

Supplemental Digital Content is available in the text.
Keywords: alleles, apoptosis, brain ischemia, cerebral hemorrhage, prognosis, stroke
Abstract
Background and Purpose—
The E3 ubiquitin ligase MDM2 (murine double minute 2) is the main negative regulator of the p53 protein—a key player in neuronal apoptosis after ischemia. A functional single-nucleotide polymorphism in the human MDM2 gene promoter (rs2279744) regulates MDM2 protein expression. We investigated whether the MDM2 SNP309, by controlling p53-mediated apoptosis, determines functional outcome after stroke.
Methods—
Primary cortical neurons were subjected to oxygen and glucose deprivation. Mice were subjected to ischemic (transient middle cerebral artery occlusion) or hemorrhagic (collagenase injection) stroke models. Protein and mRNA levels of MDM2 and p53 were measured in both neuronal and brain extracts. The interaction of MDM2 with p53 was disrupted by neuronal treatment with nutlin-3a. siRNA was used to knockdown MDM2 expression. We analyzed the link between the MDM2 SNP309 and functional outcome, measured by the modified Rankin Scale scores, in 2 independent hospital-based stroke cohorts: ischemic stroke cohort (408 patients) and intracerebral hemorrhage cohort (128 patients).
Results—
Experimental stroke and oxygen and glucose deprivation induced the expression of MDM2 in the brain and neurons, respectively. Moreover, oxygen and glucose deprivation promoted MDM2 binding with p53 in neurons. Disruption of the MDM2-p53 interaction with nutlin-3a, or MDM2 knockdown by siRNA, triggered p53 accumulation, which increased neuronal susceptibility to oxygen and glucose deprivation-induced apoptosis. Finally, we showed that patients harboring the G allele in the MDM2 promoter had higher MDM2 protein levels and showed better functional outcome after stroke than those harboring the T/T genotype. The T/T genotype was also associated with large infarct volume in ischemic stroke and increased lesion volume in patients with intracerebral hemorrhage.
Conclusions—
Our results reveal a novel role for the MDM2-p53 interaction in neuronal apoptosis after ischemia and show that the MDM2 SNP309 determines the functional outcome of patients after stroke.
Stroke is the leading neurological cause of morbidity and mortality in developed countries.1 Stroke is either because of cerebral ischemia or hemorrhage, which activates a cascade of complex biochemical events leading to cellular integrity loss and cell death.2,3 Although the precise molecular mechanisms involved in stroke are not yet fully understood, the tumor suppressor protein p53 plays a key role in neuronal death after cerebral ischemia and intracerebral hemorrhage (ICH).4–6 The p53 protein rapidly accumulates in the damaged brain and activates the program of necrotic and apoptotic pathways of neuronal death, through both transcriptional dependent and independent mechanisms.4–8
The E3 ubiquitin ligase MDM2 (murine double minute 2) is the main cellular negative regulator of p53 stabilization and activity. The interaction of the N-terminal domain of MDM2 with the transactivating domain of p53 blocks its transcriptional activity.9 The E3 activity of MDM2 is dependent on its C-terminal RING finger domain, which polyubiquitinates p53 and targets the protein for proteasome-mediated degradation; however, MDM2 activity also monoubiquitinates p53 promoting its nuclear export.10,11 There is a p53-MDM2 negative feedback loop in which p53 transcriptionally upregulates MDM2, which in turn leads to increased proteasome degradation of p53.10
Previously, we described that the Arg72Pro single-nucleotide polymorphism of the p53 gene, which modulates the apoptotic activity of p53,5,12 controls neuronal susceptibility to ischemia-induced apoptosis and dictates functional outcome of patients with stroke.5,6 It is thus conceivable that variations in key regulators of the p53 pathway, such as MDM2, might affect p53 activity and neuronal susceptibility to ischemia. A functional single-nucleotide polymorphism in the MDM2 intronic promoter (rs2279744; SNP309T>G or SNP309) regulates MDM2 protein expression13 and modulates early development of solid tumors and cancer drug resistance.13,14 However, the impact of MDM2 in stroke is largely unknown. Here, we described that experimental ischemia both in vitro, induced by oxygen and glucose deprivation (OGD),15 and in vivo rapidly upregulates MDM2 expression, which controls p53 levels in neurons. Moreover, we demonstrate that the MDM2-p53 interaction modulates neuronal susceptibility to ischemia-induced apoptosis. Finally, we show that the MDM2 SNP309 regulates MDM2 level expression, which determines the functional outcome of patients after stroke. Thus, we reveal a novel function of the MDM2 SNP309 in neuronal damage and neurological outcome after stroke.
Materials and Methods
The authors declare that all supporting data are available within the article (and the online-only Data Supplement).
Expanded methods are provided in the online-only Data Supplement.
Animals
Animals were maintained in specific-pathogen free facilities at the University of Salamanca, in accordance with Spanish legislation (RD53/2013) under license from the European Union (2010/63/EU). Protocols were approved by the Bioethics Committee of the Institute of Biomedical Research of Salamanca. All efforts were made to minimize the numbers of animals used and ensure minimal suffering.
Primary Neuronal Cultures, Transfections, and OGD
Neuronal cultures prepared from C57BL/6J and p53-null mouse embryo (E14.5) cortices16 were subjected to OGD followed by reoxygenation.15 Neurons were transfected with siRNA against MDM2. Incubations were performed in the presence of 10 µmol/L MG132 (Sigma) or 1 µmol/L nutlin-3a (Sigma). Sequences and procedures are detailed in the online-only Data Supplement.
Transient Middle Cerebral Artery Occlusion and Collagenase Injection in Mice
Mice were subjected to experimental stroke through either transient middle cerebral artery occlusion, by using an intraluminal filament,17 or collagenase injection,6 as described in the online-only Data Supplement. MDM2 and p53 protein expression were assessed 24 hours after experimental stroke.
Western Blot Analysis, Coimmunoprecipitation Assay, and Immunocytochemistry
Immunoblot analyses,5 coimmunoprecipitation assay, and immunocytochemistry18 were performed as previously done and detailed in the online-only Data Supplement.
Patients and Clinical Variables
An observational prospective study was performed on 2 independent cohorts of consecutive patients with either a first ischemic stroke (IS; the IS cohort; 408 patients) or nontraumatic ICH (the ICH cohort; 128 patients) at <12 hours after symptom onset. Inclusion criteria are detailed in the online-only Data Supplement. Baseline demographic and clinical features of patients are shown in Table I in the online-only Data Supplement. Patients were treated according to the guidelines of the Cerebrovascular Disease Study Group of the Spanish Society of Neurology.19,20 Functional outcome was evaluated at 3 and 12 months with the modified Rankin Scale (mRS) by internationally certified neurologists.21 mRS score >2 was considered poor functional outcome.5
Human Polymorphism Analysis
Genotyping of human MDM2 polymorphism was performed by authors blinded to the clinical status of patients, as detailed in the online-only Data Supplement. When indicated, 2 groups were considered: T group, T/T genotype; G group, T/G+G/G genotypes.
Statistical Analysis
The results are expressed as percentages for categorical variables and as either the mean±SD or median (25th and 75th percentiles) for the continuous variables, depending on normal distribution determined by the Kolmogorov-Smirnov test. The Student t test (normal data) or the Mann-Whitney U test (non-normal data) was used to compare continuous variables between 2 groups. Proportions were compared using the χ2 test. Spearman analysis was used for bivariate correlations. The influence of the single-nucleotide polymorphism on functional outcome was assessed by logistic regression analysis. The results are expressed as adjusted odds ratio (AOR) with the corresponding 95% CI. Experimental results are expressed as mean±SEM. A 1-way ANOVA with a least significant difference post hoc test was used to compare values between multiple groups, and a 2-tailed, unpaired Student t test was used for 2-group comparisons. P<0.05 was considered significant.
Results
MDM2-p53 Interaction Regulates Neuronal Susceptibility to OGD
MDM2 mRNA and protein levels rapidly increased after OGD, remaining high at 24 hours of reoxygenation. Although p53 mRNA expression remained unchanged, p53 protein levels increased at 4 hours after OGD (Figure 1A; Figure I in the online-only Data Supplement), indicating an effect on the protein expression rather than on transcription, as described previously.5,22 This effect was mimicked in vivo. Mouse models of IS (transient middle cerebral artery occlusion) and ICH (collagenase injection) rapidly (24 hours) enhanced MDM2 and p53 protein expression in the damaged ipsilateral hemisphere, as compared with the contralateral hemisphere or sham mice (Figure 1B; Figure II in the online-only Data Supplement). Interestingly, OGD-induced p53 accumulation was reversible, as judged by the decreased protein levels after 24 hours of reoxygenation (Figure 1A; Figure I in the online-only Data Supplement) likely suggesting p53 degradation. To test a possible p53 degradation, neurons were incubated with MG132—an inhibitor of the proteasome, and p53 protein abundance was analyzed after OGD. MG132 promoted a rapid (0 hours) accumulation of p53, which further increased at 4 hours after OGD (Figure 1C; Figure I in the online-only Data Supplement), indicating that p53 protein stability is subjected to control by the proteasomal degradation pathway. We further investigated whether MDM2 and p53 interacted together. MDM2 was immunoprecipitated from neuronal protein extracts, followed by immunoblotting against MDM2 and p53. Figure 1D shows that p53 coimmunoprecipitated with MDM2—an effect that was potentiated by OGD. This interaction was confirmed by immunocytochemistry (Figure 1D). These data indicate that OGD upregulates MDM2 expression, which interacts with p53.
Figure 1.
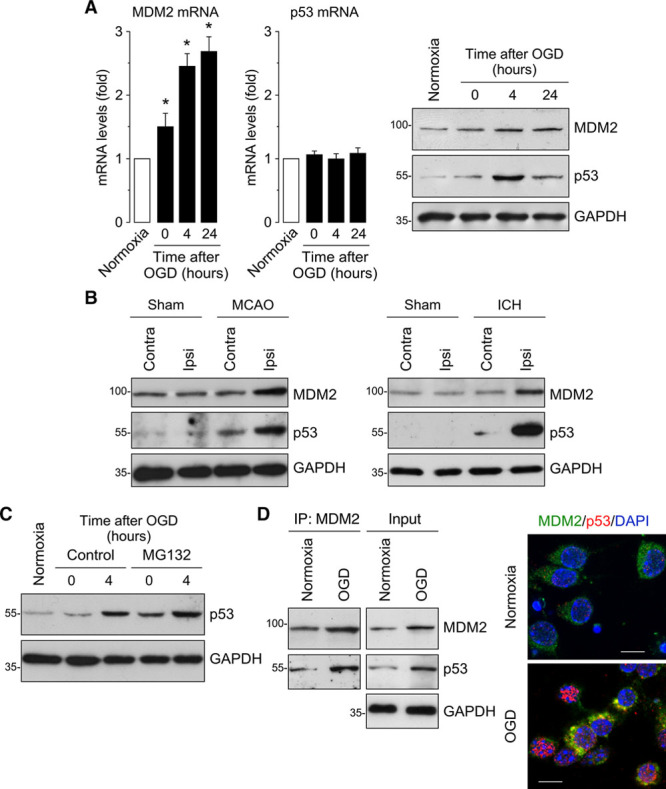
Ischemia upregulates MDM2 expression, which interacts with p53. A, Time course of MDM2 (murine double minute 2) and p53 mRNA levels measured by RT-qPCR (quantitative reverse transcription-polymerase chain reaction) showing increased MDM2 mRNA levels in primary neurons after oxygen and glucose deprivation (OGD), whereas p53 mRNA remained unchanged. Representative Western blot images showing MDM2 and p53 protein levels in neurons after OGD. Quantification of protein levels is shown in Figure I in the online-only Data Supplement (n=4 neuronal cultures). B, Mice were subjected to ischemic (transient middle cerebral artery occlusion [tMCAO]) or intracerebral hemorrhage (ICH; collagenase injection) stroke models. Representative Western blot images showing MDM2 and p53 protein levels in the contralateral (contra) and ipsilateral (ipsi) hemispheres at 24 h after tMCAO, ICH, or Sham surgery (n=4 mice). Quantification of protein levels is shown in Figure II in the online-only Data Supplement (n=4 mice). C, Neuronal treatment with the proteasome inhibitor MG132 (10 µmol/L) promoted a time-dependent accumulation of p53. Quantification of protein levels is shown in Figure I in the online-only Data Supplement (n=4 neuronal cultures). D, MDM2 and p53 proteins interact at 4 h after OGD, as shown by both coimmunoprecipitation assay and immunocytochemistry (GAPDH as loading control). Representative images are shown (green, MDM2; red, p53; blue, DAPI (4’,6-diamidino-2-phenylindole); scale bar=10 µmol/L). Data are expressed as mean±SEM from 4 different neuronal cultures. MCAO indicates middle cerebral artery occlusion. *P<0.05 compared with normoxia.
Next, we evaluated the function of this MDM2-p53 interaction on p53 stability after OGD. Neurons were incubated with nutlin-3a—a small molecule that targets the p53-binding pocket of MDM2, thus preventing an interaction between these 2 proteins.23 Nutlin-3a disrupted MDM2-p53 interaction leading to p53 accumulation (Figure 2A). Moreover, nutlin-3a potentiated OGD-induced p53 accumulation at 4 hours after OGD (Figure 2B, left; Figure III in the online-only Data Supplement). To validate this effect, we knocked down MDM2 by siRNA, which led p53 protein stabilization (Figure IV in the online-only Data Supplement, left). Confirming our previous data (Figure 1A, right), OGD in control neurons induced a biphasic effect on p53 protein abundance, that is, an increase at 4 hours followed by a decrease after 24 hours of OGD. Interestingly, knock down of MDM2 by siRNA prevented the destabilization of p53 protein that occurred at 24 hours after OGD (Figure 2B, right; Figure IV in the online-only Data Supplement). These data indicate that MDM2 interacts with p53 to regulate its protein abundance in neurons under ischemic conditions.
We next evaluated the potential impact of MDM2-p53 interaction on neuronal susceptibility to OGD damage. Nutlin-3a treatment and MDM2 knockdown by siRNA enhanced the increase in caspase 3 activity, as revealed by immunoblotting and immunocytochemistry (Figure 2C; Figure V in the online-only Data Supplement), and in neuronal apoptosis (Figure 2D) after OGD. Neuronal apoptosis caused by nutlin-3a was dependent on the presence of p53 because no apoptosis was observed by incubation of p53-null neurons (p53 KO) with nutlin-3a (Figure 2D, right). Moreover, p53 KO neurons were resistant to OGD-induced apoptosis (Figure 2D, right), confirming the key role played by p53 in the ischemic process. Thus, the disruption of the MDM2-p53 interaction increases neuronal susceptibility to ischemia. In sum, OGD upregulates MDM2 expression, which interacts with p53, thus promoting its degradation. Our results pose MDM2 as a key molecule in neuronal survival after ischemia.
MDM2 SNP309 Regulates MDM2 Levels and Functional Outcome After Stroke
We first analyzed the impact of the MDM2 SNP309 on MDM2 mRNA and protein expression in human monocytes from healthy individuals. Homozygous G/G and heterozygous T/G individuals for SNP309 showed heightened levels of MDM2 mRNA when compared with individuals harboring the T/T genotype (Figure VI in the online-only Data Supplement), as previously described in tumor cells.13,14 Because of its similar effect and the low frequency of the GG genotype (7%–8%), we grouped individuals from G/G and T/G genotypes (G group). Individuals with the T/T genotype were named T group. We found that individuals of the G group had increased levels of both MDM2 mRNA and protein (Figure VI in the online-only Data Supplement) levels than those of the T group.
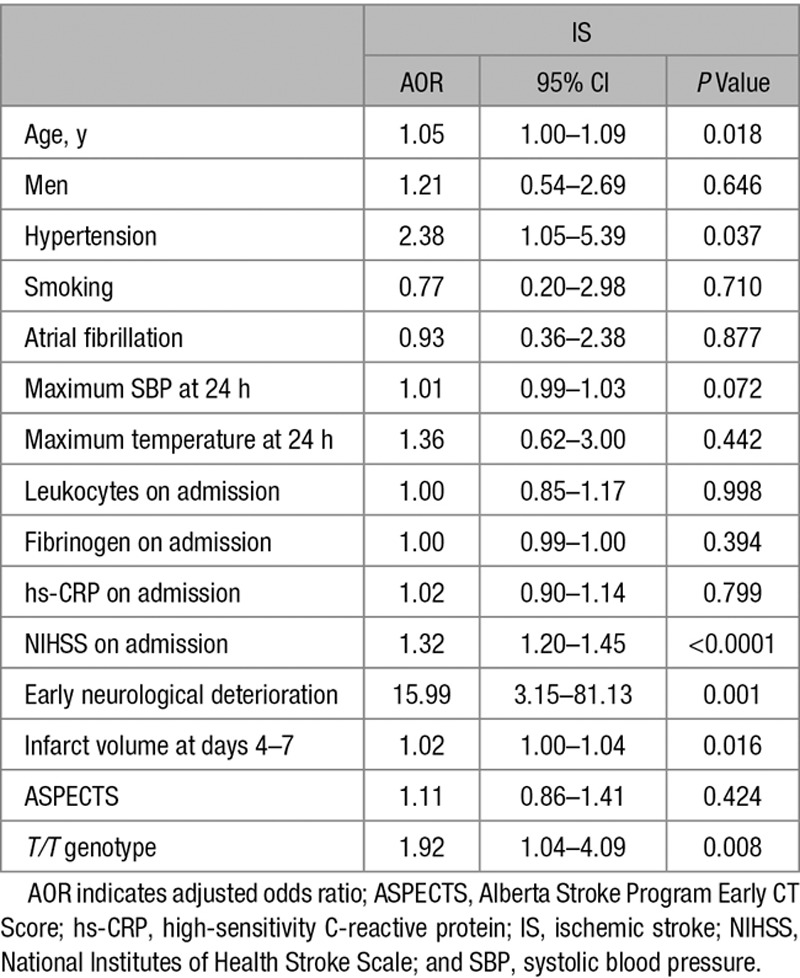
Although the MDM2 309SNP did not significantly affect initial stroke severity, as revealed by the National Institutes of Health Stroke Scale (NIHSS) scores on admission (Figure VII in the online-only Data Supplement, top), we found that patients with IS harboring the T/T genotype showed higher median mRS scores, at 3 (Figure 3A, top) and 12 months (Figure VIII in the online-only Data Supplement, top) after IS, than those with T/G and G/G genotypes. However, no significant differences were found between patients heterozygous and homozygous for the G allele (Figure 3A). This effect was maintained when patients were grouped according to the G allele: G (G/G+T/G genotypes) and T (T/T genotype; Figure 3A, bottom). Then, we analyzed the distribution of patients with IS (%) in each of the 6 mRS outcome categories, according to SNP309 genotype. We found a different pattern of mRS distribution of patients depending on the genotype. The majority of patients harboring the G allele had mRS scores ≤2 (good functional outcome). In contrast, we observed the opposite trend in patients with the T/T genotype, who were mainly associated with mRS scores >2 (poor functional outcome; Figure IX in the online-only Data Supplement, left). In fact, 74.5% of patients with poor functional outcome at 3 months after IS harbored the T/T genotype, whereas a smaller proportion (42.5%) showed good outcome (Table II in the online-only Data Supplement). Finally, infarct volume was 2-fold higher in patients of the T group than in those of the G group (Figure 3B). Interestingly, MDM2 mRNA and protein (Figure VI in the online-only Data Supplement) levels were higher in individuals with the G allele than those with the T/T genotype. The logistic regression analysis revealed that the T/T genotype was an independent marker of poor functional outcome (AOR, 1.92; 95% CI, 1.04–4.09; P=0.008) at 3 months after IS, after adjustment by age, NIHSS score on admission, infarct volume, and early neurological deterioration (Table 1).
Figure 3.
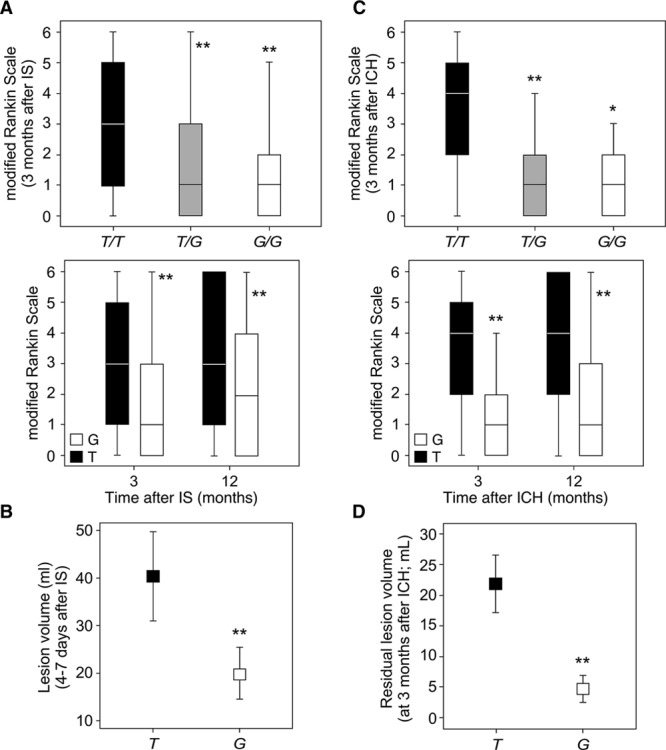
MDM2 SNP309 determines volume lesion and functional outcome after stroke. A and B, The ischemic stroke (IS) cohort included 408 patients (T/T: 225; T/G: 149; G/G: 34). A, Patients harboring the T/T genotype (T group) showed higher median mRS scores at 3 and 12 mo after IS, when compared with T/G and G/G (G group) patients. B, The infarct volume was higher in T/T patients (T group) than in the G group (T/G+G/G). C and D, The intracerebral hemorrhage (ICH) cohort included 128 patients (T/T: 76; T/G: 44; G/G: 8). C, Patients harboring the T/T genotype (T group) showed higher median mRS scores at 3 and 12 mo after ICH, when compared with T/G and G/G (G group) patients. D, The residual lesion volume was higher in T/T patients (T group) than in the G group (T/G+G/G). A and C, Box plots show median values (horizontal line inside the box), quartiles (box boundaries), and the largest and smallest observed values (error bars). B and D, Data are expressed as mean±SD. *P<0.05, **P<0.0001 compared with T/T or T group.
Table 1.
Multiple Logistic Regression Analysis Showing Independent Variables Associated With Poor Functional Outcome at 3 Months (Modified Rankin Scale, >2) After IS
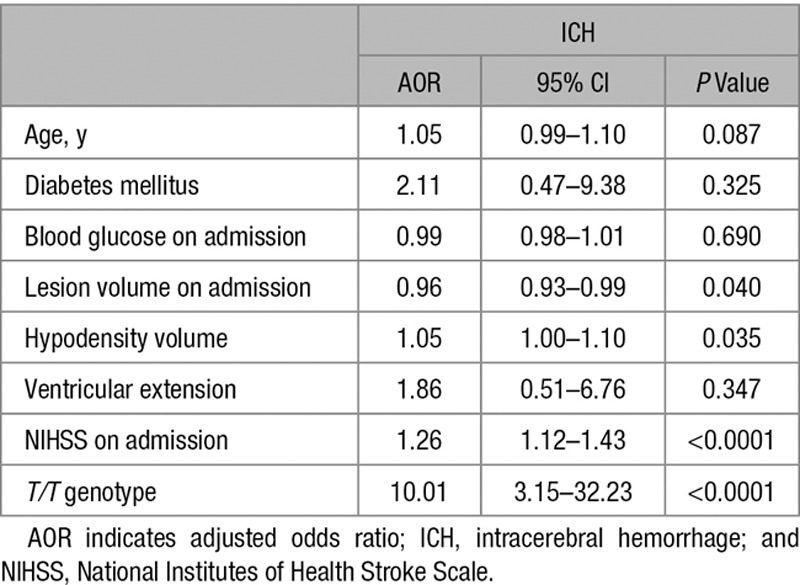
Regarding the ICH cohort, we also found no significant differences between patients harboring the G allele in homozygosity or heterozygosity. However, we found that T/T patients had higher median mRS scores, at both 3 (Figure 3C, top) and 12 months (Figure VIII in the online-only Data Supplement, middle) after ICH, than T/G and G/G patients, despite NIHSS scores and ICH volume on admission (Figure VII in the online-only Data Supplement) were similar in all genotypes, indicating similar initial hemorrhagic damage. The effect was also observed when genotypes were grouped as G and T patients (Figure 3C, top). The majority of patients harboring the G allele had mRS scores ≤2, whereas T/T patients were mainly associated with mRS scores >2 (Figure IX in the online-only Data Supplement, left). In this context, the 86.4% of patients with poor functional outcome at 3 months after ICH had the T/T genotype, whereas only a small percentage (13.8%) showed good prognosis (Table III in the online-only Data Supplement). Furthermore, a 4-fold higher residual lesion volume was found in T/T patients (T group) when compared with those of the G group, at 3 months after ICH (Figure 3D). The reduction in the lesion volume was then lower in patients with the T/T genotype than in patients carrying the G allele (Figure VIII in the online-only Data Supplement, bottom). The logistic regression analysis revealed that the T/T genotype was an independent marker of poor functional outcome (AOR, 10.01; 95% CI, 3.15–32.23; P<0.0001) at 3 months after ICH, after adjustment for hematoma volume, perihematoma hypodensity volume, and NIHSS on admission (Table 2). Surprisingly, this analysis (Table 2) revealed that the ICH lesion volume on admission seems to be associated with a good functional outcome (AOR, 0.96; 95% CI, 0.93–0.99; P<0.040), which is confusing because it is a well-known predictor of poor outcome after stroke. Moreover, as shown in Table III in the online-only Data Supplement, ICH volume on admission was significantly higher in patients with poor functional outcome at 3 months than in those with good outcome. Furthermore, when the variable MDM2SNP309 is not included in the multivariate analysis, we found that ICH volume on admission is a productive predictor of functional outcome (AOR, 1.06; 95% CI, 1.02–1.09; P<0.0001) after adjustment for age, diabetes mellitus, blood glucose on admission, ventricular extension, perihematoma hypodensity volume, and NIHSS on admission. Then, from a clinical point of view, this suggests that the MDM2 SNP309 might be implicated in mechanisms that mediate the association of ICH volume with the functional outcome of patients with stroke.
Table 2.
Multiple Logistic Regression Analysis Showing Independent Variables Associated With Poor Functional Outcome at 3 Months (Modified Rankin Scale, >2) After ICH
Finally, we evaluated possible differences of the predictive potential of the MDM2 promoter genotype according to the different stroke subtypes. We found no statistical differences between stroke subtypes and MDM2 SNP309 distribution, in both the IS (P=0.228) and ICH (P=0.457) cohorts (Table IV in the online-only Data Supplement). All these results demonstrate that the T/T genotype is linked with poor functional outcome after IS or ICH, good prognosis being mainly restricted to patients with stroke with the T/G and G/G genotypes. Thus, our results reveal that the MDM2 SNP309 regulates protein levels and determines functional outcome after stroke, regardless of whether the origin is ischemic or hemorrhagic.
Discussion
Here, we describe that the E3 ubiquitin ligase MDM2 promotes neuronal survival after an ischemic insult by destabilizing the proapoptotic p53 protein. Thus, the disruption of the MDM2-p53 interaction by pharmacological treatment with nutlin-3a or MDM2 knockdown with siRNA promoted p53 stabilization and enhanced neuronal vulnerability against an ischemic insult. Furthermore, we identify the SNP309 in the MDM2 gene promoter, which regulates MDM2 level expression, as a biomarker of functional outcome of patients with stroke. Thus, the G allele is associated with heightened MDM2 mRNA and protein levels, which reflects good prognosis of the patients. We, therefore, demonstrate the key role of the MDM2-p53 interaction in neuronal survival after ischemia and identify the SNP309 in the MDM2 gene promoter as an independent factor for predicting functional prognosis of patients with stroke.
The p53 protein is effectively antagonized by its direct interaction with MDM2, which represses transcription of p53 target genes and promotes p53 degradation via ubiquitination, all leading to p53 inactivation. The MDM2-p53 interaction plays a key function in cancer progression, thus representing an important target for the development of new anticancer therapies.11 Here, we found that this interaction is also essential for neuronal survival after ischemia. Then, the disruption of the MDM2-p53 triggers the accumulation of p53 and increases the susceptibility of neurons to an ischemic damage. In an earlier study, it was reported that in vivo ischemia induced MDM2 expression in the ischemic brain,24 thus suggesting a possible neuroprotective role for the ligase. Our results demonstrate that ischemia in vitro induces the upregulation of MDM2 expression, which interacts with p53 to promote its degradation. Thereby, the disruption of the MDM2-p53 interaction with nutlin-3a potentiates p53 accumulation and caspase 3-mediated apoptosis after ischemia. Moreover, knockdown of MDM2 and treatment with the inhibitor of proteasome degradation MG132 mimicked the accumulation and neurotoxicity of p53. Altogether, our results indicate that the MDM2-p53 interaction has a neuroprotective function and actively participates in neuronal susceptibility to ischemia-induced apoptosis.
The functional SNP309 in the MDM2 intronic promoter consists in a T>G nucleotide change, leading to an increased affinity of the transcriptional activator Sp1, which results in a constitutive enhanced MDM2 transcription.13,25 Moreover, the MDM2 SNP309 G allele has been associated with increased cancer risk in several human tumors that express wild-type p53,14,26 probably as a consequence of altered homeostasis of the MDM2-p53 pathway resulting in decreased basal steady-state levels of p53.25 We now describe that the MDM2 SNP309 G allele is associated with good functional outcome after IS. Thus, patients harboring the G allele possess higher MDM2 protein levels, which would increase p53 protein degradation, resulting in decreased neuronal apoptosis5 and, therefore, good prognosis after an ischemic damage. In contrast, lower levels of MDM2 in patients with the T/T genotype result in poor prognosis after stroke. Given that ischemia in vitro induced MDM2 expression in neurons, these data suggest that G allele exerts protection. Interestingly, Sp1 expression has been described to be induced in neurons after ischemia.27,28 Given that patients carrying the SNP309 G allele show higher affinity to Sp1 than patients with the T/T genotype,13 our data suggest that the increased Sp1 expression would be responsible for the increase in MDM2 levels.
The association of the MDM2 SNP309 G allele with good functional outcome was also observed in patients experiencing ICH. Although several injury mechanisms are involved,29 secondary ischemic lesions, within both the perihematoma region and remote from the hematoma, have been found in patients with ICH30 that may contribute to cytotoxicity after ICH.31,32 These ischemic lesions might promote MDM2 expression, particularly in patients carrying the SNP309 G allele, which shows good functional outcome after ICH. However, such a relationship would need to be confirmed in future studies.
Although the logistic regression models are adjusted by the NIHSS at admission, and the prognostic value of the MDM2 SNP309 is independent on the severity of initial neurological affectation in patients with both IS and ICH, as well as the ICH volume on admission, interestingly there is a tendency of the T/T genotype toward a greater stroke severity at the beginning of the process compared with T/G and G/G genotypes. This effect may also contribute to the poor functional outcome of patients with stroke with the T/T genotype.
In conclusion, here, we describe that the MDM2 SNP309 determines functional outcome after stroke, probably as a consequence of the modulation of the MDM2-p53 interaction. Therefore, the MDM2 SNP309 G allele increases MDM2 protein expression, leading to enhanced MDM2-mediated p53 degradation, which reduces p53-induced apoptotic responses to ischemia, resulting in good functional outcome after stroke.
Figure 2.
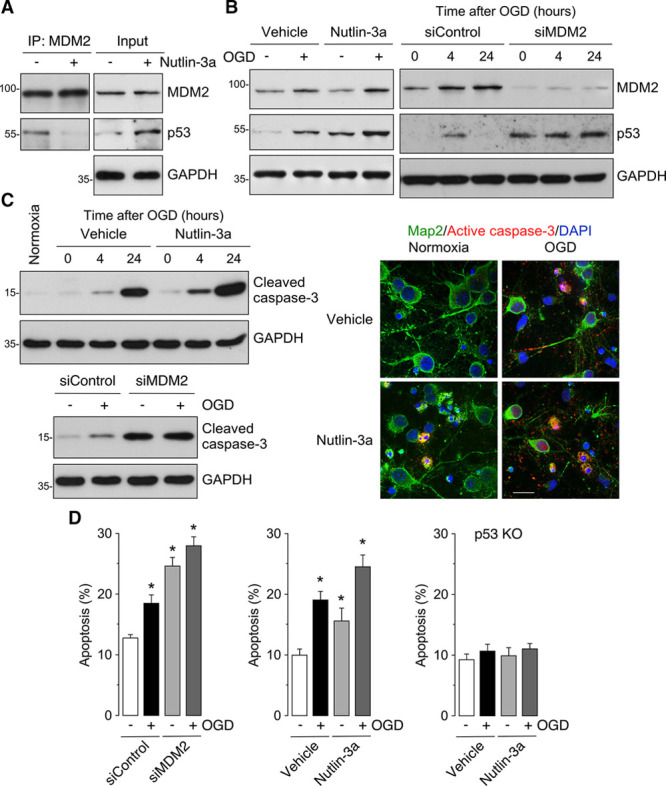
MDM2-p53 interaction modulates neuronal survival after ischemia. A, Coimmunoprecipitation assay revealing that nutlin-3a (1 µmol/L; 4 h of incubation) disrupted MDM2 (murine double minute 2) and p53 interaction in neurons. B, Western blot images showing that nutlin-3a enhanced oxygen and glucose deprivation (OGD)-induced p53 accumulation in neurons at 4 h of reoxygenation. MDM2 knockdown by siRNA (siMDM2) treatment for 2 d enhanced OGD-induced p53 accumulation and prevented the decrease in p53 levels observed in control neurons (siControl) at 24 h after OGD. C, Western blot images showing that nutlin-3a (1 µmol/L) increased OGD-induced caspase 3 activation at 4 and 24 h after OGD. Representative images confirming that nutlin-3a enhanced OGD-induced active caspase 3 expression at 4 h after OGD (green, Map2; red, active caspase 3; blue, nuclear DAPI (4’,6-diamidino-2-phenylindole); right; scale bar=20 µmol/L). siMDM2 promoted caspase 3 activation in neurons at 4 h after OGD (GAPDH as loading control). Quantification of protein levels is shown in (A) Figure III in the online-only Data Supplement, (B) Figure IV in the online-only Data Supplement, and (C) Figure V in the online-only Data Supplement (n=4 neuronal cultures). D, Measurement of neuronal apoptosis (annexinV+/7AAD− neurons) by flow cytometry revealed that treatment of neurons with siMDM2 or nutlin-3a promoted neuronal apoptosis at 4 h after OGD. Genetic deletion of p53 (p53 KO) prevented apoptosis induced by OGD or nutlin-3a. Data are expressed as mean±SEM from 4 different neuronal cultures. *P<0.05 compared with siControl normoxia (−OGD) or vehicle normoxia (−OGD) conditions.
Acknowledgments
The technical assistance of Lucia Martin, Estefania Prieto, Monica Carabias, and Monica Resch is acknowledged. We are grateful to D. Veroz for nursing assistance. We thank the healthy donors and patients for their participation in this research study.
Sources of Funding
This work was funded by Instituto de Salud Carlos III (PI15/00473, RD16/0019/0018, and RD16/0019/0001), European Regional Development Fund, Ministerio de Economía y Competitividad (SAF2016-78114-R), European Union’s Horizon 2020 Research and Innovation Programme (Grant Agreement 686009), and Junta de Castilla y León (IES007P17). Drs Rodríguez (RD16/0019/0018), Delgado-Esteban (CP0014/00010), and Sobrino (CPII17/00027) are supported by Instituto de Salud Carlos III.
Disclosures
None.
Supplementary Material
Footnotes
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/STROKEAHA.118.022529.
References
- 1.Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480–486. doi: 10.1161/CIRCULATIONAHA.108.191259. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- 2.Lo EH, Moskowitz MA, Jacobs TP. Exciting, radical, suicidal: how brain cells die after stroke. Stroke. 2005;36:189–192. doi: 10.1161/01.STR.0000153069.96296.fd. doi: 10.1161/01.STR.0000153069.96296.fd. [DOI] [PubMed] [Google Scholar]
- 3.Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–e339. doi: 10.1161/STROKEAHA.108.531632. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- 4.Hong LZ, Zhao XY, Zhang HL. p53-mediated neuronal cell death in ischemic brain injury. Neurosci Bull. 2010;26:232–240. doi: 10.1007/s12264-010-1111-0. doi: 10.1007/s12264-010-1111-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez-Sanchez JC, Delgado-Esteban M, Rodriguez-Hernandez I, Sobrino T, Perez de la Ossa N, Reverte S, et al. The human Tp53 Arg72Pro polymorphism explains different functional prognosis in stroke. J Exp Med. 2011;208:429–437. doi: 10.1084/jem.20101523. doi: 10.1084/jem.20101523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodríguez C, Sobrino T, Agulla J, Bobo-Jiménez V, Ramos-Araque ME, Duarte JJ, et al. Neovascularization and functional recovery after intracerebral hemorrhage is conditioned by the Tp53 Arg72Pro single-nucleotide polymorphism. Cell Death Differ. 2017;24:144–154. doi: 10.1038/cdd.2016.109. doi: 10.1038/cdd.2016.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo Y, Kuo CC, Shen H, Chou J, Greig NH, Hoffer BJ, et al. Delayed treatment with a p53 inhibitor enhances recovery in stroke brain. Ann Neurol. 2009;65:520–530. doi: 10.1002/ana.21592. doi: 10.1002/ana.21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geyer RK, Yu ZK, Maki CG. The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat Cell Biol. 2000;2:569–573. doi: 10.1038/35023507. doi: 10.1038/35023507. [DOI] [PubMed] [Google Scholar]
- 10.Perry ME. The regulation of the p53-mediated stress response by MDM2 and MDM4. Cold Spring Harb Perspect Biol. 2010;2:a000968. doi: 10.1101/cshperspect.a000968. doi: 10.1101/cshperspect.a000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen D, Liao W, Zeng SX, Lu H. Reviving the guardian of the genome: Small molecule activators of p53. Pharmacol Ther. 2017;178:92–108. doi: 10.1016/j.pharmthera.2017.03.013. doi: 10.1016/j.pharmthera.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dumont P, Leu JI, Della Pietra AC, III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33:357–365. doi: 10.1038/ng1093. doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- 13.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 14.Basu S, Murphy ME. Genetic modifiers of the p53 pathway. Cold Spring Harb Perspect Med. 2016;6:a026302. doi: 10.1101/cshperspect.a026302. doi: 10.1101/cshperspect.a026302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Almeida A, Delgado-Esteban M, Bolaños JP, Medina JM. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem. 2002;81:207–217. doi: 10.1046/j.1471-4159.2002.00827.x. [DOI] [PubMed] [Google Scholar]
- 16.Delgado-Esteban M, García-Higuera I, Maestre C, Moreno S, Almeida A. APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nat Commun. 2013;4:2879. doi: 10.1038/ncomms3879. doi: 10.1038/ncomms3879. [DOI] [PubMed] [Google Scholar]
- 17.Vecino R, Burguete MC, Jover-Mengual T, Agulla J, Bobo-Jiménez V, Salom JB, et al. The MDM2-p53 pathway is involved in preconditioning-induced neuronal tolerance to ischemia. Sci Rep. 2018;8:1610. doi: 10.1038/s41598-018-19921-x. doi: 10.1038/s41598-018-19921-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veas-Perez de Tudela M, Delgado-Esteban M, Maestre C, Bobo-Jimenez V, Jimenez-Blasco D, Vecino R, et al. Regulation of bcl-xl-atp synthase interaction by mitochondrial cyclin b1-cyclin-dependent kinase-1 determines neuronal survival. J Neurosci. 2015;35:9287–9301.. doi: 10.1523/JNEUROSCI.4712-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alonso de Lecinana M, Egido JA, Casado I, Ribo M, Davalos A, Masjuan J, et al. Guidelines for the treatment of acute ischaemic stroke. Neurologia. 2014;29:102–122.. doi: 10.1016/j.nrl.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 20.Rodríguez-Yáñez M, Castellanos M, Freijo MM, López Fernández JC, Martí-Fàbregas J, Nombela F, et al. Comitéad hoc del Grupo de Estudio de Enfermedades Cerebrovasculares de la SEN. Clinical practice guidelines in intracerebral haemorrhage. Neurologia. 2013;28:236–249. doi: 10.1016/j.nrl.2011.03.010. doi: 10.1016/j.nrl.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Banks JL, Marotta CA. Outcomes validity and reliability of the modified Rankin scale: implications for stroke clinical trials: a literature review and synthesis. Stroke. 2007;38:1091–1096. doi: 10.1161/01.STR.0000258355.23810.c6. doi: 10.1161/01.STR.0000258355.23810.c6. [DOI] [PubMed] [Google Scholar]
- 22.Leker RR, Aharonowiz M, Greig NH, Ovadia H. The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin alpha. Exp Neurol. 2004;187:478–486. doi: 10.1016/j.expneurol.2004.01.030. doi: 10.1016/j.expneurol.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 23.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 24.Tu Y, Hou ST, Huang Z, Robertson GS, MacManus JP. Increased Mdm2 expression in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1998;18:658–669. doi: 10.1097/00004647-199806000-00008. doi: 10.1097/00004647-199806000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Post SM, Quintás-Cardama A, Pant V, Iwakuma T, Hamir A, Jackson JG, et al. A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer Cell. 2010;18:220–230. doi: 10.1016/j.ccr.2010.07.010. doi: 10.1016/j.ccr.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cox J, Jackson AP, Bond J, Woods CG. What primary microcephaly can tell us about brain growth. Trends Mol Med. 2006;12:358–366. doi: 10.1016/j.molmed.2006.06.006. doi: 10.1016/j.molmed.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 27.Woo SK, Kwon MS, Geng Z, Chen Z, Ivanov A, Bhatta S, et al. Sequential activation of hypoxia-inducible factor 1 and specificity protein 1 is required for hypoxia-induced transcriptional stimulation of Abcc8. J Cereb Blood Flow Metab. 2012;32:525–536. doi: 10.1038/jcbfm.2011.159. doi: 10.1038/jcbfm.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Formisano L, Guida N, Valsecchi V, Cantile M, Cuomo O, Vinciguerra A, et al. Sp3/REST/HDAC1/HDAC2 complex represses and Sp1/HIF-1/p300 complex activates ncx1 gene transcription, in brain ischemia and in ischemic brain preconditioning, by epigenetic mechanism. J Neurosci. 2015;35:7332–7348. doi: 10.1523/JNEUROSCI.2174-14.2015. doi: 10.1523/JNEUROSCI.2174-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilkinson DA, Pandey AS, Thompson BG, Keep RF, Hua Y, Xi G. Injury mechanisms in acute intracerebral hemorrhage. Neuropharmacology. 2018;134(pt B):240–248. doi: 10.1016/j.neuropharm.2017.09.033. doi: 10.1016/j.neuropharm.2017.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gioia LC, Kate M, Choi V, Sivakumar L, Jeerakathil T, Kosior J, et al. Ischemia in intracerebral hemorrhage is associated with leukoaraiosis and hematoma volume, not blood pressure reduction. Stroke. 2015;46:1541–1547. doi: 10.1161/STROKEAHA.114.008304. doi: 10.1161/STROKEAHA.114.008304. [DOI] [PubMed] [Google Scholar]
- 31.Menon RS, Burgess RE, Wing JJ, Gibbons MC, Shara NM, Fernandez S, et al. Predictors of highly prevalent brain ischemia in intracerebral hemorrhage. Ann Neurol. 2012;71:199–205. doi: 10.1002/ana.22668. doi: 10.1002/ana.22668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prabhakaran S, Naidech AM. Ischemic brain injury after intracerebral hemorrhage: a critical review. Stroke. 2012;43:2258–2263. doi: 10.1161/STROKEAHA.112.655910. doi: 10.1161/STROKEAHA.112.655910. [DOI] [PubMed] [Google Scholar]