluoresence imaging of human ASM cells labeled for sm-α-actin (green) at d 0 and after 7-d serum deprivation (d 7). Nuclei were labeled with Hoechst dye (blue). Cells with condensed or fragmented nuclei (indicated by asterisks) are considered apoptotic. Scale bar = 50 μm. E) Representative TUNEL photographs of human ASM cells at d 0 and after 7-d serum deprivation (d 7). Cells treated with staurosporine (0.1 μm, 7 h) were used as positive control for apoptosis. Cells incubated with labeling solution only (without TdT) were used as negative control. Cells at d 7 show limited TUNEL-positive staining, as indicated by asterisks. Scale bar = 20 μm. F) Representative blots showing effect of sm-α-actin protein abundance and corresponding caspase 3 activity following cotreatment with staurosporine (St; 10−7 M) at the time of serum deprivation. G) Representative blots showing effect of sm-α-actin protein abundance and corresponding caspase 3 activity with staurosporine (St; 10−7 M, 10−6 M) added 3 d after 7-d serum deprivation.
To address whether acquisition of a contractile phenotype was associated with a change in ASM cell apoptosis, we assessed the activity of caspase 3, an effector of apoptosis. ASM cells exhibited significantly reduced caspase 3 activity following serum deprivation, and this response was prevented by YIGSR (Fig. 1B). This observation demonstrates that laminin-integrin binding is required for the concomitant acquisition of a contractile phenotype and suppression of proapoptotic caspase activity.
To confirm the activation state of caspase 3, we measured the content of PARP, a downstream substrate of caspase 3 (Fig. 1C). PARP abundance was markedly increased, indicating reduced cleavage by caspase-3, during prolonged serum deprivation. Notably, as for caspase 3 activity, PARP accumulation was prevented by YIGSR, confirming a role for endogenous laminin in facilitating apoptosis suppression (Fig. 1C).
We next directly assessed the degree of cellular apoptosis in ASM cells by staining nuclear DNA and examining nuclear morphology (Fig. 1D). The level of cell apoptosis, marked by nuclear condensation/fragmentation, was reduced after serum deprivation, and this correlated with increased numbers of ASM cells that were immunoreactive for sm-α-actin. Consistent with this, we also performed TUNEL staining on the ASM cells. As shown in Fig. 1E, TUNEL-positive signals were predominantly detected in nuclei of human ASM cells at d 0. In contrast, less TUNEL-positive signals were detected in human ASM cells that were serum deprived for 7 d (Fig. 1E).
As further proof of concept, we also performed experiments whereby mild apoptosis was induced, and its effect on the level of sm-α-actin was measured. Two experiments were performed with staurosporine, a well-characterized inducer of cell apoptosis (13). In the first experiment, using a relatively low concentration of staurosporine (10−7 M) added at the time of serum deprivation, we observed that loss of caspase 3 activity was prevented, and this was associated with markedly reduced accumulation of sm-α-actin protein (Fig. 1F). Of note, we also tested staurosporine at a 10-fold higher concentration (10−6 M), but this induced cell detachment (anoikis), precluding our ability to acquire protein lysates of adherent cells and confirming the feasibility of using 10−7 M staurosporine to invoke mild apoptosis. In the second experiment, staurosporine (10−7 M) was added for 3 d after 7-d serum deprivation. In this setting, sm-α-actin protein abundance and caspase 3 activity were refractory to staurosporine (Fig. 1G). However, a 10-fold higher concentration of staurosporine (which induced anoikis in the first set of experiments; Fig. 1F), was required to reduce sm-α-actin protein abundance and increase caspase 3 activity to levels similar to that seen in cells prior to serum deprivation. These observations suggest that ASM cells that acquire a contractile phenotype are more resistant to apoptotic agents.
Together, the results indicate that endogenously synthesized laminin is not only required for a contractile ASM phenotype but is also a critical upstream antiapoptotic factor in ASM cells.
Laminin is sufficient to suppress ASM cell apoptosis
We next assessed whether exogenously applied laminin was sufficient to increase the survival of ASM cells. Concomitant with an increase in sm-α-actin abundance (Fig. 2A), serum withdrawal suppressed caspase 3 activity. This effect was significantly augmented in ASM cells seeded onto α2-chain laminin but not in ASM cells seeded onto Engelbreth-Holm-Swarm (EHS) murine sarcoma laminin that lacks α2-chain laminin (Fig. 2B). We also observed that full-length PARP abundance was higher in ASM cells cultured on α2-chain laminin but was unaffected by EHS laminin (Fig. 2C). These results suggest that exogenous laminin that specifically contains α2-chain laminin is sufficient to reduce activation of ASM apoptosis.
Figure 2.

Laminin is sufficient to induce ASM cell survival signaling. A–C) Human ASM cells were seeded onto plastic or laminin-coated plates and then serum deprived for 7 d before protein abundance for sm-α-actin (A), caspase 3 activity (B), and protein abundance for PARP (C), were determined. α2, affinity-purified α2-chain laminin from human placenta, which includes laminin-211 and -221; EHS, laminin from Engelbreth-Holm-Swarm murine sarcoma consisting mainly of laminin-111. Data are expressed as fold increment over basal (d 0) relative to β-actin protein abundance. *P < 0.05 vs. d 0; †P < 0.05 vs. d 7 response in the absence of laminin coating. D) Representative blots showing effect of α2-chain laminin siRNA on sm-α-actin protein abundance and corresponding caspase 3 activity. TA served as vehicle control. NT, nontargeting siRNA; LN, laminin. Results are representative of 3 independent experiments.
From our previous studies (8), we showed that following serum deprivation both mRNA and protein levels for α2-chain laminin were elevated. Also important was the observation that all the chains that make up laminin-211 were elevated following serum deprivation. To further confirm that α2-chain laminin is responsible for the observed effects in culture, we performed experiments in which we silenced α2-chain laminin using siRNA. Using this approach, we observed that by reducing α2-chain laminin protein, we suppressed sm-α-actin accumulation during prolonged serum withdrawal, and this was associated with increased caspase 3 activity (Fig. 2D).
Laminin-binding integrin α7β1 is required for ASM cell survival
We showed that siRNA against integrin α7 reduced integrin α7 protein by 97 ± 0.4% and suppressed sm-α-actin accumulation during prolonged serum withdrawal (Fig. 3A, B). Concomitantly, integrin α7 silencing promoted apoptotic signaling and made ASM cells refractory to the suppressive effects of serum deprivation on caspase 3 activity (Fig. 3C). Similarly, the abundance of PARP was also reduced (an indicator of increased apoptosis signaling) in ASM cells treated with integrin α7 siRNA (Fig. 3D). For all experiments, we included a vehicle control (TA only) and GFP siRNA to confirm the specificity of integrin α7β1 effects. Experiments were terminated following 6 d serum deprivation based on preliminary experiments, which showed effective silencing of integrin α7 mRNA and protein expression for ≥6 d for siRNA experiments. These results reveal that the binding of matrix to laminin-selective integrin α7β1 is necessary to mediate extracellular cues that underpin the suppression of physiological apoptosis in ASM cells as they undergo maturation to a contractile phenotype following serum deprivation.
Figure 3.

Laminin is sufficient to promote ASM cell survival via integrin α7β1. Human ASM cells were treated in the absence or presence of integrin α7 siRNA and then serum-deprived for 6 d before protein abundance for sm-α-actin (A) and integrin α7 (B), caspase 3 activity (C), and protein abundance for PARP (D) were determined. Experiments were terminated following 6-d serum deprivation based on preliminary experiments, which showed effective silencing of integrin α7 mRNA and protein expression for ≥6 d. GFP served as negative control; TA served as vehicle control. Results are representative of 3 independent experiments. Data are expressed as fold increment over basal (d 0) relative to β-actin protein abundance. *P < 0.05 vs. d 0; †P < 0.05 vs. d 6 response without integrin α7 siRNA.
Bcl-2 but not Bax is involved in regulating laminin-induced ASM cell survival
The Bcl-2 family of proteins are critical determinants for cell survival or death. We monitored two stereotypical family members: antiapoptotic Bcl-2 and proapoptotic Bax. Under basal conditions, ASM cells expressed low levels of Bcl-2; however, after 7-d serum deprivation, the abundance of Bcl-2 was markedly increased (Fig. 4A–C). This observation is consistent with our evidence for concomitant enhancement of cell survival signaling and ASM contractile phenotype acquisition. Notably, we observed that YIGSR reduced Bcl-2 accumulation (Fig. 4A). Furthermore, seeding cells onto α2-chain laminin was sufficient to augment Bcl-2 accumulation (Fig. 4B). Unlike our data on caspase activity, EHS laminin also induced a statistically significant increase in Bcl-2 compared to cells seeded directly onto plastic, though the magnitude of the effect was markedly lower than the response of ASM cells seeded onto α2-chain laminin. Finally, integrin α7 siRNA prevented accumulation of antiapoptotic Bcl-2 during ASM maturation in prolonged serum-free conditions (Fig. 4C).
Figure 4.
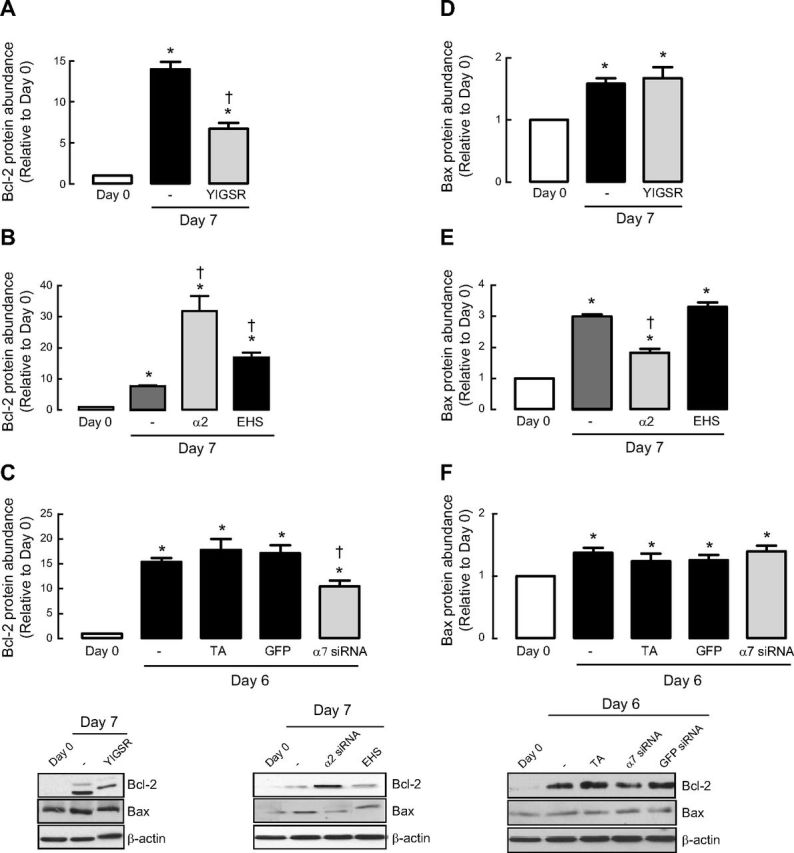
Bcl-2 but not Bax is involved in regulating laminin-induced ASM cell survival. Bcl-2 (A–C) or Bax (D–F) protein abundance was measured following 7- or 6-d serum deprivation in the presence or absence of selective laminin-competing peptide YIGSR (A, D), various types of laminin coating (B, E) or integrin α7 siRNA (C, F). Experiments were terminated after 6-d serum deprivation based on preliminary experiments, which showed effective silencing of integrin α7 mRNA and protein expression for ≥6 d for siRNA experiments. Results are representative of 3 independent experiments. α2, affinity-purified α2-chain laminin from human placenta, which includes laminin-211 and -221; EHS, laminin from EHS murine sarcoma consisting mainly of laminin-111. GFP served as negative control; TA served as vehicle control. Data are expressed as fold increment over basal (d 0) relative to β-actin protein abundance. *P < 0.05 vs. d 0; †P < 0.05 vs. d 7 response in absence of peptide or laminin coating or d 6 response without integrin α7 siRNA.
We next examined the abundance of Bax. Under basal conditions, Bax abundance was quite low but increased after 7-d serum deprivation (Fig. 4D–F). In striking contrast to Bcl-2, accumulation of proapoptotic Bax was not prevented by treatment with YIGSR, indicating that the balance of proapoptotic and antiapoptotic Bcl2 family members is skewed in favor of the former if laminin binding to ASM cells is blocked (Fig. 4D). Interestingly, Bax abundance was reduced when ASM cells were seeded onto α2-chain laminin extracts, but unlike our findings for Bcl-2, plating onto EHS laminin, which lacks α2-chain laminin, was without effect (Fig. 4E). Consistent with our YIGSR data, accumulation of Bax occurred independently of integrin α7β1 expression (Fig. 4F).
Collectively, we show that endogenous and exogenous α2-chain laminin, via a signaling axis that requires integrin α7β1, selectively promotes accumulation of antiapoptotic Bcl-2, but is without effect on proapoptotic Bax, which is in relatively low abundance in ASM cells. This reveals that laminin skews the balance of Bcl-2 family proteins in a manner that promotes cell survival under the same conditions that promote ASM cells to acquire a contractile phenotype.
Phosphoinositide-3-kinase (PI3K) links ASM contractile phenotype with cell survival
Our previous studies and those of others demonstrate a critical role for PI3K signaling in ASM phenotype maturation and cellular hypertrophy (6, 14). PI3K is also involved in promoting survival in multiple cell types; thus, we hypothesized that it may be a signaling mechanism that both inhibits physiological apoptosis and underpins ASM maturation and hypertrophy. To test this hypothesis, we incubated ASM cells with two pharmacological inhibitors of PI3K, LY294002 or wortmannin. We first confirmed the biological activity of these agents using Western blot analysis to measure Akt phosphorylation, a downstream effector of the PI3K cascade. In the presence of either LY294002 (20 μM; ref. 15) or wortmannin (100 nM; ref. 15), Akt phosphorylation was significantly reduced (Fig. 5A). This effect correlated with suppressed accumulation of sm-α-actin, confirming the requirement of this pathway for phenotype maturation. PI3K inhibition also inhibited accumulation of Bcl-2 (Fig. 5A). These findings suggest that PI3K activity is required for both augmented prosurvival (i.e., antiapoptotic) signaling and phenotype maturation of ASM cells in primary culture. To determine whether this may be an effector of laminin effects, we assessed PI3K signaling in response to YIGSR and found that the laminin-selective peptide was sufficient to reduce Akt phosphorylation (Fig. 5B).
Figure 5.
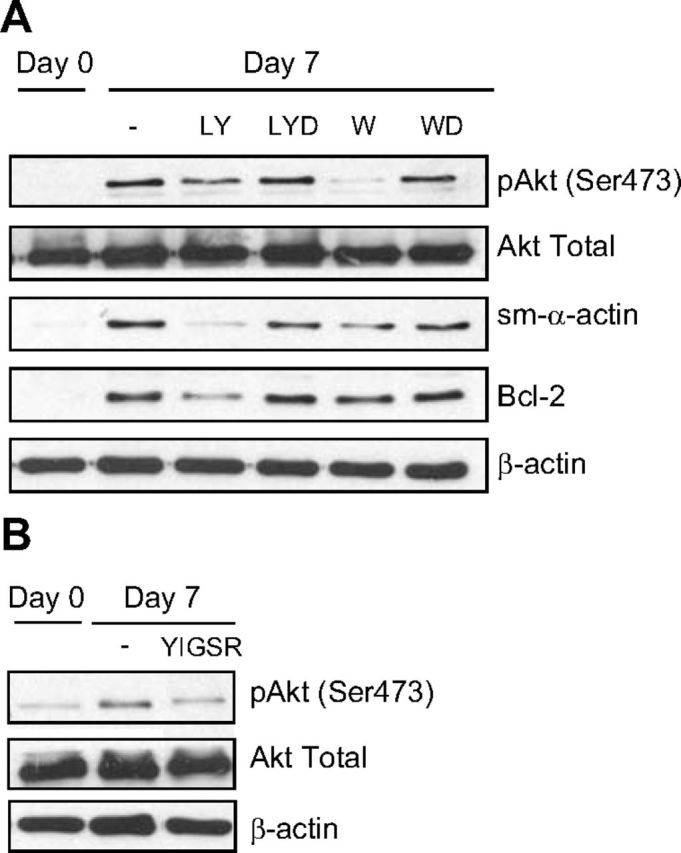
PI3K links ASM contractile phenotype signaling with cell survival. A) Effect of PI3K inhibitors, LY294002 (LY, 20 μM) and wortmannin (W, 100 nM), on the protein abundance of Akt, sm-α-actin, and Bcl-2. LYD and WD are vehicle controls for LY294002 (0.2% DMSO) and wortmannin (0.01% DMSO), respectively. B) Effect of selective laminin-competing peptide YIGSR on the protein abundance of Akt. Results are representative of 3 independent experiments.
ASM cells from humans with asthma exhibit a prosurvival phenotype
To extend the relevance of our studies, we examined whether asthmatic and nonasthmatic primary cultured ASM cells differ in expression of proapoptotic and antiapoptotic proteins. Basal expression of sm-α-actin and Bcl-2 was greater in confluent ASM cell cultures from subjects with asthma (Fig. 6A). During prolonged serum deprivation, ASM cells from donors with asthma also accumulated markedly greater Bcl-2 compared with ASM cells from nonasthmatic donors. Furthermore, Bax abundance and caspase 3 activity (Fig. 6A, B) were lower in ASM cells from donors with asthma both prior to and after serum deprivation compared with nonasthmatic ASM cells. These data indicate that ASM cells from subjects with asthma exhibit a protein fingerprint that supports a prosurvival phenotype. Moreover, using immunoblotting, we found that these differences are associated with differences in α2-chain laminin and integrin α7 protein abundance, with ASM cells from donors with asthma showing 2- to 3-fold greater abundance of α2-chain laminin (2.96±1.13) and integrin α7 (2.04±0.6) compared with ASM cells from nonasthmatic donors.
Figure 6.
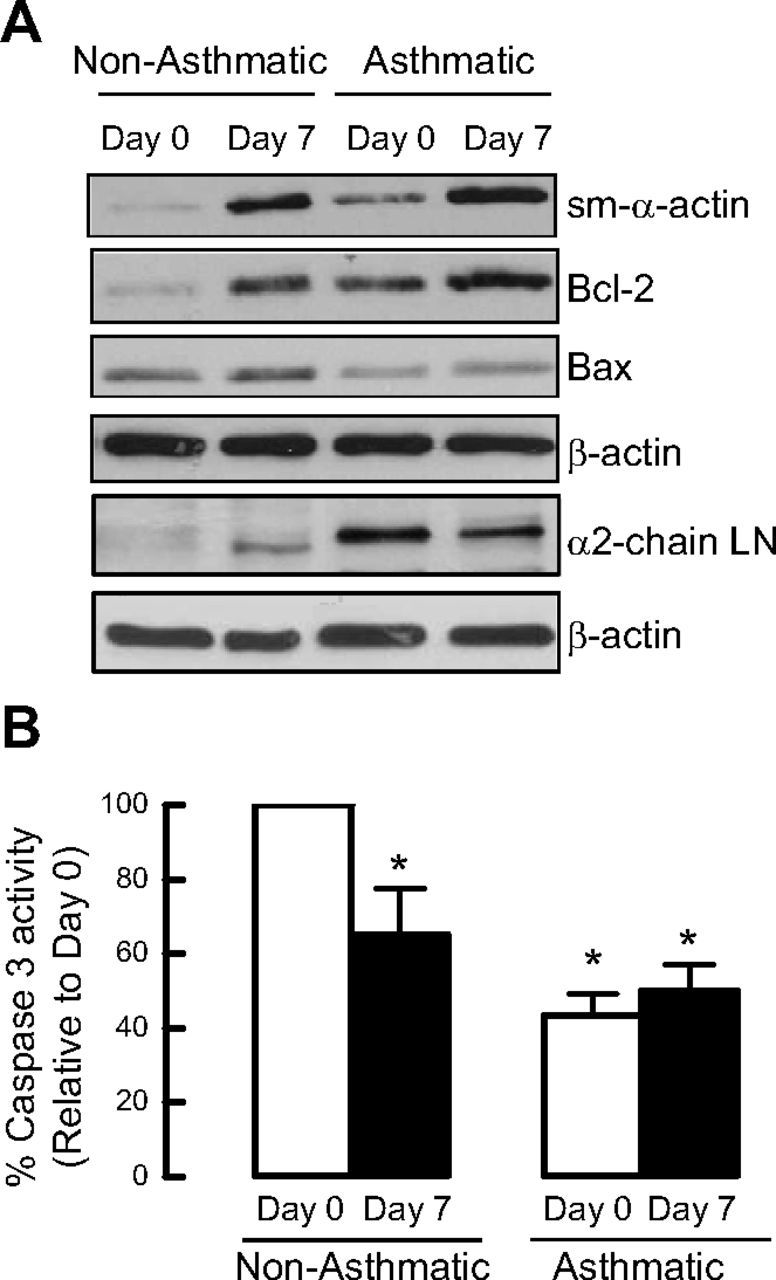
Asthmatic primary human ASM cells express higher levels of cell survival proteins than nonasthmatic primary human ASM cells. Comparison of the protein abundance levels for sm-α-actin, Bcl-2, Bax, and α2-chain laminin (LN) (A), and caspase 3 activity between primary cultured human ASM cells from asthmatic vs. healthy nonasthmatic airways (B). Results are representative of 4 independent experiments. *P < 0.05 vs. d 0 nonasthmatic.
Lama2−/− mice do not develop allergen challenge-induced airway hyperreactivity
To investigate whether prosurvival laminin signaling observed in vitro may contribute to the development of primary features of asthma, we measured the effect of OVA sensitization and challenge on airway reactivity to methacholine in mice that spontaneously express a mutated and functionally deficient form of α2-chain laminin (Lama2−/−). Airway responsiveness of wild-type mice sensitized and challenged with OVA was significantly higher than that of saline-exposed mice, indicating that OVA aeroallergen induces AHR (Fig. 7A). Notably, lung function of Lama2−/− mice was unaffected by OVA sensitization and challenge, indicating that mice lacking α2-chain laminin are refractory to allergen-induced airway hyperreactivity. The magnitude of airway responses in OVA- and saline-exposed Lama2−/− mice did not differ from that of saline-exposed wild-type mice. In addition, when mouse lung samples were analyzed, caspase 3 activity was reduced in OVA-sensitized and -challenged wild-type mice, but this was not the case for OVA- and saline-exposed Lama2−/− mice (Fig. 7B). Moreover, the total area of sm-α-actin in Lama2−/− OVA-sensitized and -challenged mice were significantly reduced as compared to wild-type OVA-sensitized and -challenged mice (Fig. 7C, D). Consistent with this, by TUNEL staining of mouse lung samples, wild-type saline-exposed mice had a higher number of ASM cells stained with TUNEL (indicated by asterisks in Fig. 7D) as compared to wild-type OVA-sensitized and -challenged mice. In Lama2−/− saline-exposed mice, ASM cells were predominantly stained with TUNEL, as compared with wild-type, saline-exposed mice. Moreover, there is an inverse correlation between the abundance of sm-α-actin-positive cells and TUNEL-positive staining (Fig. 7D).
Figure 7.
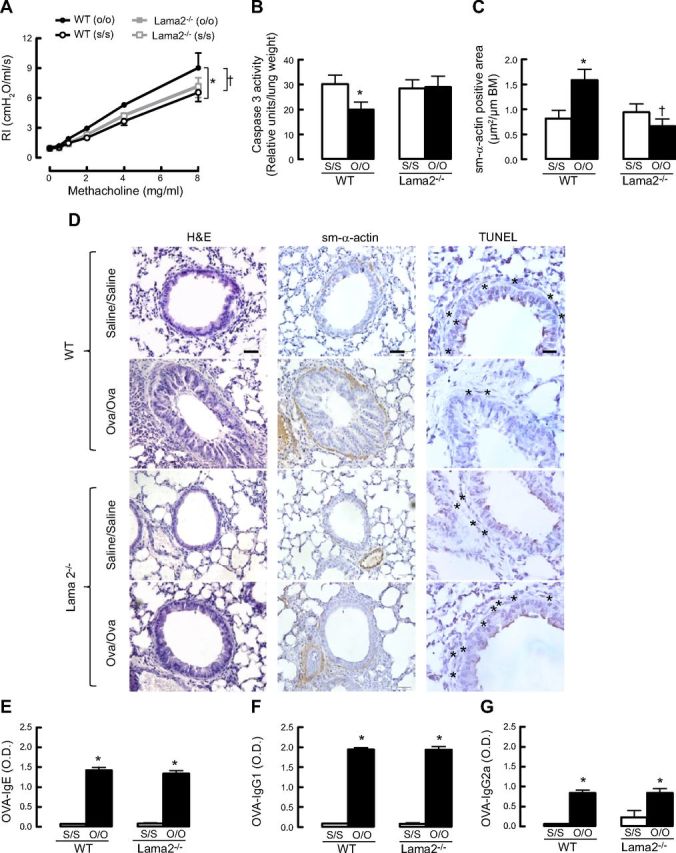
Lama2−/− mice do not develop allergen challenge-induced airway hyperreactivity. A) Comparison of airway reactivity between wild-type (WT) and Lama2−/− mice (n=4–7/group); results for Lama2−/− saline sensitization and saline challenge (s/s) and Lama2−/− OVA sensitization and OVA challenge (o/o) are superimposed. *P < 0.05 vs. WT (s/s)-treated mice; †P < 0.05 vs. WT (o/o). B) Comparison of caspase 3 activity between WT and Lama2−/− mice (n=3–4/group). *P < 0.05 vs. WT (s/s)-treated mice. C) Comparison of sm-α-actin-positive area relative to basement membrane (BM) between WT and Lama2−/− mice (n=3–4/group). *P < 0.05 vs. WT (s/s); †P < 0.05 vs. WT (o/o). D) H&E and immunohistochemistry staining for sm-α-actin and TUNEL staining in mouse whole-lung slices. Asterisks indicate TUNEL-positive apoptotic cells. Scale bars = 20 μm. E–G) Levels of OVA-specific IgE (E), IgG1 (F), and IgG2a (G) in mouse serum that was collected 24 h after the last OVA aerosol challenge. O.D., optical density. *P < 0.05 vs. WT (s/s)-treated mice.
Next, we determined anti-OVA antibody production in wild-type and Lama2−/− mice. OVA-specific IgG2a antibodies, which are typically produced during Th1 response, and IgG1 and IgE antibodies, which are associated with Th2 development, were measured by ELISA. Serum levels of OVA-specific IgE, IgG1, and IgG2a antibodies were not significantly different between wild-type OVA-sensitized and -challenged mice and Lama2−/− OVA-sensitized and -challenged mice, indicating that reduced airway hyperreactivity in the Lama2−/− mice is unlikely due to modification of an ongoing anti-OVA antibody cell response in vivo (Fig. 7E–G).
DISCUSSION
We provide new mechanistic insights linking expression of α2-chain laminin to AHR. We show that activation of PI3K signaling that governs accumulation of contractile ASM marker proteins and ASM hypertrophy correlates with cell survival effects of α2-chain laminin. Moreover, laminin, via integrin α7β1, is both required and sufficient to promote accumulation of prosurvival proteins and reduce levels of proapoptotic proteins. These studies highlight the importance of laminin-associated ECM proteins and mechanisms for effects on smooth muscle phenotype. Notably, we demonstrate the importance of these effects with respect to asthma, as ASM cells from donors with asthma demonstrated a prosurvival phenotype that correlated with elevated α2-chain laminin expression.
Novel findings of this study are that effects on ASM survival are induced exclusively by α2-chain laminins; these are mediated via integrin α7β1, and involve signaling pathways that concomitantly regulate cell survival and ASM cell maturation. This suggests that suppressed cell apoptosis (increased cell survival signals) is a plausible mechanism to mediate augmented ASM function in asthma. Indeed, Lama2−/− mice exhibit lower airway reactivity than their wild-type control littermates. Consistent with this finding, using a rat model of OVA-induced ASM remodeling, Ramos-Barbon et al. (16) showed that antigen-specific CD4+ T cells can drive increased ASM mass, in part, by inhibiting apoptosis. It is not clear whether these results were linked with concomitant effects on local laminin expression, but they suggest a link with our current study. In an asthma-like equine disease, heaves, this group also showed a coordinated increase in the number of apoptotic and proliferating ASM cells, suggesting the existence of a complex and variable relationship between survival, apoptosis, and trophic signals in the physiological turnover and accumulation of ASM cells (17).
The effect of asthma-associated biomolecules on ASM apoptosis remains poorly understood. We provide evidence that primary cultured ASM cells from donors with asthma express a prosurvival phenotype marked by high and low levels of antiapoptotic and proapoptotic proteins, respectively. Our experiments have indicated the need to assess both cell survival and cell apoptosis markers in comparing samples from asthmatic and nonasthmatic donors, as in the former, proapoptotic proteins are present at low levels, making detection and quantification difficult. This scenario may be at the root of differences in our data and that from Kaur et al. (18), who reported a lack of difference in percentage apoptosis between ASM cells from subjects with and without asthma. Another possible reason for the difference is that they used asthmatic-donor ASM cells from endobronchial biopsies obtained during fiberoptic bronchoscopy, and nonasthmatic-donor ASM cells from subjects undergoing lung resection. However, both our asthmatic- and nonasthmatic-donor ASM cells were derived from samples taken from subjects undergoing lung resection. Future analysis of physiological apoptosis of human ASM in situ in airways of different sizes and from subjects of different ages is needed in healthy control subjects alongside subjects with asthma of a well-defined subtype, controlling for medication use and duration of disease.
Freyer et al. (19) showed that treatment of ASM with arginine-glycine-aspartic acid (RGD) peptides to inhibit ASM-ECM interaction led to increased cell death in a concentration-dependent manner. Fibronectin, laminin, and collagens I and IV were identified as important antiapoptotic elements, with integrin α5β1 being an important transducer of survival signals in ASM cells from explants of trachealis muscle. We reveal that ASM cells, prepared from enzymatic digestion of bronchial muscle show marked dependence on laminin for survival. Notably, we show that YIGSR, a competing peptide that is more selective for laminin than RGD peptides, completely prevents the accumulation of Bcl-2 and loss of caspase 3 activity associated with phenotype maturation. This demonstrates that endogenously expressed laminin drives ASM cell survival, as well as being essential for contractile phenotype acquisition (8). Moreover, the addition of exogenous laminin is sufficient to further increase cell survival markers and reduce proapoptotic markers. Consistent with this idea, Pullan et al. (20) reported that collagen I had no impact on primary mouse mammary epithelial cell death, whereas laminin-rich basement membrane was essential for survival.
Dekkers et al. (21) showed that exogenous laminin sustains a contractile phenotype and prevents proliferation of bovine tracheal smooth muscle cells. Using a guinea pig model of chronic asthma to further explore the role of laminins in ASM remodeling in vivo, the researchers treated the animals with the specific soluble laminin competing peptide YIGSR and showed that YIGSR did inhibit allergen-induced ASM accumulation (22). However, in contrast to their previous in vitro studies, soluble YIGSR promoted a hypercontractile phenotype. The researchers reasoned that the microenvironment of the peptide is an important factor; thus, even though the effects of the peptide are consistent with disrupting laminin-ASM interactions in vitro and ex vivo conditions, the response to artificial laminin-competing mimetic in tissue (in vivo), may not be as straightforward, as there are multiple cell types involved and the in vivo system is subject to immune regulation. In our study, we showed that in mice that spontaneously express a mutated and functionally deficient form of the α2-chain laminin, allergen-induced AHR was not observed. Thus, as with any pharmacological intervention, the development of peptide compounds that target ASM-laminin interactions will require careful assessment of effects on all aspects of airway inflammation and remodeling.
There are some limitations to our study: allergen challenge was for a short duration (3 d); thus, although it induces inflammation-induced AHR, the duration of time is too short to cause airway remodeling; and we limited examination of signaling effectors that correlate with laminin effects to PI3K. Although there are many other pathways that could also be important (e.g., Src, ERK1/2), we chose to first focus on PI3K, as it has been directly implicated in ASM phenotype maturation, and it is a recognized upstream signaling effector in cell survival.
In summary, our results provide strong new evidence causally linking laminin expression to maintenance and regulation of a contractile ASM phenotype and cell survival, effects that appear to contribute to AHR and could support the enlargement of muscle bundles encircling the airways, a cardinal feature of AWR. This suggests that targeting the laminin-integrin α7β1 signaling axis may offer new avenues for the development of therapies to reduce dysfunction associated with contractile ASM cells in patients with asthma. This study also has implications for other lung diseases where AHR and/or tissue remodeling are features, such as cystic fibrosis, idiopulmonary fibrosis, acute lung injury, and chronic obstructive pulmonary disease. Notably, although our studies are focused on smooth muscle cells from one organ, the findings may be applicable to other smooth muscle-containing organs, where smooth muscle remodeling features in development and disease, such as in atherosclerotic plaques, resistance arterioles in hypertension, gastrointestinal remodeling in inflammatory bowel disease, and urinary bladder hypertrophy, in which the laminin-integrin α7β1 signaling axis could also play a role.
Acknowledgments
The authors thank Ms. Karol McNeill and Ms. Tze Khee Chan (TUNEL assay) for excellent technical assistance.
This study received financial support from the Singapore Ministry of Education's Academic Research Fund (T13-0702-P26), as well as support from a Singapore Deputy President (Research and Technology) startup grant to T.T. A.J.H. is supported through the Canada Research Chairs Program.
Footnotes
- ASM
- airway smooth muscle
- AWR
- airway wall remodeling
- AHR
- airway hyperresponsiveness
- ECM
- extracellular matrix
- EHS
- Engelbreth-Holm-Swarm
- GFP
- green fluorescent protein
- H&E
- hematoxylin and eosin
- Ig
- immunoglobulin
- NUS
- National University of Singapore
- OVA
- ovalbumin
- PARP
- poly-(ADP-ribose) polymerase
- PI3K
- phosphoinositide-3-kinase
- RGD
- arginine-glycine-aspartic acid
- siRNA
- small interfering RNA
- TA
- transfection agent
- TdT
- terminal deoxynucleotidyl transferase
- TUNEL
- terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling
- YIGSR
- tyrosine-isoleucine-glycine-serine-arginine
REFERENCES
- 1.Benayoun L., Druilhe A., Dombret M. C., Aubier M., Pretolani M. (2003) Airway structural alterations selectively associated with severe asthma. Am. J. Respir. Crit. Care Med. , 1360–1368 [DOI] [PubMed] [Google Scholar]
- 2.Lambert R. K., Wiggs B. R., Kuwano K., Hogg J. C., Pare P. D. (1993) Functional significance of increased airway smooth muscle in asthma and COPD. J. Appl. Physiol. , 2771–2781 [DOI] [PubMed] [Google Scholar]
- 3.Stewart A. G. (2004) Emigration and immigration of mesenchymal cells: a multicultural airway wall. Eur. Respir. J. , 515–517 [DOI] [PubMed] [Google Scholar]
- 4.Ward J. E., Harris T., Bamford T., Mast A., Pain M. C., Robertson C., Smallwood D., Tran T., Wilson J., Stewart A. G. (2008) Proliferation is not increased in airway myofibroblasts isolated from asthmatics. Eur. Respir. J. , 362–371 [DOI] [PubMed] [Google Scholar]
- 5.Bentley J. K., Deng H., Linn M. J., Lei J., Dokshin G. A., Fingar D. C., Bitar K. N., Henderson W. R., Hershenson M. B. (2009) Airway smooth muscle hyperplasia and hypertrophy correlate with glycogen synthase kinase-3(beta) phosphorylation in a mouse model of asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. , L176–L184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halayko A. J., Kartha S., Stelmack G. L., McConville J., Tam J., Camoretti-Mercado B., Forsythe S. M., Hershenson M. B., Solway J. (2004) Phophatidylinositol-3 kinase/mammalian target of rapamycin/p70S6K regulates contractile protein accumulation in airway myocyte differentiation. Am. J. Respir. Cell Mol. Biol. , 266–275 [DOI] [PubMed] [Google Scholar]
- 7.Altraja A., Laitinen A., Virtanen I., Kampe M., Simonsson B. G., Karlsson S. E., Hakansson L., Venge P., Sillastu H., Laitinen L. A. (1996) Expression of laminins in the airways in various types of asthmatic patients: a morphometric study. Am. J. Respir. Cell Mol. Biol. , 482–488 [DOI] [PubMed] [Google Scholar]
- 8.Tran T., McNeill K. D., Gerthoffer W. T., Unruh H., Halayko A. J. (2006) Endogenous laminin is required for human airway smooth muscle cell maturation. Respir. Res. , 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran T., Ens-Blackie K., Rector E. S., Stelmack G. L., McNeill K. D., Tarone G., Gerthoffer W. T., Unruh H., Halayko A. J. (2007) Laminin-binding Integrin α7 is required for contractile phenotype expression by human airway myocyte. Am. J. Respir. Cell Mol. Biol. , 668–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bao Z., Guan S., Cheng C., Wu S., Wong S. H., Kemeny D. M., Leung B. P., Wong W. S. (2009) A novel anti-inflammatory role for andrographolide in asthma via inhibition of the nuclear factor-κB pathway. Am. J. Respir. Crit. Care Med. , 657–665 [DOI] [PubMed] [Google Scholar]
- 11.Robinson P. J., Hegele R. G., Schellenberg R. R. (1996) Increased airway reactivity in human RSV bronchiolitis in the guinea pig is not due to increased wall thickness. Pediatr. Pulmonol. , 248–254 [DOI] [PubMed] [Google Scholar]
- 12.Iwamoto Y., Robey F. A., Graf J., Sasaki M., Kleinman H. K., Yamada Y., Martin G. R. (1987) YIGSR, a synthetic laminin pentapeptide, inhibits experimental metastasis formation. Science , 1132–1134 [DOI] [PubMed] [Google Scholar]
- 13.Tamaoki T., Nomoto H., Takahashi I., Kato Y., Morimoto M., Tomita F. (1986) Staurosporine, a potent inhibitor of phospholipid/Ca2+-dependent protein kinase. Biochem. Biophys. Res. Commun. , 397–402 [DOI] [PubMed] [Google Scholar]
- 14.Deng H., Hershenson M. B., Lei J., Bitar K. N., Fingar D. C., Solway J., Bentley J. K. (2010) p70 Ribosomal S6 kinase is required for airway smooth muscle cell size enlargement but not increased contractile protein expression. Am. J. Respir. Cell Mol. Biol. , 744–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma P., Tran T., Stelmack G. L., McNeill K., Gosens R., Mutawe M. M., Unruh H., Gerthoffer W. T., Halayko A. J. (2008) Expression of the dystrophin-glycoprotein complex is a marker for human airway smooth muscle phenotype maturation. Am. J. Physiol. Lung Cell. Mol. Physiol. , L57–L68 [DOI] [PubMed] [Google Scholar]
- 16.Ramos-Barbon D., Presley J. F., Hamid Q. A., Fixman E. D., Martin J. G. (2005) Antigen-specific CD4+ T cells drive airway smooth muscle remodeling in experimental asthma. J. Clin. Invest. , 1580–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herszberg B., Ramos-Barbon D., Tamaoka M., Martin J. G., Lavoie J. P. (2006) Heaves, an asthma-like equine disease, involves airway smooth muscle remodeling. J. Allergy Clin. Immunol. , 382–388 [DOI] [PubMed] [Google Scholar]
- 18.Kaur D., Hollins F., Saunders R., Woodman L., Sutcliffe A., Cruse G., Bradding P., Brightling C. (2010) Airway smooth muscle proliferation and survival is not modulated by mast cells. Clin. Exp. Allergy , 279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freyer A. M., Johnson S. R., Hall I. P. (2001) Effects of growth factors and extracellular matrix on survival of human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. , 569–576 [DOI] [PubMed] [Google Scholar]
- 20.Pullan S., Wilson J., Metcalfe A., Edwards G. M., Goberdhan N., Tilly J., Hickman J. A., Dive C., Streuli C. H. (1996) Requirement of basement membrane for the suppression of programmed cell death in mammary epithelium. J. Cell Sci. , 631–642 [DOI] [PubMed] [Google Scholar]
- 21.Dekkers B. G., Schaafsma D., Nelemans S. A., Zaagsma J., Meurs H. (2007) Extracellular matrix proteins differentially regulate airway smooth muscle phenotype and function. Am. J. Physiol. Lung Cell. Mol. Physiol. , L1405–L1413 [DOI] [PubMed] [Google Scholar]
- 22.Dekkers B. G., Bos I. S., Halayko A. J., Zaagsma J., Meurs H. (2010) The laminin beta1-competing peptide YIGSR induces a hypercontractile, hypoproliferative airway smooth muscle phenotype in an animal model of allergic asthma. Respir. Res. , 170. [DOI] [PMC free article] [PubMed] [Google Scholar]