

Abstract
Alzheimer's disease (AD) is a disease of aging that results in cognitive impairment, dementia and death. Pathognomonic features of AD are amyloid plaques composed of proteolytic fragments of the amyloid precursor protein (APP) and neurofibrillary tangles composed of hyperphosphorylated tau protein. One type of familial Alzheimer's disease (FAD) occurs when mutant forms of APP are inherited. Both APP and tau are components of the microtubule-based axonal transport system, which prompts the hypothesis that axonal transport is disrupted in AD, and that such disruption impacts cognitive function. Transgenic mice expressing mutated forms of APP provide preclinical experimental systems to study AD. Here we perform manganese-enhanced magnetic resonance imaging (MEMRI) to study transport from hippocampus to forebrain in four cohorts of living mice: young and old wild-type and transgenic mice expressing a mutant APP with both Swedish and Indiana mutations (APPSwInd). We find that transport is decreased in normal aging and further altered in aged APPSwInd plaque-bearing mice. These findings support the hypothesis that transport deficits are a component of AD pathology and thus may contribute to cognitive deficits.
Keywords: Amyloid precursor protein (APP), fast axonal transport, CA3 of the hippocampus, dentate gyrus, transgenic mice for Alzheimer's disease investigation, aging, Manganese-enhanced magnetic resonance imaging (MEMRI), cholinergic neurons, septal nuclei
1. Introduction
The aging brain has a heightened susceptibility to neurodegenerative diseases that produce cognitive impairment and dementia, such as Alzheimer's disease. Alzheimer’s disease (AD) accounts for half of all dementias, with associated cognitive decline characterized by deficits in episodic memory, short-term memory, impaired judgment and synapse loss (Keller, 2006; Reitz et al., 2011; Sonnen et al., 2011). The neuropathological hallmark of AD is the presence of neuritic plaques (SPs) composed primarily of aggregates of amyloid-β (Aβ), and neurofibrillary tangles (NFTs) formed from hyper-phosphorylated tau (p-tau), both of whose precursors have putative roles in axonal transport (Kamal et al., 2001; Kamal et al., 2000; Vershinin et al., 2007). Aβ is produced by proteolytic cleavage of the amyloid precursor protein (APP), an evolutionarily conserved integral membrane protein expressed in many tissues and found at particularly high concentrations at neuronal synapses (Tharp and Sarkar, 2013; Walsh and Selkoe, 2007). APP has many putative functions, including cell adhesion, copper homeostasis, iron export, synapse formation and neural plasticity (Duce et al., 2010; Heber et al., 2000; Herms et al., 2004; Muller et al., 1994; Priller et al., 2006; Satpute-Krishnan et al., 2006; Turner et al., 2003; Young-Pearse et al., 2007). One form of familial Alzheimer's disease (FAD) is a consequence of mutations in the gene encoding APP (Bertram and Tanzi, 2008). Other mutations that cause FAD are in genes for APP processing, such as the gamma secretase enzymes, PSEN-1 or ×2, that produce the toxic Aβ fragments found in plaques (Campion et al., 1999). These findings indicate that APP integrity has a causal relationship with AD clinical symptoms. To study the biology of the mutant APP within the brain, transgenic mice have been generated that express a mutant form of APP found in FAD (Jankowsky and Zheng, 2017; Webster et al., 2014). Overexpression in mouse of mutant APP from either or both Swedish or Indiana FAD families produces Aβ plaques resembling those in humans (Chen et al., 2000; Dodart et al., 2000; Elder et al., 2010; Jankowsky et al., 2005).
Both APP and tau are involved in axonal transport, leading to the hypothesis that transport may be defective in Alzheimer's disease (Encalada and Goldstein, 2014). APP is a cargo-receptor for the fast axonal transport of organelles (Satpute-Krishnan et al., 2006; Seamster et al., 2012), probably mediated through the kinesins (Kamal et al., 2001; Kamal et al., 2000; Seamster et al., 2012). Knockout of the APP gene leads to defects in transport within axons in Drosophila and mouse peripheral nerves (Gunawardena and Goldstein, 2001; Salehi et al., 2006; Schindowski et al., 2008; Stokin et al., 2005) and in the hippocampal-forebrain circuit within the mouse brain (Gallagher et al., 2012). Tau binds microtubules, stabilizing them and regulating interactions with molecular motors, such as the kinesins and dynein (Wang and Mandelkow, 2016). A minor increase in APP (1.5-fold) is also seen in a mouse model for Down's Syndrome, in which disruption of retrograde transport of nerve growth factor (NGF) from the hippocampus to cholinergic neurons in the medial septal nucleus (MSN) (Salehi et al., 2006) and paradoxically an increase anterograde transport of Mn2+ to the basal forebrain (Bearer et al., 2007b) are found. Alternatively, when the APP gene is deleted, mice have impaired hippocampal long-term potentiation, deficits in passive avoidance learning (Dawson et al., 1999; Senechal et al., 2008) and delayed transport from CA3 of the hippocampus to both the contralateral hippocampus and the septal region (Gallagher et al., 2012). Transgenic mice offer the opportunity to test the hypothesis that transport deficits occur in AD.
We have developed a new approach to witness and measure axonal transport in the living brains of mice over time: Manganese-enhanced Magnetic Resonance Imaging (MEMRI) (Bearer et al., 2007b; Bearer et al., 2009; Gallagher et al., 2013; Gallagher et al., 2012; Medina et al., 2017a; Zhang et al., 2010). MEMRI takes advantage of the paramagnetic ion manganese (Mn2+) as a contrast agent which gives a hyper-intense signal in MRI (Bearer et al., 2009; Lin and Koretsky, 1997; Merritt et al., 1989; Pautler, 2004; Silva et al., 2004; Zhang et al., 2010). Because Mn2+ enters neurons via voltage-gated calcium channels (Lin and Koretsky, 1997; Merritt et al., 1989), MEMRI directly indicates active circuitry rather than being a proxy of neural activity as is provided by blood oxygen level dependent (BOLD) imaging used in human fMRI. After entry into active neurons, Mn2+ is transported within axons by fast axonal transport, driven in part by kinesin-1, a microtubulebased motor (Bearer et al., 2007a; Medina et al., 2017b), and then is released to cross active synapses (Bearer et al., 2007a). Thus localized injection of Mn2+ followed by sequential MR imaging reveals functional, anatomically connected, circuitry. MRI is non-destructive and mice are imaged live. Thus each mouse can be imaged, returned to the cage, and re-imaged multiple times over hours and days. Mn2+ transport occurs in between the imaging sessions in the awake, behaving mouse and images are captured at intervals, like snapshots. By applying powerful computational statistic parametric mapping to compare images of multiple mice between time points, we obtain a unique unbiased comprehensive view of the transport dynamics across the whole brain in threedimensions (Bearer et al., 2007b; Medina et al., 2017b).
Injection of Mn2+ into the cornu ammonis field 3 (CA3) region of the hippocampus results in measureable transport to known distal sites over time (Bearer et al., 2007b; Gallagher et al., 2012; Medina et al., 2017b), allowing us to follow the dynamics of Mn2+ transport and its accumulation in living animals in three-dimensions by MEMRI. Major projections of neurons in CA3 are to the basal forebrain via fimbria, fornix and septal nuclei (SN) (Paxinos, 2004; Swanson and Cowan, 1977) and to the contralateral hippocampus (Gallagher et al., 2012). To monitor transport dynamics, we collect whole brain images at four time points: prior to injection and at 0.5, 6 and 24 hours after injection (Diagram 1). We use automated whole brain, unbiased, voxel-wise statistical analysis within group between time points to identify the progression of Mn2+-induced intensity patterns between and across these time points (Bearer et al., 2007b; Gallagher et al., 2012; Medina et al., 2017a). This analysis reveals the anatomy of the circuit, the rate of transport within it and subsequent accumulation at distal regions (Gallagher et al., 2012; Medina et al., 2017a). To compare between genotypes, we use a region of interest analysis, selecting those anatomical locations with statistically significant intensity increases for intensity measurements and between group comparisons (Medina et al., 2017a). Histopathology of the same mice that were imaged live by MRI provides correlation of anatomy and AD pathologies with the differential transport and accumulation along hippocampal projections.
Diagram 1. Time course of time-lapse imaging.

A pre-injection image is captured to provide a non-enhanced image for alignments of post-injection images. Next, 3–5 nL of Mn2+ with RDA is injected into CA3 of the hippocampus and then timelapse MR images are captured at 0.5h, 6h and 24h post-injection. Animals are returned to their cage for 3 weeks to allow the RDA to transport distally from the injection site and then the animals are sacrificed for histologic and biochemical analysis.
Here we deploy MEMRI to quantify transport dynamics in a transgenic mouse which expresses a mutant form of hAPP with both Swedish and Indiana familial mutations (Jankowsky et al., 2005). These mice have significant plaque deposition by mid-life, progressive atrophy of the granule cells in the hippocampal dentate gyrus, behavioral changes and deficits in spatial learning and memory (Han et al., 2012; Jankowsky et al., 2005).
2. Material and methods
2.1. Animals
To study the effect of mutant APP expression on transport, we used transgenic mice that overexpress the human APP695 gene carrying both the Swedish and Indiana familial Alzheimer’s disease mutations (SwInd) with expression under control of the Tet-off promoter (Jankowsky et al., 2005). These mice were only available as frozen embryos at JAX, deposited by the Borchelt lab, B6.Cg-Tg(tetO-APPSwInd, strain 34845), and will be referred to here as APPSwInd. We recovered breeding pairs and back-crossed them to C57/B6 (also referred to as C57Bl/6J, JAX strain 000664) to obtain a monogenic stock congenic with C57/B6.
Monogenic APPSwInd were bred to monogenic tTA mice (JAX 007004B6.Cg-Tg (CaMKIIa-tTA) 1Mmay-DboJ), a transgenic line that expresses the TetO transcription activator (tTA) in a C57/B6 background driven by the CaMKIIa promoter (Mayford et al., 1996), using IACUC-approved breeding protocols at UNM to produce double transgenics. The genotypes were monitored in all mice at each generation via tail snip genotyping by PCR at Transnetyx. This breeding protocol produced offspring with four genotypes: no transgene, one or the other transgene (monogenic tTA or monogenic APP) and double transgenic mice in each litter from each breeding pair, thus providing "wild-type" (WT) littermates for all double transgenics. We found no detectable impact on phenotype of either the monotransgenic tTA or APPSwInd in behavior, histology, biochemical analysis of APP expression or in MEMRI transport dynamics. We thus grouped these non-expressing genotypes for analysis and refer them as "wild-type (WT)" throughout. Double transgenics will be referred to as "APPSwInd", as this is the only genotype expressing the APPSwInd transgene. Mice were either aged to 5–6 months (young WT, n = 12; young APPSwInd, n = 11) or 13–17 months (old WT, n = 11; old APPSwInd, n = 6) (Table 1). Institutional Animal Care and Use Committee (IACUC) both of the California Institute of Technology and of the University of New Mexico approved all protocols involving animals.
Table 1:
Mouse genotypes for MR imaging
| Cohort | Age | Strain/genotype/numbers of animals |
|---|---|---|
| Young WT | 4–6 months | C57/B6 (n = 12) |
| Young APPSwInd | 5–6 months | C57/B6, APPSwInd (n = 11) |
| Old WT | 13–17 months | C57/B6 WT (n = 1) and monogenics: tTA (n = 4) APP (n = 6) (Total n = 11) |
| Old APPSwInd | 13–17 months | C57/B6, APPSwInd (n = 6) |
Additional animals for each cohort were used for histology (see Materials and Methods).
2.2. Stereotactic injection
Stereotactic injections were performed as previously described (Bearer et al., 2009; Gallagher et al., 2012; Medina et al., 2017a). Briefly, mice were anesthetized and secured in a stereotaxic frame (Stoelting, Wood Dale, IL), and 3–5 nL of 200 mM Mn2+ in sterile water, was coinjected with 0.5 mg/ml rhodamine dextran-amine (RDA; 3k; Molecular Probes/Invitrogen, OR, USA) over a 5 min period using a quartz micropipette guided by a computer-assisted stereotaxic injector (myNeuroLAB.com, IL, USA) into the right hippocampus (target coordinates × = 3.2 mm right of midline, −4.1 mm posterior of bregma, 3.4 mm down from the surface). Animals were transferred to the MR scanner and maintained under anesthesia with 1–1.5% isoflurane. The RDA was co-injected with the Mn2+ so that we could identify the injection site and monitor the precise location for any injury by histology. This co-injection also allowed us to determine that Mn2+ was delivered into the expected hippocampal circuit, as determined by following the RDA transport along hippocampal projections by fluorescence microscopy of histologic sections obtained after the conclusion of the MR imaging sessions. Because we aimed for precise localization, the volume of injectate was purposefully kept very small. Thus the amount of RDA that could be injected was too low for fluorescence-microscopy-based tract-tracing beyond the most robust connections, such as from CA3 to the septum or to the contralateral hippocampus.
Injection sites were determined from the MR images by recording coordinates of the hypointense region in the 0.5h aligned images and plotting the coordinates onto the physical dimensions of our 3D mouse brain template atlas (Medina et al., 2017b), as determined by relative position to bregma defined in Paxinos and Franklin (Paxinos, 2001). Visualization of 3D-rendered injection site coordinates for each mouse were created using AMIRA software (FEI, Hillsboro, OR). The injection site was also identified in individual mice by fluorescence microscopy of the coinjected rhodamine-dextran in histologic sections prepared after sacrifice and fixation (see below).
2.3. Time-lapse MR imaging of axonal transport
Animals were imaged prior to administration of Mn2+ to acquire a pre-injection image and then at 0.5h, 6h and 24h after injection (See Diagram 1). There was no significant difference in timing of the imaging within or between the experimental groups. The midpoint of each 46-minute scan was identified as the “scan time”. MR imaging was performed in an 11.7T 89 mm vertical bore Bruker BioSpin Avance DRX500 scanner (Bruker BioSpin Inc., Billerica, MA) equipped with a Micro2.5 gradient system with a 35 mm linear birdcage radio frequency (RF) coil. During imaging the animal's head was secured in a Teflon stereotaxic unit within the RF coil to minimize movement and to aid in reproducible placement within the column. Temperature and respiration were continuously monitored during data acquisition and maintained within normal ranges. Similar to previous MEMRI studies carried out by this group (Bearer et al., 2007b; Gallagher et al., 2012; Medina et al., 2017a), we employed a 3D multiecho RARE imaging sequence that is both T1- and T2-weighted, with RARE factor of 4 and the following parameters: 4 averages, TR/TE eff = 250 ms/12 ms; matrix size of 160 × 128 × 88; FOV 16 mm × 12.8mm × 8.8 mm; yielding 100 μm isotropic voxels with a 46 minute scan time. We thus capture both the Mn2+ intensity in the T1-weighting and increased anatomical detail with T2-weighting. This imaging protocol produces higher intensity signal from the ventricles than T1-weighting alone (see Supplemental Fig. 1. Some animals were scanned with “flipback ON” (i.e. water resonance returned to B0 at end of each acquisition) which enhanced ventricles but did not change parenchymal signal or Mn2+ enhancement. We cannot rule out that some Mn2+ may leak into the vascular or ventricular systems, but with this small amount injected, dilution into those large compartments would cause the signal to be undetectable and not contribute to our data.
2.4. Histology and immunohistochemistry.
We used histological approaches for four aspects of this study: 1) To confirm that expression of the APPSwInd transgene had produced plaques not present in WT; 2) To identify the locations of the injection; 3) To analyze the cholinergic neurons in a target destination; and 4) To determine the histopathology of the hippocampus. Three weeks after MR imaging, mice were sacrificed, fixed and submitted for histologic analysis (see detailed protocol below). Three weeks were necessary to allow the RDA to reach distal destinations (Bearer et al., 2007b). Additional mice not imaged by MR from our transgenic colonies were also sacrificed, fixed and submitted for histology. Histologic sections of wild-type young and old C57/B6 mice from other studies performed by this lab were also included (Gallagher et al., 2013; Gallagher et al., 2012; Medina et al., 2017a; Zhang et al., 2010), as well as monogenic tTA mice at each age (young, n = 3, and old, n = 6), young WT (nontransgenics), n = 12, young APPSwInd, n = 11; old WT (APP monogenics as well as nontransgenics) n=15; old APPSwInd, n = 8). Subsets of these sections were selected for microscopic examination and analyses as specified in figure legends.
Mice were anesthetized and sacrificed by cardiac perfusion of 30 ml of warm heparinized phosphate buffered saline (PBS), followed by 30 ml of room temperature 4% paraformaldehyde (PFA) in PBS as previously described (Bearer et al., 2007b; Bearer et al., 2009; Gallagher et al., 2013; Gallagher et al., 2012; Tyszka et al., 2006; Zhang et al., 2010). After decapitation, the head is rocked in 4% PFA and PBS overnight at 4 °C and stored in PBS-azide. Prior to processing, each brain is released from the skull, fixed for an additional 1–3 days, and sent to Neuroscience Associates (NeuroScience Associates, Knoxville, TN) for gelatin embedding and serial sectioning in register at 35 μm thickness. Typically up to 25 brains are embedded together in a single block and sectioned coronally in register. Sections are collected in 24 sequential cups such that each cup contains serial sections across the whole brain at 840 μm intervals. To image the injection site, individual sections through the predicted location of the injection site are mounted unstained in Dapi-containing antiquench (Vector Lab, Burlingame, CA) and imaged for Dapi and rhodamine by fluorescence microscopy. Because of the tiny volume injected (3–5 nL), only a very small amount of RDA can be delivered without clogging the micropipette. This amount is not sufficient to trace tracts by fluorescent microscopy beyond the most robustly transported regions. Hence we only examined sections for RDA through the medial septum and the contrlateral hippocampus, known projections of CA3 of the hippocampus. The presence of RDA in these areas provides further confirmation that the injection site is well-placed in CA3. Since the injection sites aggregated in our MR analysis, we only checked a subset of animals for RDA injection site. For histochemistry and immunochemistry, alternate cups containing serial sections were stained in a variety of ways: Thionine-Nissl for microscopic anatomy, Campbell Switzer silver stain (Switzer, 2000) for plaques and tangles, or immunohistochemistry. Cholinergic neurons were stained with anti-ChAT antibodies and phosphotau with the AT8 monoclonal antibody (NeuroScience Associates, Knoxville, TN) (Bearer et al., 2007b). A subset of stained sections for each marker were analyzed by sterology (see below).
Microscopy of brain sections was performed using a Zeiss V8 stereoscope (equipped with an AxioCam) running AxioVision 4.8 for low magnification, and a Zeiss Axioscope Z1 with a MRM and HRC Axiocam digital cameras running AxioVision 4.6 software. For histochemical stains, images were captured on a Nikon Eclipse Ci brightfield clinical microscope with a DS L3 cooled digital camera. Digital images were cropped and prepared for publication using Adobe Photoshop (Adobe Systems Incorporated, San Jose, CA). The position of images relative to bregma are according to the C57Bl/6J atlas: http://www.mbl.org/atlas170/atlas170_frame.html and Paxinos and Frankin (Paxinos, 2001).
2.5. Western blots and dot blots
Mouse brains were harvested within 90 sec of sacrifice and rapidly frozen by plunging into liquid N2. Brain tissue was harvested from each frozen brain individually, weighed, and 10–20 mg thawed and extracted according to established protocols (Bhaskar et al., 2010). Briefly, 10 mg of brain are first homogenized in ice-cold Tissue-Protein Extraction Reagent (T-PER, 78510; Pierce, now Thermo Fisher Scientific, Waltham, MA) with protease (P8340; Sigma-Aldrich) and phosphatase (P5726; Sigma-Aldrich) inhibitor cocktails with a Dounce homogenizer for 1 minute, and then sonicated in a Bronson sonifier set at 20% for 20 sec on ice. After microfuging at 14,000 rpm in an Eppendorf microfuge for 15 min, the supernatant and pellet are separated and stored at - 80o C. For SDS protein gels, 20–50 μg of protein-containing extract (supernatant) is boiled in Laemmle gel sample buffer and run on a 4–20% gradient gel (Thermo Fisher Scientific, Waltham, MA) (Seamster et al., 2012). Proteins were transferred to nitrocellulose in a Hoeffer tank transfer device overnight in transfer buffer (Seamster et al., 2012). For dot blots, 3 μl of brain extract was dotted onto nitrocellulose allowed to try for 20 min, washed in blocking buffer (Seamster et al., 2012) for 20 min, and then probed with primary anti-Aβ monoclonal antibody MOAB (#MABN254, Millipore, Burlington, MA) at 1:1000; or for SOD1 with polyclonal anti-SOD1, 1:1000, (#AB5482, Millipore, Burlington, MA). HRP-conjugated secondary antibodies were goat anti-mouse (#AP308P, Millipore, Burlington, MA) and goat anti-rabbit (#AP307P, Millipore, Burlington, MA) diluted at 1:1000 in blocking buffer. Western blots were probed with the SOD1 antibodies and 6E10 anti-APP monoclonal antibody #SIG-39320 at 1:1000 (Covance, Brea, CA). To compare mouse and human APP expression, Western blots were stained in parallel with monoclonal 22C10, a polyclonal rabbit antibody raised against the conserved C-terminus of APP with pan-species reactivity at 1:1000 (#MAB348. Millipore, Burlngton, MA), and 6E10, a monoclonal against the non-conserved Nterminus of human APP which is non-reactive with mouse APP, at 1:1000 (Covance, Brea, CA). Stained blots were imaged with ECL Clarity (BioRad #170–5060, Hercules, CA) on BioRad ChemiDoc XR+ using the Magic Mark ladder (Invitrogen #LC5602, Carlsbad, CA) and Kaleidoscope ladder (BioRad #161–0375, Hercules, CA). Intensity of bands was calculated using ImageJ (https://imagej.nih.gov/ij/).
2.6. MR image analysis
We capture images from living animals, which thus include non-brain structures. To perform unbiased voxel-wise statistical analysis, the brain images must be aligned. A first step is to extract the brain image from non-brain structures. Next, all extracted brain images in the datasets must be the same type, resolution, dimension, intensity scale and then the anatomy aligned. Our protocols for brain extraction, "skull-stripping" (Delora et al., 2016), and alignments are published elsewhere (Medina et al., 2017b). Briefly for this study, 3D MR images were converted to 32-bit FLOAT format. Images were cropped to equal image dimensions of 158 × 116 × 78 voxels using the Medical Image Processing, Analysis and Visualization (MIPAV) program, developed by NIH (McAuliffe et al., 2001) (https://mipav.cit.nih.gov). Header file geometry is uniformly copied across all images for all mouse groups and time points using FSL (FMRIB software library) (Analysis Group, Oxford, UK) (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/) (Jenkinson et al., 2012; Smith et al., 2004; Woolrich et al., 2009). Images are aligned by rigid body transformation (6 degrees of freedom), to our anatomical MR 3D mouse template brain atlas (Medina et al., 2017b) using NiftyReg (Modat et al., 2010) from the Translational Imaging Group, University College London, (http://cmictig.cs.ucl.ac.uk/wiki/index.php/NiftyReg), and corrected for B1 inhomogeneity using MIPAV. The mode of the intensity histogram is then scaled to be equivalent across all images using MATLAB (MathWorks, Natick, MA) script developed by the Jacobs lab at the California Institute of Technology, the code is posted as supplemental materials for Medina et al 2017 (Medina et al., 2017b). This sets the mode of the intensity histogram to 1 for all images. A template mask is generated using our mouse brain atlas (Mackenzie-Graham et al., 2007) as template and the Brain Surface Extractor within BrainSuite11 with manual refinement (Shattuck and Leahy, 2001). The rest of the images are stripped of extra-meningeal head structures using our automation program (Delora et al., 2016). After skull-stripping, all images are registered to our mouse brain atlas (Medina et al., 2017a) using the Oxford Centre for Functional MRI of the Brain’s (FMRIB) Linear Image Registration Tool (FLIRT) (FMRIB, Oxford, UK) (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/) (Jenkinson et al., 2012; Smith et al., 2004; Woolrich et al., 2009), and then again corrected for field inhomogeneity. The modes of the grayscale intensity histogram of the extracted brain images are again re-scaled across all images (Medina et al., 2017b). Resultant aligned images are smoothed with full-width half-maximum (FWHM) Gaussian kernel (0.3 mm) for statistical processing, using statistical parametric mapping software (SPM) (UCL, London, UK) (http://www.fil.ion.ucl.ac.uk/spm/doc/) (Ashburner, 2012; Penny et al., 2006). For this project, group-specific averaged images were created using FSL (Analysis Group, Oxford, UK) (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/) (Jenkinson et al., 2012; Smith et al., 2004; Woolrich et al., 2009) to enable visual assessment of signal enhancement due to Mn2+ presence and transport following intra-hippocampal Mn2+ injection. To align post-injection images, a warp-field (control point grid) was created for each mouse by aligning its pre-injection image to the atlas. The resultant warp field is then applied to the post-injection images (Medina et al., 2017a; Medina et al., 2017b). This two-step alignment for post-injection images is necessary since the high intensity of the Mn2+ in the injection site interferes with intensity-based alignment processing.
To focus on Mn2+-enhancement at successive time points as a measure of its transport from the injection site, we performed statistical parametric mapping (SPM) analysis (http://www.fil.ion.ucl.ac.uk/spm/doc/biblio/) (Ashburner, 2012; Friston, 2005; Penny et al., 2006), following a similar computational paradigm as in our previous publications (Bearer et al., 2007b; Bearer et al., 2009; Gallagher et al., 2012; Medina et al., 2017a; Zhang et al., 2010). Within group between time points after injection were compared using pairwise t-tests (6h < 0.5h, and 24h > 0.5h) with an FDR corrected p value of 0.01 in SPM to obtain an unbaised, whole brain, voxel-wise map of transport dynamics.
2.7. Segmentation and region of interest analysis
The hippocampus and septal regions were segmented with reference to the Waxholm space atlas (http://www.civm.duhs.duke.edu/neuro201001/) (Badea et al., 2007; Johnson et al., 2010). This atlas was created from MR images of 14 different mice captured with three different pulse sequences and annotated to Nissl-stained histologic sections with reference to the Paxinos and Franklin mouse brain atlas (Paxinos, 2001), the expression atlas (Hawrylycz et al 2011; Cell Centered Database (CCDB, http://www.ccdb.ucsd.edu/) and the Mouse Brain Reference Atlas from Allen Institute for Brain Science (http://mouse.brain-map.org/static/atlas).
We created masks for contralateral hippocampus and septal nuclei using the Waxholm space scalable brain atlas and its associated anatomical label map (Badea et al., 2007; Bakker et al., 2015; Johnson et al., 2010; Lein et al., 2007). Because this atlas is based on MRI of mouse brain rather than histologic sections, its grayscale allows us to register it with our MR images. To generate the masks, the Waxholm space atlas was first rigid body aligned using NiftyReg, and then affine linear transformed (12 degrees of freedom) using FLIRT (FMRIB, Oxford, UK), onto our mouse brain template, to which all the unsmoothed images had previously been aligned (see above). Using the same control point grid generated through these transformations, the anatomical label map was registered to our mouse brain template image. Anatomical masks for the contralateral hippocampus and septal nuclei were generated using the newly aligned annotated atlas and refined with the Brain Surface Extractor tool within BrainSuite (Shattuck and Leahy, 2001). The segmented anatomical regions were used to measure average intensity and to calculate the volume occupied by statistically significant voxels within the segmented region of each image. Segmentation also assisted in the selection of sites for region of interest analyses (see below).
Subregions, 0.5 mm3 (5 × 5 × 5 voxels), within the segmented structures (medial septal nucleus, lateral septal nucleus, and cortex) were selected based on the SPM signal in one or the other cohort and propogated across all aligned images in both cohorts. Intensity values were measured for each animal at each time point after injection using FSL in the same anatomical region as specified by coordinates across all aligned images (Analysis Group, Oxford, UK). Mean intensity from a cortical ROI in each animal, selected based on identical anatomical location and lack of statistically significant voxel intensity change, was used to background-correct other ROI regions (mean intensity/background mean intensity) and all background corrected intensities were normalized by setting the intensity at 0.5h post-injection image equal to 1 and applying that adjustment to the other time points, as previously described (Medina et al., 2017a). Statistical analyses of the ROIs were performed in GraphPad Prism (GraphPad Software Inc., La Jolla, CA) with a two-way, withinsubjects ANOVA, and confirmed with Tukey’s post-hoc test. 3D segmented regions, SPM maps within these regions, and our mouse brain template atlas were loaded together into Amira and superimposed to create a 3D image and generate a video.
2.8. Stereology of ChAT neuron counts and morphology of the dentate gyrus
The medial septal nuclei (MSN) in histologic sections (1–3 sections per mouse) was segmented based on classical landmarks (Allen Mouse Brain Reference Atlas) (Lein et al., 2007) and Paxinos and Franklin (Paxinos, 2001). ChAT-positive cells identified by ChAT immunostaining were counted using the Stereo Investigator software (Microbrightfield Stero Investigator Software, Williston, VT) for design-based stereology. We used the fractionator probe (Microbrightfield Stero Investigator Software, Williston, VT) to count all ChAT-stained cells in the MSN within a contour, with the anterior commisure as landmark. Cells were only included if the entire cell body was inside the contour. Both the number of cholinergic neurons counted and the density of cells were averaged for each group. The density of the ChAT-staining was determined based on the average number of neurons counted divided by the average area occupied by the counted cells. Results were grouped according to genotype and age and analyzed with GraphPad Prism (GraphPad Software Inc., La Jolla, CA) using a one-way ANOVA and Tukey’s post-hoc tests.
For the dentate gyrus, we measured the width of the granule cell layer rather than counting cells because the cells overlapped in our 35 μm thick sections and could not be reliably distinguished from each other and thus counted. Widths of the granule cell layer (GCL) of the dentate gyrus were estimated using Nissl-stained sections in the dorsal region of the hippocampus. Locations of measurements were determined using the orthogonal fractionator probe (Microbrightfield Stero Investigator Software, Williston, VT) and taken orthogonally to the midline of the GCL on a 50 μm × 50 μm grid. Edges were determined based on visual inspection and density of Nissl-stained cell bodies. Also see Supplemental Fig. 2.
Results were grouped into 4 cohorts according to age and genotype, and analyzed with GraphPad Prism (GraphPad Software Inc., La Jolla, CA) using a one-way ANOVA, and Tukey’s post-hoc tests. All stereological protocols were performed on an Olympus DSU inverted microscope at the University of New Mexico Fluorescence Microscopy Shared Resource Center.
3. Results
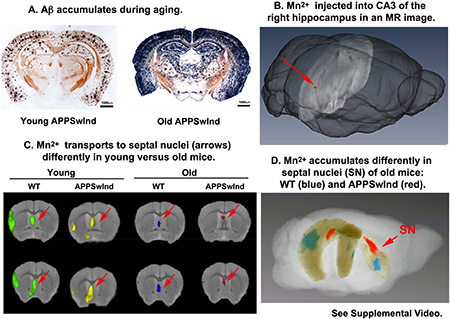
3.1. . Expression of APPSwInd in mice produces Aβ deposition that increases with aging
Both young (6 months old) and old (13–17 months old) APPSwInd mice had silver-stained AD pathology throughout the cortex, hippocampus, and lateral septal regions of the brain, with the degree of staining and the density of neuritic plaques increasing with age (Fig. 1A and C), as previously reported (Jankowsky et al., 2005). The Campbell-Switzer silver stain, black in the histologic sections, detects both Aβ and p-tau aggregates. Staining was relatively much lighter in young mice (Fig. 1A) compared to older animals (Fig 1C). As expected age-matched WT littermates, whether monogenic or non-transgenic, had no black silver staining (Fig. 1B and D). Inspection of silver-stained sections through the septal nuclei (Fig, 1E-H) demonstrated that young APPSwInd mice had few plaques (3–4 on average) (Fig. 1E) compared to older animals (Fig. 1H). In older mice, silver staining was heaviest in the lateral septal area, with apparent sparing of the medial septal nuclei (Fig. 1G). Monogenic and non-transgenic littermates had no staining, regardless of age (Fig. 1F and H). At higher magnification, silver-stained plaques in young APPSwInd mice appeared more loosely aggregated, diffuse and smaller than those in the older APPSwInd mice (Fig, 1I and J). Also of note is the silver-stained material not in defined plaques, suggesting that abnormal
Figure 1. Analysis of plaques and Aβ/APPSwInd expression in transgenic and WT mouse brains.

(A-D) Examples of Campbell-Switzer silver stains of coronal brain sections (×2.2 mm posterior to bregma) from (A) 6 month-old APPSwInd; (B) 6 month-old WT age-matched littermate; (C) 13 month-old APPSwInd; (D) an age-matched old WT littermate. Brains were embedded in the same block, serially sectioned and sections containing many brains stained together. Note that the young APPSwInd has less staining for plaques than the old APPSwInd, and that WT have no black silver staining regardless of age.
(E-H) Examples of Campbell-Switzer silver-stained coronal sections through the septal nuclei (+0.4 mm anterior to bregma) from young APPSwInd (E) and WT (F) (6 month-old); and old APPSwInd (G) and age-matched old WT littermate (H) (13 month-old). Note few plaques in the septum of young APPSwInd (E, example indicated by arrowhead) and a lot of silver staining in the lateral septum of the old APPSwInd, with both discrete plaque formation and diffuse staining (G). No black silver staining in WT at either age (F and H).
(I-J) Examples of cortical plaques such as those within the boxed regions in (A and C): (I) a young APPSwInd mouse and (J) an old APPSwInd mouse. Note the differences in size and central brown staining of the plaques between the age groups.
(K) Examples of dot blots probed for Aβ and Western blots probed for human APP (hAPP) and SOD1 (a loading control). Shown are results for six young (3 each WT and APPSwInd) and 5 old (3 WT littermates and 2 APPSwInd) mice. Note the difference in Aβ between the young and old APPSwInd which compares to the plaque burden between these age groups in (A) and (B). The WT mice display no plaques and do not express hAPP. Faint bands detected with the anti-hAPP antibody in WT mice likely represent weak cross-reactivity to endogenous mouse APP. Quantitative comparisons of blots for human APP with those for pan-APP demonstrated a 3.6 fold increase in total APP in the APPSwInd compared to WT (not shown). Also note the increased size of the Aβ-stained dot in the older mice, consistent with the increased silver staining in the histologic analysis.
Aβ fibrils aggregate throughout the brain. Western blots and dot blots for human APP (hAPP) and Aβ, respectively, confirmed high protein levels for both in brain extracts derived from APPSwInd compared to age-matched WT controls (Fig. 1K; faint bands in the WT mouse lanes are likely due to weak cross-reactivity with mouse APP). The dot blot for Aβ is larger in the older mice relative to a SOD1 loading control, indicating a higher amount of Aβ in the older brains analogous to the darker silver staining in older mice and consistent with a slow accumulation of Aβ during aging. Comparison of Western blots stained for the pan-specific 22C10 monoclonal and for the human specific 6E10 monoclonal demonstrated an approximate 3.6 fold increase of total APP in the APPSwInd mice (not shown).
3.2. Placement of the injection site
To map functional transport circuitry from the hippocampus to the contralateral hippocampus and basal forebrain, we co-injected the tract-tracer rhodamine-dextran (RDA) together with the Mn2+ into CA3 of the right hippocampus. RDA is a traditional tract tracer that can be imaged by fluorescence microscopy, which allows precise identification of the injection site in histologic sections together with microscopic anatomy (Bearer et al., 2007b; Bearer et al., 2009). Thus, by post-mortem fluorescence microscopy of mice after imaging by MR, we obtain detailed histology of the injection site at higher resolution than afforded by MRI, and thus can determine placement and detect any injury (Bearer et al., 2007b; Gallagher et al., 2012; Medina et al., 2017a). In addition, RDA transport to expected locations confirms that the injection site was placed appropriately. The volume of the injectate (3–5 nL at a concentration of 0.5 mg/ml) is not sufficient to deliver enough RDA for tracing of tracts other than the most robust fibers. In this study, fluorescence imaging of the RDA showed that the injection site was located in CA3 of the hippocampus as intended, with no apparent injury (Fig. 2A, white arrow), and that RDA is transported to both the contralateral hippocampus (Fig. 2B) and to the septal nuclei of the basal forebrain (Fig. 2C). Quantifying differences in fluorescence intensities has proven challenging, especially at this low level of signal. Hence quantification of transport dynamics and distal accumulation was done by statistical analysis of time-lapse MR images obtained from multiple individuals, as described below.
Figure 2. Rhodamine-dextran (RDA) co-injection identifies the injection site, and demonstrates introduction into expected circuits: Contralateral hippocampus and septal nuclei.

(A) Example of a fluorescent image of a coronal section at ~4.1 mm posterior to bregma showing co-injected RDA in CA3 of the right hippocampus, the expected injection site (arrow).
(B) Example of the contralateral left hippocampus showing rhodamine fluorescence (red) from the RDA in CA3, an expected projection from injection site counterstained with DAPI (blue) for nuclei in a coronal section (~2.5 mm posterior to bregma). Little fluorescence is present in the dentate gyrus (DG).
(C) An example of a section through the septal nuclei (~0.4 mm anterior of bregma), showing strong rhodamine fluorescence in cell bodies of the medial septal nuclei (MSN) and some signal in the lateral septal region (LSr). Inset shows higher magnification of red fluorescing cells in the MSN with rhodamine fluorescent puncta surrounding the DAPI-stained blue nuclei, as expected for transported dextran.
Review of the hypo-intense injection site in MR images captured immediately after injection allows us to compare the position of each across all living animals (Fig. 3A, 3B and 3C) (Gallagher et al., 2012). The average injection sites clustered in CA3 of the right hippocampus (see Table 2). Distance from the centroid of all injections was 0.9 mm or less in all three dimensions. The mean location varied 0.12 mm lateral to midline, 0.14 mm antero-posterior and 0.56 mm deep. The largest standard deviation from the mean was 0.66 mm due to one animal in one of the cohorts. Analysis with and without this outlier made no difference to the SPM maps.
Figure 3. Locations of injection sites.

(A) Sagittal, (B) coronal, (C) axial and (D) axial/coronal tilted projections of 3D images with injection sites indicated. Mouse template brain atlas (grayscale brain image), young WT (green), young APPSwInd (yellow), old WT (blue) and old APPSwInd (red). In (D) note that all injections fall within the hippocampus, as visualized through this semi-transparent, rendering of the highresolution 3D mouse brain template atlas. All injections fell within 0.38 × 0.53 × 0.9 mm3 of the centroid (see Table 2).
Table 2.
Injection site location
| Cohort | Age (mo) | Injection sites measured in MR image 0.5h post-injection | ||
|---|---|---|---|---|
| X (L-R) | Y (front-back) | Z (deep) | ||
| WT | 6 | 3.10 ± 0.30 | −4.44 ± 0.35 | 2.83 ± 0.44 |
| APPSwInd | 6 | 2.84 ± 0.37 | −4.81 ± 0.21 | 2.41 ± 0.40 |
| WT | 13–15 | 3.10 ± 0.31 | −4.28 ± 0.45 | 2.75 ± 0.66 |
| APPSwInd | 13–15 | 3.22 ± 0.44 | −4.33 ± 0.45 | 3.13 ± 0.53 |
| Distance from Centroid (mm) | 0.38 | 0.53 | 0.9 | |
Planned injection site × = −3.2 mm (midline), y = −4.1 mm (bregma), z = 3.4 mm (down)
3.3. . Old wild-type and old APPSwInd mice display altered circuitry when compared to young wildtype mice of either genotype
To assess differences in transport dynamics and circuitry between young and old mice with and without the APPSwInd overexpression, we first aligned all images for each cohort captured at three time points after injection into the same 3D atlas. Comparison of raw images from each cohort and its group average demonstrated preservation of anatomical detail (Fig. 4A, also see Supplemental Fig. 1). As expected for this multiecho pulse sequence, the CSF in the ventricles is brighter than un-enhanced parenchyma in pre-Mn2+-injected images (Supplemental Fig. 1) and ventricles remain similarly bright after Mn2+ injection throughout the time series. The hyperintensity from the Mn2+ can be seen in the injection site in the hippocampus and perhaps in the septal nuclei at later time points (Fig. 4A).
Figure 4. Functional circuitry is altered in old mice and in APPSwInd mice.

(A) Representative examples of individual raw images for young WT, young APPSwInd, old WT and old APPSwInd mice (top four images), and within-group averaged images from young WT, young APPSwInd, old WT and old APPSwInd mice (bottom four images). Images are from the 24h post-injection time point. White arrows, injection site; red arrowheads, septum. Also see Supplemental Figure 1.
(B) Statistical parametric maps generated by automated, unbiased, voxel-wise whole brain pairwise within group t-tests are projected onto a grayscale image of our 3D mouse brain template atlas. Selected slices from 3D images show statistically significant Mn2+-enhanced intensity at 24h compared to 0.5h for each genotype in coronal (left panel), sagittal and axial slices (right panel) as indicated: young WT (green), young APPSwInd (yellow), old WT (blue), and old APPSwInd (red). Injection site is on the mouse's right, left side of the images. Arrowheads indicate contralateral hippocampus. Arrows point to septal regions. Viewing planes of these slices are indicated by blue lines on the 3D mouse brain template atlas shown in the bottom left panel. Note the robust transport in the young mice, and the altered location of the most significant voxels in the old WT and old APPSwInd, p<0.01 FDR corr.
Voxel-wise statistical parametric maps comparing Mn2+-induced intensities between images captured at 24h with those captured at 0.5h post-injection by a pair-wise Students t-test (p<0.01 FDR corr.) revealed that young mice of either genotype had more statistically significant intensity increases, i.e. Mn2+ accumulation, in distal projections (the contralateral hippocampus and the septal nuclei) than old mice at 24h post-injection (Fig. 4B). Furthermore in old mice, the distal location of voxels with significantly increased intensity differed between genotypes--while old WT mice had Mn2+-induced increased intensity in the medial septal nuclei (MSN), old age-matched APPSwInd littermates had significantly increased intensity predominantly in the lateral septal region (LSr). Only the expected circuitry from hippocampus to forebrain was statistically enhanced by SPM at 24h>0.5h--no generalized enhancement was detected of parenchyma or ventricles.
To determine the dynamics of accumulation in the septum after injection, we compared statistical maps of images captured at 6h after injection with immediate post-injection images captured at 0.5h; images captured at 24h after injection with images at 6h; and 24h images with 0.5h images using pair-wise, within group t-tests (p<0.01 FDR corr.) (Fig. 5). These comparisons reveal differences in dynamics and location of accumulation between young WT (green) and young APPSwInd (yellow) that presage larger differences between old WT (blue) and old APPSwInd (red). In young APPSwInd, Mn2+ makes less progress at 6h after injection and at 24h compared to 6h, and accumulation in the septum overall is patchy at 24h compared to 0.5h, as compared to WT. In the old mice, less intensity appears in the septal region and the anatomical location of accumulation differs between genotypes.
Figure 5. The voxels with the most significant intensity changes differ in anatomic position in young and old WT and APPSwInd mice.

Sagittal slices from 3D brain images showing voxels with significantly increased intensity changes between time points after Mn2+ injection in each group are overlaid on a grayscale image of the same slice from our 3D template atlas: young WT (green), young APPSwInd (yellow), old WT (blue), old APPSwInd (red). In the last panel on the right, old WT (blue) and old APPSwInd (red) are overlaid together on the same grayscale slice to show anatomic relationships of most-significant voxels in the two datasets. Slices are taken at 0.6 mm right of midline. Statistical parametric maps were generated using pairwise within-group between time points Student's t-test, with intensity at 6h greater than 0.5h (top row), 24h greater than 6h (middle row) and 24h greater than 0.5h (bottom row) p<0.01 FDR corr. Note differences in the anatomical positions of the Mn2+-enhanced voxels, particularly obvious in the double overlay of the two old mouse genotypes (far right column). Also see Supplemental Video.
For direct anatomical comparisons between the two old genotypes, we projected maps for both old mice cohorts onto the same grayscale atlas (Fig. 5, last column on the right). Visual inspection of these images revealed that the anatomical areas with significantly increased intensity differed between these two old genotypes at each time point. Differences between genotypes were detected regardless of the statistical approach used to generate the maps. These observations suggest that aging decreases transport, and that the increasing presence of plaques further delays transport and changes the ultimate destination of transported Mn2+.
3.4. Segmentation and region of interest (ROI) analysis reveals differences in Mn2+ accumulation between groups
We segmented the hippocampus and the septum, generated a mask so that only those regions would be visible, and projected the masked segmented region together with masked statistical maps (24h > 0.5h) for each old genotype onto a whole brain grayscale image of our atlas (Fig 6; also see Supplemental video). Selected still-frames from the 3D video show the transparent whole brain image with segmentation of hippocampus and septal region (yellow) and statistically significant voxels for old WT (blue) and old APPSwInd (red). Note the difference in locations of significantly enhanced voxels between old WT and old APPSwInd. We calculated the volume occupied by statistically significant voxels (p<0.01 FDR) at 24h>0.5h in the contralateral hippocampus (old WT, 0.51 mm3; Old APPSwInd, 0.04 mm3) and septal nuclei (old WT, 1.1 mm3; old APPSwInd, 0.27mm3). Thus the volume of accumulation of transported Mn2+ was greatly reduced by expression of APPSwInd and the presence of amyloid plaques as compared to old wild-type littermates.
Figure 6. 3D rendering of the segmented hippocampal and septal nuclei with overlay of statistically enhanced voxels in old WT and old APPSwInd brains.

Shown are still frames from a counter-clockwise horizontal rotation of a 3D rendering of the mouse brain template segmented for both hippocampus and septal nuclei (yellow), with statistical maps of significantly enhanced voxels in old WT (blue) and old APPSwInd (red) at 24h greater than 0.5h as in Fig. 4 and 5 (p<0.01 FDR corr.). Brain is rotating counter-clockwise in the horizontal plane. At 90o the nose is pointing to the back and at 270o towards the viewer. The injection site is in the right hippocampus. Note the difference between the two genotypes in the 3D anatomy of the Mn2+ - induced intensity changes. See Supplemental video.
We compared intensity changes between groups in two ways: 1) by measuring the average intensity within the segmented contralateral hippocampus and the septal nuclei; and 2) by region of interest (ROI) analysis within segmented regions (Fig. 7). These analyses allowed us to quantify the degree of intensity change and to compare between groups, which is different from statistical mapping, which detects the significance of change between time points within each group. We selected 0.5 mm3 ROI within the LSr highlighted in the old APPSwInd SPM for one measurement, and the MSN highlighted in the old WT SPM for a second measurement. Each selected region was propagated across all aligned images in both datasets to ensure that the same anatomical region would be measured in all animals. Intensities were background-corrected by calculating ratio of intensity in the ROI divided by the intensity measurement in the cortex, a region not expected to be enhanced by Mn2+ transport, for each image (Fig. 7C, red box). This step was designed to correct for any differences in generalized parenchymal intensity, which could have happened if appreciable Mn2+ had leaked into blood or CSF/ventricles. No significant difference between time points in cortical parenchyma or ventricular intensity was found by the SPM analyses shown above, nor in these cortical measurements used for background correction.
Figure 7. Quantification of Mn2+-induced intensity changes over time after injection in the contralateral hippocampus and septal nuclei of old WT and old APPSwInd.

(A-B) Shown are anatomical segmentations of the right hippocampus, (A, green), the left hippocampus (A, yellow) and the septal nuclei (B, yellow) projected onto a semi-transparent grayscale rendering of our 3D mouse brain template. Red boxes in (B) indicate the locations of the 5 × 5 × 5 voxel cubes (0.5 mm3) selected for region of interest (ROI) intensity measurements in the MSN and LSr. (C) shows a 3D rendering of the mouse template in the axial orientation. The red box indicates the position of the 0.5 mm3 ROI used for measurements in the cortex which had stable intensity over time as it is not an expected destination in this circuit, and thus useful for background correction. Locations of ROIs were chosen based on SPM maps and propagated through all aligned images such that the same anatomical region was measured across all animals and time points. Measurements are made at successive time points after injection (0.5, 6 and 24h).
(D-F) Graphs showing changes in mean intensity over time after Mn2+ injection for indicated regions at each imaging time point for each cohort, with old WT (blue lines) and old APPSwInd (red lines). Error bars represent standard error of the mean. All measurements were back-ground corrected and normalized. Statistics represent a two-way ANOVA with repeated measures for time (group × time; F(4,50) = 2.658, p≤0.04), with Tukey’s post-hoc test (* p≤0.5; ** p≤0.01). (D) Intensity changes in the segmented contralateral hippocampus. (E) Mean intensity changes in the ROI in the medial septal region (MSN, red box in B).
(F) Mean intensities in the lateral septal region (LSr, red box in B).
Comparisons of average intensities within either the segmented contralateral hippocampus (Fig. 7 A and D) or the septal nuclei (not shown) found no significant difference between genotypes. In contrast ROI analyses of 0.5 mm3 regions within the MSN and LSr, selected based on statistically significant voxels from the SPMs, demonstrated significant differences between genotypes (Fig. 7 B, E and F). In the MSN intensity change was significantly greater in the old WT compared to the old APPSwInd (Fig. 7E). And old APPSwInd mice had larger increases in Mn2+-induced intensity in their LSr at 6h post-injection than the old WT mice (Fig. 7F). These differences are statistically significant using two-way ANOVA with repeated measures for time (F(4,50) = 2.658, p < 0.04), with Tukey’s post-hoc test (* p ≤ 0.5; ** p ≤ 0.01). Thus, Mn2+ transport to the septum is abnormal in old mice, accumulating in the LSr of old APPSwInd plaque-burdened mice.
3.5. . Histologic examination of the septal region
To determine whether this differential transport was due to death of cholinergic neurons in the MSN, coronal brain sections through the MSN were stained by immuno-histochemistry for choline acetyl transferase (ChAT), a marker of cholinergic neurons, (Fig. 8A). Using unbiased stereological methods, we determined the effect of age and genotype on the number of ChATpositive neurons in the MSN, and calculated the density of ChAT neurons therein. The total number of ChAT-positive cells was not significantly different between the four groups of mice (Fig. 8B). Old WT mice have statistically significant larger MSN areas averaged over serial sections than other mice in our cohort (Fig. 8C). However, interestingly, while young and old WT mice have no significant difference in the number of ChAT-positive neurons, APPSwInd mice have a significantly higher cell density, probably because the same number of neurons is crowded into a smaller space (Fig. 8D). Thus, loss of ChAT neurons cannot explain the altered transport/accumulation of Mn2+ in old APPSwInd mice -- although loss of some other component in the MSN causing reduced volume could contribute to decreased accumulation in old APPSwInd.
Figure 8. Cholinergic neuron differences in the MSN.

(A) Representative histological section through the MSN stained for choline-acetyl transferase (ChAT), with yellow outline indicating the region used for counts (0.38 mm anterior to bregma).
(B) Counts of ChAT-stained neurons in the MSN, 90.3–0.4 anterior to bregma, of young WT (4–8 months old, n = 15), young APPSwInd (5–6 months old, n=11), old WT (1013–17 months old, n = 7) and old APPSwInd (1317 months old, n = 8) mice. Cross-hatch pattern indicates APPSwInd expressors. No significant difference was found between cohorts.
(C) The area of the MSN is significantly diminished in APPSwInd compared to young and old WT. (** p≤0.006; *** p≤0.001).
(D) ChAT neuron density calculated from the numbers of neurons as in (B) divided by the area of the MSN as in (C) demonstrates significantly increased density of ChAT neurons in the APPSwInd mice compared to young or old WT. * p≤0.04, *** p≤0.0001, **** p≤0.0001. Statistics represent mixed model ANOVA, with Tukey’s post-hoc test. Error bars are represented as SEM.
3.6. . Histologic examination of the hippocampus
To determine whether altered transport in the older mice with and without APPSwInd expression is due to anatomical changes in the hippocampus, we also examined the hippocampus of the same mice imaged by MR in histologic sections. We were particularly interested in the dentate gyrus as it is diminished in tTA-expressing mice with a mixed strain background, but not in congenic C57/B6 as used in our experiments (Han et al., 2012). To confirm tTA expression alone did not affect the microscopic anatomy of the hippocampus in the congenic C57/B6 tTA monogenic mice studied here, we fixed, embedded, sectioned and stained brains of six cohorts of mice: young and old wild-type, young and old tTA monogenics (a sub-type of wild-type that does not express APPSwInd), and young and old APPSwInd after MR imaging and reviewed hippocampal morphology in Nissl-stained coronal sections (Fig. 9A - F). We found no effect of tTA expression on hippocampus in our C57/B6 congenic tTA monogenic mice at either age by histology (Fig. 9B & E). However old APPSwInd expressing mice had thinner dentate gyri obvious in microscopic images (Fig. 9C & F). To quantitate this visual impression, we performed stereological measurements of the thickness of the granule cell layer in the hippocampal dentate gyrus between four groups of mice: young WT, young APPSwInd, old monogenic tTA and old APPSwInd (Fig. 9G and Supplemental Fig. 2). We found no significant difference in the average thickness of the dentate gyrus granule cell layer between young WT, young APPSwInd and old tTA monogenic mice, while old APPSwInd have significantly decreased width compared to all other cohorts, young or old. Thus this effect is apparently not due to tTA expression and appears to be dependent on both age and APPSwInd genotype (Fig. 9G), although a synergistic effect on the dentate gyrus by co-expression of both APPSwInd and tTA cannot be ruled out.
Figure 9. Hippocampal histology.

A-F: Representative sections through the hippocampus stained with Thionine-Nissl from six cohorts of mice: (A) young WT; (B) young monogenic tTA; (C) old APPSwInd expressors; (D) old WT; (E) old monogenic tTA; (F) old APPSwInd expressors. DG: Dentate gyrus; CA3: Cornu Ammonis field 3. Mag. bar lower right, 200 μm.
(G) Quantification and statistical analysis of granule cell layer thickness of the dentate gyrus in young WT with no transgene (n = 7), young APPSwInd (n = 6), old tTA monogenic (n=6) and old APPSwInd expressing (n = 5) mice. No statistical difference between the dentate thickness in old tTA monogenics and young WT mice was found. ANOVA between group comparisons with Tukey’s post-hoc testing revealed a significant difference between old APPSwInd and all three other groups, including the old tTA (**** p≤0.0001). Error bars represent standard error of the mean.
(H-K) Representative sections through the hippocampus of young (H and J) and old (I and K) APPSwInd mice showing plaques and dystrophic neurites by silver stain (H and I) and parallel sections stained for phospho-tau with AT8 antibody (J-K). Red arrow: CA1, Cornu Ammonis field 1; CA 3, Cornu Ammonis field 3. Mag bar lower right, 200 μm.
(L, M) Boxed regions in J and K shown at higher magnification (~6×).
We next examined the plaques in the hippocampus by silver stain and probed for phosphotau (p-tau) by immunohistochemistry. As previously reported and shown in Fig. 1, in young APPSwInd mice silver-stained plaques are few, whereas in older APPSwInd mice plaques are so abundant as to become almost confluent (Fig. 9H and I). In addition to plaque, silver-stained material appears throughout the interstitium in both young and old APPSwInd age groups, albeit much more heavily in the older animals. This material likely represents Aβ aggregates and fibrils. The plaques in young mice are fewer (3–5 plaques per hippocampal section in young mice, versus 10–15 in the old APPSwInd mice (n = 6 per group)). Silver staining lines the basement membrane of the dentate gyrus in both young and old APPSwInd mice, with heavier deposition in the older mice on both sides of the granule-cell layer and along CA1, with less deposition in CA3, the site of our injection.
Next we examined young and old APPSwInd mice for phosphorylated tau (p-tau), since p-tau produces aggregates that also stain for silver by the Campbell-Switzer procedure (Switzer, 2000). Although phosphorylation of mouse tau is not commonly described in mice expressing FADassociated APP mutations, dystrophic neurites containing mouse p-tau were present in Aβ plaques in two lines of mice overexpressing the Swedish mutation of human APP (Sturchler-Pierrat et al., 1997), one of the two mutations present in the APPSwInd variant described here. In parallel sections of the same young and old APPSwInd mice shown in Fig. H and I, plaques also stained for p-tau in both young and old APPSwInd (Fig. 9J and K). At higher magnification, these p-tau stained structures appear to be dystrophic neurites surrounding plaques, and were more symmetric and welldefined in the older mice, as if plaques mature during aging. Since these mice only carry the transgene for mutant human APP, the p-tau detected here must represent endogenous mouse tau. The p-tau deposits appear clumped rather than filamentous. In both mice, as for silver staining, CA3 seems relatively spared from p-tau staining. We did not find neurofibrillary tangles, which would be aggregates of p-tau forming a cage around neuronal nuclei.
4. Discussion
Here we explore the effect of aging with and without expression of the APP mutant protein, APPSwInd, on transport dynamics and functional connections between CA3 in the hippocampus and the septal nuclei using magnetic resonance imaging of Mn2+ in living transgenic mice and their wildtype littermates. We applied two statistical approaches to investigate and compare the transport of Mn2+ in these four cohorts: statistical parametric mapping between time points within group followed by visual inspection of the SPM map's anatomy; and segmentation with region of interest (ROI) analysis followed by between group ANOVA statistical comparisons of intensity values. Our findings suggest that Mn2+ transport in this system decreases with aging and arrives at different destinations within the septum in mice expressing mutant APP who also carry Aβ plaques. The most striking difference between the aging WT and age-matched APPSwInd transgenic littermates was the location of accumulation of the transported Mn2+, with more accumulation in the lateral septal region and less in the medial septal nuclei in old APPSwInd compared to either age-matched old mice, or younger animals. Taken together these results show altered functional circuitry naturally arising in aging that is further compromised by expression of mutant APP and the presence of Aβ plaques.
Here we consider various explanations for alterations in the dynamics of transport and accumulation. First, what variance might the experimental manipulations contribute; and second what biological process or processes may be responsible?
Differences in experimental procedures such as placement of the injection site and the exact volume of Mn2+ injectate could play a role, which might be expected to randomly influence different individuals and not create differences between cohorts. To rule this out, we examined the injection site location both by MR and by histology and found only small variance in location and diameter insufficient to explain the significant differences in distal distribution.
Differences in mouse strain genetics could introduce variance if our mice were not all congenic C57/B6. This possibility is excluded since we compared APPSwInd expressing mice with their littermates. Some residual genetic variance in the Dbo/Mmx background in which the original APPSwInd transgenic was generated could affect the impact of tTA expression in the double transgenics on the dentate gyris. This is unlikely since no effect is detected in the tTA monogenics and only a minor effect in the young APPSwInd. C57/B6 congenic tTA monogenics have been reported previously to display a normal dentate gyrus (Han et al., 2012).
Could the Mn2+ be traveling by some mechanism other than by fast axonal transport within neurons, such as diffusion, vascular uptake or migration of glial cells? The 24h time course of transport accumulation negates these possibilities. For example, Mn2+ begins to diffuse around the injection site in a spherical radius immediately after injection. Our data reported here confirm this, since only a small diameter of signal around the injection site is detected in statistical comparisons between time points. Vascular uptake is similarly rapid and would not result in the increasing accumulation witnessed in comparisons of the 6h and 24h images, which occurs after the injection site signal has dissipated. The tiny amount of Mn2+ injected here (3–5 nL) once diluted in parenchyma or blood is likely too little to increase MR signal. Indeed no enhancement of the brain parenchyma or the ventricles was found in either SPM or ROI analyses, demonstrating that Mn2+ is not redistributing to these compartments at any detectable levels. Finally, glial and immune cells do not accumulate at the injection site (Gallagher et al., 2012), and their migration would take much longer than 24h to travel from the right hippocampus to the contralateral hippocampus or to the septum, each of which are almost a centimeter distant from the injection site. Since Mn2+ travels in these circuits by vesicular microtubule-based transport (Medina et al., 2017b), it would be predicted to travel at a rate of 0.2–1 μm/sec per vesicle (Brady et al., 1982; Seamster et al., 2012; Vale et al., 1985a; Vale et al., 1985b), although the actual rate of vesicular transport in the central nervous system has not been measured. Bulk transport of radioactive tracers moves within axons by active transport at a predicted rate of 0.7–3.6 mm/h, which fits well with our observed accumulation of Mn2+ signal at distal sites more than a centimeter away at 6h post-injection.
Decreased transport to the medial septal nuclei in aging mice cannot be attributed to loss of cholinergic neurons in the septum. Although cholinergic neurons are decreased or otherwise altered in both human Alzheimer's disease and transgenic mice expressing FAD-associated mutated proteins (Contestabile, 2011; Salehi et al., 2006; Schliebs and Arendt, 2011), we did not find this in our APPSwInd mice. Amyloid plaques are associated with dystrophic cholinergic neuronal processes (Sturchler-Pierrat et al., 1997), including varicosities that appear when a subunit of kinesin is deleted in the context of mutant APP expression (Stokin et al., 2005), which could also impact transport dynamics. Such changes were not found here and thus cannot explain the transport defects.
Over-expression of APP (3.6-fold) in these APPSwInd mice could reduce the impact of Aβ on transport. APP is an anterograde motor receptor (Gunawardena and Goldstein, 2001; Kamal et al., 2001; Satpute-Krishnan et al., 2006) that when over-expressed, even at only 1.5 fold as in mice carrying the Down Syndrome extra chromosome, increases anterograde transport of Mn2+ (Bearer et al., 2007b), and when deleted diminishes anterograde transport of Mn2+ (Gallagher et al., 2012). Thus over-expression of APPSwInd may increase transport and lead to under-estimation of the impact of Aβ deposition. Mice with normal levels of endogenous APP, together with Aβ fibrils and plaques will need to be generated to decouple the effects of plaque from APP over-expression.
Increased accumulation in the lateral septum in the old APPSwInd, where increased silver staining occurs, suggests that uptake in CA3 is normal and transport may even be accelerated, even while progress beyond the septum seems decreased. Since Mn2+ crosses active synapses (Bearer et al., 2007a), one explanation for this antithetical finding is that the Aβ deposits in the LSr disrupts synaptic connections. In this scenario, either Mn2+ is released and sequestered in the plaque, or the synapses are silent and Mn2+ is not released but accumulates in pre-synaptic termini. The slight bu significant decreased accumulation in the medial septum, where little silver staining is detected even in these old APPSwInd, could also be secondary to Aβ deposition at the injection site that may interfere with Mn2+ uptake, or to overall decreased transport from hippocampus to medial septum.
The reduction of volume in the granular cell layer of the dentate could secondarily affect transport from CA3, since granule cells project unidirectionally to CA3, primarily to interneurons but also to pyramidal cells, and are the principal excitatory cells of the dentate gyrus. Lack of excitation would reduce Mn2+ uptake via voltage-gated calcium channels, and thereby reduce the amount that could be transported. Thus reduction of the granule cell layer may be a consequence of Aβ fibril deposition rather than plaque formation, and could underlie the findings of reduced transport from CA3 to distal regions. This possibility will need further exploration.
The finding of dystrophic neurites containing hyper-phosphorylated tau encircling Aβ plaques suggests that the microtubule transport system may be compromised. Tau stabilizes microtubules and helps them serve as tracks for molecular motors to carry cargo along them (Wang and Mandelkow, 2016). Phospho-tau aggregates and obstructs transport. Mouse p-tau in dystrophic neurites in the hippocampus may impact uptake and transport of Mn2+, despite CA3 being relatively spared. In human AD, p-tau is hypothesized to be an early indication of disease, where its deposition progresses from entorhinal cortex to CA1 and only at late stages to CA3 (Braak and Braak, 1997). These mice appear to have the same sequence of deposition even though expression of the mutant APP is abnormally driven indirectly from the CamK2a promoter rather than the APP promoter as in the human case, which could alter location of plaque throughout the brain. CaMKIIa promoter was originally thought to drive expression in the forebrain (Mayford et al., 1996). Recent in situ hybridization for CAMK2a expression from the Allen Institute for Brain Science demonstrates neuron-specific expression throughout the neocortex and subcortical areas, with less in pons and cerebellum (http://mouse.brain-map.org/gene/show/12107), which is very similar to the expression of APP (http://mouse.brain-map.org/experiment/show/79591705) in human and mouse.
How well these mouse mutants, or any mouse carrying FAD mutations, exemplify the human condition is an open question. For FAD in humans with the same mutation relevance is clear, and the biological information obtained from such mice may be very helpful in understanding the disease. Whether the biological impact of over-expressing a single mutant protein explains sporadic AD, a condition for which no single mutation has been found, is less clear. Secondary effects of Aβ deposition within the brain, such as described here, are likely to accompany all types of AD, since Aβ plaques are a diagnostic feature. Indeed, the reason for plaque deposition may be less important than its consequences on transport, synapse function and neuronal survival.
Our results from MEMRI demonstrate that functional brain circuitry from hippocampus to basal forebrain is altered with age. Expression of mutant APP further affects transport, altering the degree and also the location of accumulation of transported Mn2+ at distant target sites. Many factors likely contribute to these findings, including Aβ deposition in the granule cell layer of the dentate gyrus, more plaque in the lateral than in the medial septum, and p-tau within neuritic plaques wherever they occur throughout the brain. Our work here raises new questions about how anatomically specific pathology may play a role in axonal transport disruption in learning and memory circuits in AD. These findings suggest that early transport defects and Aβ distribution contribute to clinical presentations of mild cognitive impairment in patients with AD, as has been described by the Dominantly-Inherited Alzheimer Network (Bateman et al., 2012; Ryman et al., 2014). Additionally, this mouse experimental system in combination with a novel MR imaging technique (MEMRI), has helped us provide unique insight into the importance of APP in both AD and in axonal transport, with the prospect for future studies to determine how and which of those two components of APP may contribute.
Supplementary Material
Highlights:
APPSwInd expression produces Aβ plaques that increase in number over time.
Aging decreases transport from hippocampus to basal forebrain.
Aβ plaque burden enhances the effect of aging and alters transport destination.
The number of medial septal cholinergic neurons is not changed by aging or plaques.
Aged APPSwInd mice have decreased thickness in the dentate gyrus.
Acknowledgements
The authors would like to acknowledge all members of the Bearer Lab, including Amber J. Zimmerman, Sharon Wu Lin, Kathleen Kilpatrick and Drs. Xiaowei Zhang and Joseph J. Gallagher for providing technical assistance, training and commentary, as well as Kevin P. Reagan for his administrative support. We thank Kiran Bhaskar for protocols for Western blotting, and Joanna Jankowsky for introducing us to these exciting double transgenic mice, Daniel R. Barto and Karen SantaCruz for useful discussions. We are indebted to Vince Calhoun of UNM and Art Toga and his group at Laboratory of NeuroImaging at University of California, Los Angeles and University of Southern California for inspiration on applying computational approaches for analysis of mouse brain MRI, Angela Wandinger-Ness (Director of the UNM Cancer Center Biological Imaging Facility), and the Beckman Biological Imaging Center. The work was supported by NINDS RO1 NS062184, NIMH MH096093 and NIGMS P5OGM08273, as well as by the Harvey Family Endowment and Moore Distinguished Scholar award at Caltech (ELB) and by the Beckman Institute (REJ).
Footnotes
Disclosure statement
The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashburner J, 2012. SPM: a history. Neuroimage 62(2), 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badea A, Ali-Sharief AA, Johnson GA, 2007. Morphometric analysis of the C57BL/6J mouse brain. Neuroimage 37(3), 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker R, Tiesinga P, Kotter R, 2015. The Scalable Brain Atlas: Instant Web-Based Access to Public Brain Atlases and Related Content. Neuroinformatics 13(3), 353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC, 2012. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. The New England journal of medicine 367(9), 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer EL, Falzone TL, Zhang X, Biris O, Rasin A, Jacobs RE, 2007a. Role of neuronal activity and kinesin on tract tracing by manganese-enhanced MRI (MEMRI). Neuroimage 37 Suppl 1, S37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer EL, Zhang X, Jacobs RE, 2007b. Live imaging of neuronal connections by magnetic resonance: Robust transport in the hippocampal-septal memory circuit in a mouse model of Down syndrome. Neuroimage 37(1), 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer EL, Zhang X, Janvelyan D, Boulat B, Jacobs RE, 2009. Reward circuitry is perturbed in the absence of the serotonin transporter. Neuroimage 46(4), 1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE, 2008. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nature reviews. Neuroscience 9(10), 768–778. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT, 2010. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68(1), 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, 1997. Diagnostic criteria for neuropathologic assessment of Alzheimer's disease. Neurobiology of aging 18(4 Suppl), S85–88. [DOI] [PubMed] [Google Scholar]
- Brady ST, Lasek RJ, Allen RD, 1982. Fast axonal transport in extruded axoplasm from squid giant axon. Science 218(4577), 1129–1131. [DOI] [PubMed] [Google Scholar]
- Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T, 1999. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. American journal of human genetics 65(3), 664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG, 2000. A learning deficit related to age and betaamyloid plaques in a mouse model of Alzheimer's disease. Nature 408(6815), 975–979. [DOI] [PubMed] [Google Scholar]
- Contestabile A, 2011. The history of the cholinergic hypothesis. Behav Brain Res 221(2), 334–340. [DOI] [PubMed] [Google Scholar]
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, Van der Ploeg LH, Sirinathsinghji DJ, 1999. Agerelated cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 90(1), 1–13. [DOI] [PubMed] [Google Scholar]
- Delora A, Gonzales A, Medina CS, Mitchell A, Mohed AF, Jacobs RE, Bearer EL, 2016. A simple rapid process for semi-automated brain extraction from magnetic resonance images of the whole mouse head. J Neurosci Methods 257, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Mathis C, Saura J, Bales KR, Paul SM, Ungerer A, 2000. Neuroanatomical abnormalities in behaviorally characterized APP(V717F) transgenic mice. Neurobiol Dis 7(2), 71–85. [DOI] [PubMed] [Google Scholar]
- Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, Leong SL, Perez K, Johanssen T, Greenough MA, Cho HH, Galatis D, Moir RD, Masters CL, McLean C, Tanzi RE, Cappai R, Barnham KJ, Ciccotosto GD, Rogers JT, Bush AI, 2010. Ironexport ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell 142(6), 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder GA, Gama Sosa MA, De Gasperi R, 2010. Transgenic mouse models of Alzheimer's disease. Mt Sinai J Med 77(1), 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encalada SE, Goldstein LS, 2014. Biophysical challenges to axonal transport: motor-cargo deficiencies and neurodegeneration. Annual review of biophysics 43, 141–169. [DOI] [PubMed] [Google Scholar]
- Friston KJ, 2005. Models of brain function in neuroimaging. Annual review of psychology 56, 57–87. [DOI] [PubMed] [Google Scholar]
- Gallagher JJ, Zhang X, Hall FS, Uhl GR, Bearer EL, Jacobs RE, 2013. Altered reward circuitry in the norepinephrine transporter knockout mouse. PLoS One 8(3), e57597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JJ, Zhang X, Ziomek GJ, Jacobs RE, Bearer EL, 2012. Deficits in axonal transport in hippocampal-based circuitry and the visual pathway in APP knock-out animals witnessed by manganese enhanced MRI. Neuroimage 60(3), 1856–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS, 2001. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 32(3), 389–401. [DOI] [PubMed] [Google Scholar]
- Han HJ, Allen CC, Buchovecky CM, Yetman MJ, Born HA, Marin MA, Rodgers SP, Song BJ, Lu HC, Justice MJ, Probst FJ, Jankowsky JL, 2012. Strain background influences neurotoxicity and behavioral abnormalities in mice expressing the tetracycline transactivator. J Neurosci 32(31), 10574–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP, Muller U, 2000. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci 20(21), 7951–7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S, Muller U, 2004. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J 23(20), 4106–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Gonzales V, Savonenko AV, Wen JC, Jenkins NA, Copeland NG, Younkin LH, Lester HA, Younkin SG, Borchelt DR, 2005. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med 2(12), e355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Zheng H, 2017. Practical considerations for choosing a mouse model of Alzheimer's disease. Molecular neurodegeneration 12(1), 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson M, Beckmann CF, Behrens TE, Woolrich MW, Smith SM, 2012. FSL. Neuroimage 62(2), 782–790. [DOI] [PubMed] [Google Scholar]
- Johnson GA, Badea A, Brandenburg J, Cofer G, Fubara B, Liu S, Nissanov J, 2010. Waxholm space: an image-based reference for coordinating mouse brain research. Neuroimage 53(2), 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS, 2001. Kinesinmediated axonal transport of a membrane compartment containing beta-secretase and presenilin1 requires APP. Nature 414(6864), 643–648. [DOI] [PubMed] [Google Scholar]
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS, 2000. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 28(2), 449–459. [DOI] [PubMed] [Google Scholar]
- Keller JN, 2006. Age-related neuropathology, cognitive decline, and Alzheimer's disease. Ageing Res Rev 5(1), 1–13. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, Jones AR, 2007. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445(7124), 168–176. [DOI] [PubMed] [Google Scholar]
- Lin YJ, Koretsky AP, 1997. Manganese ion enhances T1-weighted MRI during brain activation: an approach to direct imaging of brain function. Magn Reson Med 38(3), 378–388. [DOI] [PubMed] [Google Scholar]
- Mackenzie-Graham AJ, Lee EF, Dinov ID, Yuan H, Jacobs RE, Toga AW, 2007. Multimodal, multidimensional models of mouse brain. Epilepsia 48 Suppl 4, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER, 1996. Control of memory formation through regulated expression of a CaMKII transgene. Science 274(5293), 1678–1683. [DOI] [PubMed] [Google Scholar]
- McAuliffe MJ, Lalonde FM, McGarry D, Gandler W, Csaky K, Trus BL, 2001. Medical Image Processing, Analysis & Visualization in clinical research. Comp Med Sy, 381–386. [Google Scholar]
- Medina CS, Biris O, Falzone TL, Zhang X, Zimmerman AJ, Bearer EL, 2017a. Hippocampal to basal forebrain transport of Mn2+ is impaired by deletion of KLC1, a subunit of the conventional kinesin microtubule-based motor. Neuroimage 145(Pt A), 44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina CS, Manifold-Wheeler B, Gonzales A, Bearer EL, 2017b. Automated Computational Processing of 3-D MR Images of Mouse Brain for Phenotyping of Living Animals. Current protocols in molecular biology 119, 29A 25 21–29A 25 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt JE, Jacob R, Hallam TJ, 1989. Use of manganese to discriminate between calcium influx and mobilization from internal stores in stimulated human neutrophils. J Biol Chem 264(3), 1522–1527. [PubMed] [Google Scholar]
- Modat M, Ridgway GR, Taylor ZA, Lehmann M, Barnes J, Hawkes DJ, Fox NC, Ourselin S, 2010. Fast free-form deformation using graphics processing units. Comput Meth Prog Bio 98(3), 278–284. [DOI] [PubMed] [Google Scholar]
- Muller U, Cristina N, Li ZW, Wolfer DP, Lipp HP, Rulicke T, Brandner S, Aguzzi A, Weissmann C, 1994. Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell 79(5), 755–765. [DOI] [PubMed] [Google Scholar]
- Pautler RG, 2004. In vivo, trans-synaptic tract-tracing utilizing manganese-enhanced magnetic resonance imaging (MEMRI). NMR Biomed 17(8), 595–601. [DOI] [PubMed] [Google Scholar]
- Paxinos G, 2004. The rat nervous system, 3rd ed. Elsevier Academic Press, Amsterdam; Boston. [Google Scholar]
- Paxinos G, Franklin K, 2001. The Mouse Brain in Stereotaxix Coordinates. [Google Scholar]
- Penny W, Friston K, Ashburner J, Kiebel S, 2006. Statistical Parametic Mappng: The Analysis of Functional Brain Images. Academic Press, Waltham, MA. [Google Scholar]
- Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J, 2006. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci 26(27), 7212–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Brayne C, Mayeux R, 2011. Epidemiology of Alzheimer disease. Nature reviews. Neurology 7(3), 137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, Goate A, Frommelt P, Ghetti B, Langbaum JB, Lopera F, Martins R, Masters CL, Mayeux RP, McDade E, Moreno S, Reiman EM, Ringman JM, Salloway S, Schofield PR, Sperling R, Tariot PN, Xiong C, Morris JC, Bateman RJ, 2014. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 83(3), 253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC, 2006. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51(1), 29–42. [DOI] [PubMed] [Google Scholar]
- Satpute-Krishnan P, DeGiorgis JA, Conley MP, Jang M, Bearer EL, 2006. A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proc Natl Acad Sci U S A 103(44), 16532–16537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindowski K, Belarbi K, Buee L, 2008. Neurotrophic factors in Alzheimer's disease: role of axonal transport. Genes Brain Behav 7 Suppl 1, 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliebs R, Arendt T, 2011. The cholinergic system in aging and neuronal degeneration. Behav Brain Res 221(2), 555–563. [DOI] [PubMed] [Google Scholar]
- Seamster PE, Loewenberg M, Pascal J, Chauviere A, Gonzales A, Cristini V, Bearer EL, 2012. Quantitative measurements and modeling of cargo-motor interactions during fast transport in the living axon. Physical biology 9(5), 055005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senechal Y, Kelly PH, Dev KK, 2008. Amyloid precursor protein knockout mice show agedependent deficits in passive avoidance learning. Behav Brain Res 186(1), 126–132. [DOI] [PubMed] [Google Scholar]
- Shattuck DW, Leahy RM, 2001. Automated graph-based analysis and correction of cortical volume topology. IEEE transactions on medical imaging 20(11), 1167–1177. [DOI] [PubMed] [Google Scholar]
- Silva AC, Lee JH, Aoki I, Koretsky AP, 2004. Manganese-enhanced magnetic resonance imaging (MEMRI): methodological and practical considerations. NMR Biomed 17(8), 532–543. [DOI] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen-Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, Niazy RK, Saunders J, Vickers J, Zhang Y, De Stefano N, Brady JM, Matthews PM, 2004. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23 Suppl 1, S208–219. [DOI] [PubMed] [Google Scholar]
- Sonnen JA, Santa Cruz K, Hemmy LS, Woltjer R, Leverenz JB, Montine KS, Jack CR, Kaye J, Lim K, Larson EB, White L, Montine TJ, 2011. Ecology of the aging human brain. Archives of neurology 68(8), 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS, 2005. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307(5713), 1282–1288. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B, 1997. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A 94(24), 13287–13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW, Cowan WM, 1977. An autoradiographic study of the organization of the efferent connections of the hippocampal formation in the rat. J Comp Neurol 172(1), 49–84. [DOI] [PubMed] [Google Scholar]
- Switzer RC 3rd, 2000. Application of silver degeneration stains for neurotoxicity testing. Toxicol Pathol 28(1), 70–83. [DOI] [PubMed] [Google Scholar]
- Tharp WG, Sarkar IN, 2013. Origins of amyloid-beta. BMC Genomics 14, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PR, O'Connor K, Tate WP, Abraham WC, 2003. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol 70(1), 1–32. [DOI] [PubMed] [Google Scholar]
- Tyszka JM, Readhead C, Bearer EL, Pautler RG, Jacobs RE, 2006. Statistical diffusion tensor histology reveals regional dysmyelination effects in the shiverer mouse mutant. Neuroimage 29(4), 1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheetz MP, 1985a. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42(1), 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Schnapp BJ, Reese TS, Sheetz MP, 1985b. Movement of organelles along filaments dissociated from the axoplasm of the squid giant axon. Cell 40(2), 449–454. [DOI] [PubMed] [Google Scholar]
- Vershinin M, Carter BC, Razafsky DS, King SJ, Gross SP, 2007. Multiple-motor based transport and its regulation by Tau. Proc Natl Acad Sci U S A 104(1), 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ, 2007. A beta oligomers - a decade of discovery. J Neurochem 101(5), 1172–1184. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E, 2016. Tau in physiology and pathology. Nature reviews. Neuroscience 17(1), 5–21. [DOI] [PubMed] [Google Scholar]
- Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ, 2014. Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Frontiers in genetics 5, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolrich MW, Jbabdi S, Patenaude B, Chappell M, Makni S, Behrens T, Beckmann C, Jenkinson M, Smith SM, 2009. Bayesian analysis of neuroimaging data in FSL. Neuroimage 45(1 Suppl), S173–186. [DOI] [PubMed] [Google Scholar]
- Young-Pearse TL, Bai J, Chang R, Zheng JB, LoTurco JJ, Selkoe DJ, 2007. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci 27(52), 14459–14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bearer EL, Boulat B, Hall FS, Uhl GR, Jacobs RE, 2010. Altered neurocircuitry in the dopamine transporter knockout mouse brain. PLoS One 5(7), e11506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.