

Abstract
The classical crystal structure of insulin was determined in 1969 by D.C. Hodgkin and colleagues following a 35-year program of research. This structure depicted a hexamer remarkable for its self-assembly as a zinc-coordinated trimer of dimer. Prominent at the dimer interface was an “aromatic triplet” of conserved residues at consecutive positions in the B chain: PheB24, PheB25 and TyrB26. The elegance of this interface inspired the Oxford team to poetry: “A thing of beauty is a joy forever” (John Keats as quoted by Blundell, T.L., et al. Advances in Protein Chemistry 26:279–286 (1972)). Here, we revisit this aromatic triplet in light of recent advances in the structural biology of insulin bound as a monomer to fragments of the insulin receptor. Such co-crystal structures have defined how these side chains pack at the primary hormone-binding surface of the receptor ectodomain. On receptor binding, the B-chain β-strand containing the aromatic triplet (residues B24-B28) detaches from the α-helical core of the hormone. Whereas TyrB26 lies at the periphery of the receptor interface and may functionally be replaced by a diverse set of substitutions, PheB24 and PheB25 engage invariant elements of receptor domains L1 and αCT. These critical contacts were anticipated by the discovery of diabetes-associated mutations at these positions by D.F. Steiner and his colleagues at the University of Chicago. Conservation of PheB24, PheB25 and TyrB26 among vertebrate insulins reflects the striking confluence of structure-based evolutionary constraints: foldability, protective self-assembly and hormonal activity.
Keywords: protein structure, protein evolution, protein recognition, structure-activity relationships, hormone-receptor recognition
1 |. INTRODUCTION
Insulin has provided an enduring model for studies in protein chemistry and cell biology virtually since the hormone’s landmark discovery in Toronto in 1921 [1]. The sequence of bovine insulin, the first protein sequence to be determined [2, 3], established that this class of biological heteropolymers in fact had specific sequences—and so made concrete the concept of a genetic code [4]. Insulin was the second protein to be crystallized (after urease) [5, 6] and provided the first example of protein crystallization as a process that in itself can confer pharmacologic advantage (for review, see [7]). Efforts to determine the three-dimensional (3D) structure of insulin began in 1934 in the graduate studies of D.C. Hodgkin (then at Cambridge University) [6, 8]. These studies came to fruition in 1969 at the University of Oxford with the first crystal structure of a peptide hormone [9, 10]. It is fitting to begin this contribution, written in honor of the late Prof. Donald F. Steiner, at the point of intersection between his laboratory at the University of Chicago with the Hodgkin laboratory.
In an issue dedicated to Prof. Steiner, it is fitting to include a structural perspective as his studies integrated structure with enzymology, cell biology, and pathophysiology [11, 12]. His faculty career began in the 1960s, a decade of transformational discovery in molecular endocrinology. The first half of this decade, for example, witnessed international efforts to develop methods for the total chemical synthesis of insulin [13]. Major programs were led by P.G. Katsoyannis at the University of Pittsburg [14], by H. Zahn in Wool Institute of Aachen, German [15] and by the late Niu Jingyi and his colleagues at the Institute of Biochemistry, Academia Sinica (Shanghai) [16]. The success of these programs highlighted in the breach a seeming paradox posed by the low yields of insulin chain combination. Although the isolated A- and B chains contained sufficient information to direct formation of the three specific disulfide bridges identified by Sanger (cystines A7-B7, A6-A11 and A20-B19) [17], the reaction was dominated by side reactions leading to cyclic chains and peptide aggregates, including what is now understood to be peptide fibrils [18–21]. This critical barrier to chemical synthesis raised the central question of how efficiency of native disulfide pairing might be achieved in the biosynthesis of insulin in the specialized endoplasmic reticulum (ER) of pancreatic β-cells [22–24].
Such was the context for the seminal discovery of a single-chain insulin precursor by Steiner and Philip E. Oyer in 1967 [22, 23]. Designated proinsulin, the precursor contains a connecting peptide between the C-terminal residue of the B chain (ThrB30 in human insulin) and N-terminal residue of the A chain (conserved as GlyA1). The discovery of proinsulin and the C peptide quickly found medical application through development of radioimmunoassays (RIAs) to probe insulin secretory capacity in normal and pathological states, including residual β-cell function in patients with Type 1 diabetes mellitus (T1DM), β-cell failure in long-established Type 2 DM (T2DM) and inappropriate insulin secretion in β-cell neoplasia (insulinoma) (for review, see [25]). Cellular mechanisms ensuring the fidelity of disulfide pairing and their perturbation in a monogenic DM syndrome due to toxic misfolding of variant proinsulins are reviewed in an accompanying article in this issue by M. Liu et al. [26].
The discovery of proinsulin led to exchange of visitors between Chicago and Oxford, stimulated by the determination of the crystal structure of the zinc insulin hexamer in 1969 and at increasing resolution in 1971 [9, 10]. These years preceded the development of computer-graphics systems for visualization of protein structures. Intuition was facilitated through construction of physical models, hand-built in the first instances.
Such was the collegial respect between Professors Hodgkin and Steiner that one of these rare early models of the insulin monomer (now classified as a T-state protomer of the T6 hexamer) was given to the Chicago laboratory, hand-delivered by a student visiting from Oxford. For more than 40 years this model assumed pride of place in Don’s office (Figure 1). Inset at upper right are pictures of Professors Hodgkin and Steiner holding corresponding models.
Figure 1.

Office of Prof. D.F. Steiner at the University of Chicago. Shown on table at front in plexiglass box is a plastic model of insulin as a crystallographic protomer. Inset at top right, Hodgkin and Steiner with matching molecular models. This figure was originally published in a remembrance of D.F. Steiner [12] and is reprinted here with permission of the authors. The molecular model is now on display as part of an historical collection in the Division of Endocrinology at the Pritzker School of Medicine. One of the authors (MAW) recalls many pleasant and stimulating discussions in this office during which Don references one or more side chains in this model. Often these included the aromatic triplet—PheB24, PheB25 and TyrB26—in light of their special role in the Hodgkin structure.
Since 1969, the structure of insulin has been determined in a variety of crystal forms [27–31], in solution by NMR methods [32–39], and most recently, in complexes with fragments of the insulin receptor (IR) [40, 41]. Our review focuses on a conserved segment of the B chain—the conserved “aromatic triplet” PheB24, PheB25 and TyrB26 [42]—as the thread of a remarkable story. The positions of these residues in the zinc insulin hexamer (T6) are shown in Figure 2A, and their positions in the dimer are shown in Figure 2B. Interest in the contribution of the aromatic triplet to the structure and function of insulin thus began with the first crystal structure of insulin, deepened with the first discovery of mutations in the insulin gene associated with diabetes mellitus [43–46] and continues in ongoing efforts to decipher the structure of the hormone-receptor complexes [47, 48], including by single-particle cryo-electron microscopy [49].
Figure 2.
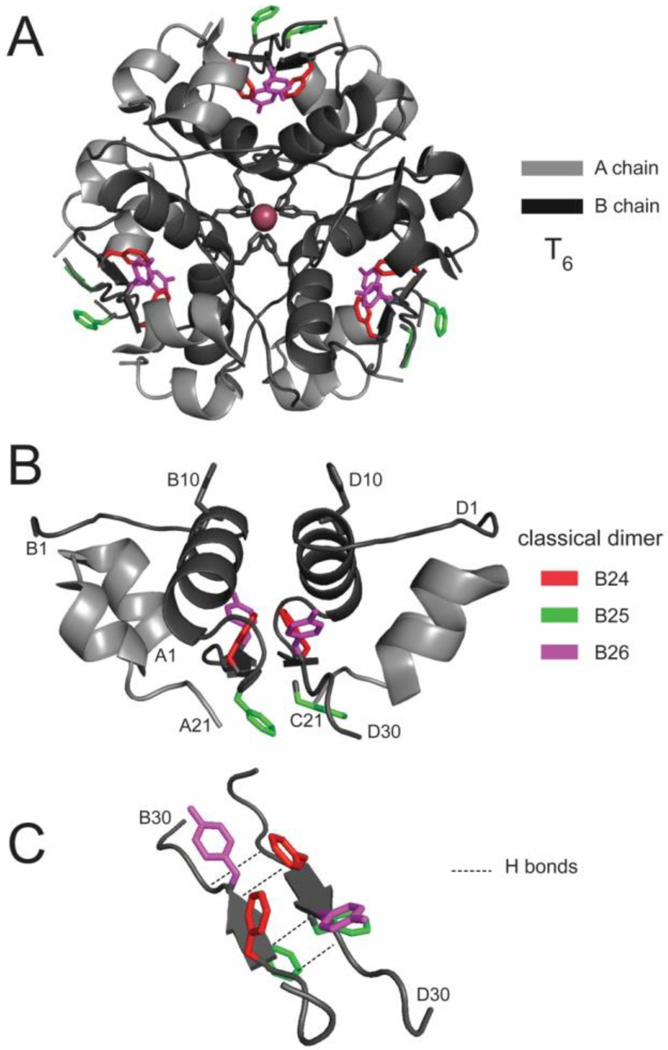
Insulin self-assembly as defined in the 1969 crystal structure of insulin [9, 10]. (A) T6 (or 2-Zn) insulin hexamer, (B) Constituent dimer and (C) anti-parallel C-terminal dimer-related β-sheet (residues B24-b28; arrows) with the side chains of residues B24-B26 shown in red, green and violet, respectively. Coordinates were obtained from Protein databank entry 4INS.
2 |. CRYSTAL STRUCTURES OF INSULIN
The 1969 crystal structure of insulin (then designated 2-Zn insulin and now classified as a T6 hexamer [50]) was the among the first set of protein structures to be determined (for reviews, see [6, 51]). Each of these early structures (spanning globins, ribonucleases and proteases) uncovered a new aspect of protein structure and its relationship to function. These features were to inform general principles of protein folding and assembly in the ensuing decades.
The 2-Zn insulin structure depicted a 32-point-group hexameric self-assembly whose dimer-related interfaces contained a short anti-parallel β-sheet (Fig. 2C) and whose trimer-related interface was stabilized by histidine-mediated 2-zinc-ion coordination [9, 10]. Formation of these interfaces concealed extensive non-polar surfaces that would otherwise be exposed to solvent in an isolated protomer. The specific role of zinc ions in the structure of the insulin hexamer motivated continuing studies of zinc transporters in the cell biology of insulin storage within the secretory granules of pancreatic β-cells [52–54].
Crystal structures of zinc-free insulin dimers by Caspar and colleagues demonstrated that the T2 substructure in the 2-Zn insulin hexamer does not require hexamer assembly or zinc-ion coordination [29, 55].
2.1 |. The TR Transition
This original T6 structure contained a dimer in the crystallographic asymmetric unit, with the hexamer defined by a crystallographic three-fold symmetry axis. The two protomers within the dimer (designated molecules 1 and 2) exhibited subtle differences. More dramatic differences were subsequently observed in the 1976 crystal structure of “4-Zn” insulin [27] in which high concentrations of NaCl induced a conformational change to yield the T3R3 hexamer [6, 56]. The T→R transition entailed N-terminal extension of the B-chain α-helix; cylinder models of the constituent TR dimer are shown in Figure 3. Subtle differences were also observed in this transition, including (i) in the conformation of the A7-B7 disulfide bridge, (ii) rotation and reorientation of the A1-A8 α-helix, and (iii) a slight displacement of the B24-B28 β-strand relative to A chain such that an inter-chain hydrogen bond (B25 NH…O=C A19) is broken. The latter feature foreshadowed the frank displacement of the B-chain β-strand on receptor binding as described below (Section 3).
Figure 3.
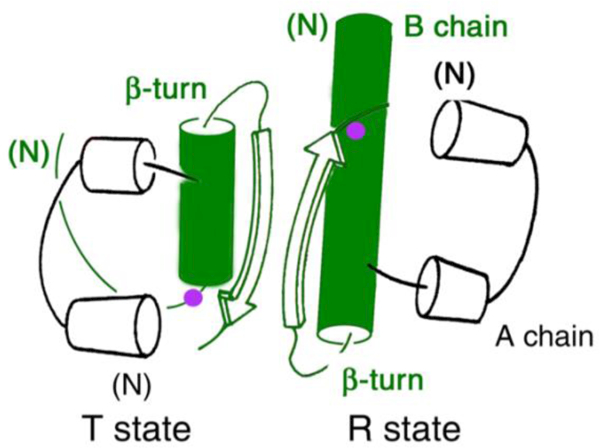
The two classical conformations of insulin: T and R. TR dimer as extracted from crystal structure of a T3R3 zinc insulin hexamer, first observed in the classical “4-Zn” crystal form [27]. The position of B8 is indicated by a purple circle within the B-chain helix (green), which comprises residues B9-B19 in the T state, B1-B19 in the R state (or B3-B19 in the “frayed” Rf state).
Comparison of the 2-Zn and 4-Zn structures provided a model for analysis of the long-range transmission of conformational change in a protein assembly [56] and inspired speculation regarding which structure (T or R) might best inform structure-activity relationships [6]. One study exploited a key aspect of this transition: a change in the conformation of an invariant glycine at position B8 (purple circles in Figure 3) from the right side of the Ramachandran plot in the T-state β-turn (B7-B10) to the left side to reside within the canonical α-helical island as in the R state [60]. Stabilization of the T state through substitution of D-Ala at B8 impaired binding to the IR by at least 100-fold [60].
The T6→T3R3 transition was “completed” in 1989 through the discovery of R6 zinc insulin hexamers by G.G. Dodson and colleagues [28]. The conformational plasticity of insulin in the crystalline state (i.e., through the series of structures T6→T3R3→R6) motivated molecular-dynamics (MD) simulations [61–64] and NMR studies in solution [32–39]. The latter demonstrated that the predominant conformation of the hormone as a monomer in solution (the physiologic state in the bloodstream) closely resembles the crystallographic T state (Figure 4). Such studies exploited engineered insulin monomers to circumvent confounding effects of self-association [59]. In Section 3 we will shall see how this flexibility is exploited to achieve the novel receptor-bound structure of the hormone.
Figure 4.
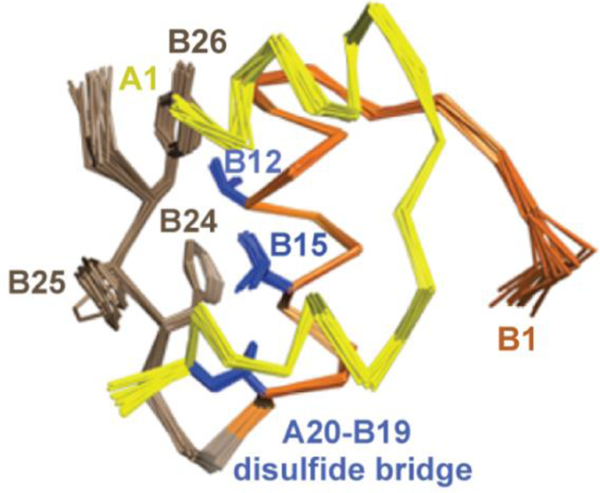
1H-NMR spectroscopy defines T-like conformation of an engineered insulin monomer. Solution structure of DKP-insulin highlighting the aromatic triplet (PheB24, PheB25 and TyrB26) in brown relative to ValB12, LeuB15 and cysteine B19-A20 in blue. In this monomeric analog dimerization was impaired by the paired substitutions LysB28 and ProB29 [57], and the trimer interface was disrupted by AspB10 [58, 59]. The A chain is shown in yellow, and B chain in orange (B1-B19) and brown (B20-B30).
2.2 |. The Aromatic Triplet
The sequence of the B chain is notable for the invariance of consecutive phenylalanine residues at positions B24 and B25 (red and green in Figure 5) and for the broad conservation of tyrosine at position B26 (violet). (Divergent rodent sequences may contain ArgB26 or SerB26, substitutions likely to preserve activity [67].) The conserved aromatic triplet is part of a β-strand within a dimer-related anti-parallel β-sheet (B24-B28; Figure 2C). In the T6→T3R3→R6 series of hexameric structures the side chains of PheB24 and TyrB26 (red and violet in Figure 6) participate in similar structural interactions whereas the position of PheB25 is variable and more peripheral to the hydrophobic core (green). The short-B-chain β-strands follow the B20-B24 β-turn, which contains conserved glycines at B20 and B23 (canonical β-turn positions 1 and 4) [68].
Figure 5.

Conservation of the “Aromatic Triplet”: sequence alignment of residues B20-B30, which encompasses a β-turn (B20-B23), β-trand (B24-B28) and less well-ordered terminal residues (B29 and B30). Phe is invariant among vertebrate insulins at positions B24 (red) and B25 (green) whereas Tyr is highly conserved at position B26 (lilac). The divergent insulin in the rodent O. degu is associated with insulin fibrillation of the islet, leading to β-cell death and senile DM [65, 66].
Figure 6.
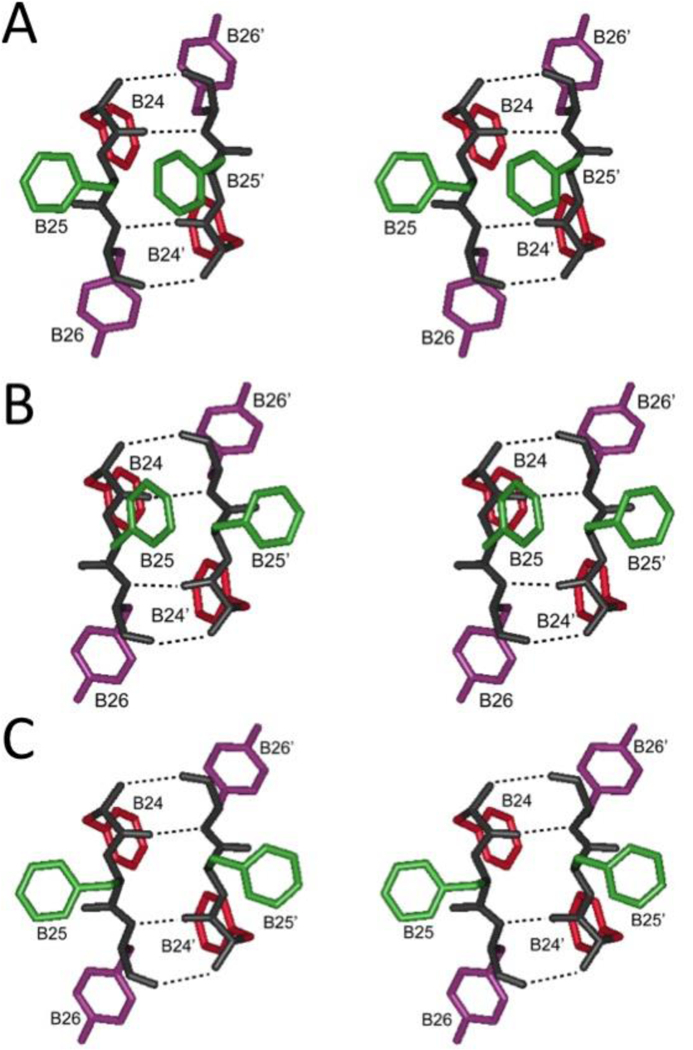
Aromatic triplet at dimer interface. Stereo pairs showing PheB24 (red), PheB25 (green) and TyrB26 (violet) in dimer-related β-sheet in (A) T6 hexamer (B) T3Rf3 hexamer and (C) R6 hexmer. Coordinates were obtained from Protein Databank entries 4INS, 1TRZ and 1ZNJ, respectively. Dotted lines indicate β-sheet-related hydrogen bonds.
The aromatic side chains of PheB24, PheB25 and TyrB26 contribute to an extensive non-polar surface of the insulin monomer (Figure 7). This surface is engaged at the dimer interface in crystallographic dimers and hexamers [6, 51]. The side chains of the aromatic triplet thus occupy distinct structural environments in the insulin monomer and are associated with marked differences in structure activity relationships. Interested in these positions was stimulated in the 1980s by the discovery (by Steiner and his colleagues [including Howard S. Tager and Arthur H. Rubenstein] at the University of Chicago) of mutations at B24 and B25 in patients with diabetes mellitus [46].
Figure 7.
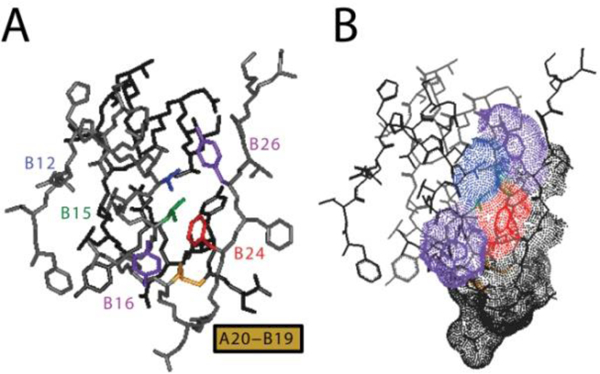
T-state structure of insulin. Aromatic triplet is part of an extensive non-polar surface of the insulin monomer. (A) Stick representation highlighting side chains of PheB24 (red), PheB25 (gray; not labeled at right), and TyrB26 (purple at upper right) relative to α-helical residues ValB12 (blue), LeuB15 (green) and TyrB16 (purple at lower left). The internal disulfide bridge of cystine B19-A20 is shown in yellow. The A chain is shown in dark gray, and B chain otherwise shown in light gray. (B) Corresponding dot-surface representation of the highlighted residues with neighboring residues B20-B23, B25 and B27 in light gray; A20-A21 is dotted in dark gray.
Position B24.
The aromatic ring of PheB24 lies within a crevice such that one face seals the hydrophobic core and the other exposed to solvent. The ring contacts the methyl groups of LeuB15 and the β-protons of CysB19 as a conserved aspect of B-chain super-secondary structure. All natural amino-acid substitutions—with the anomalous exception of Gly—impair receptor binding [69, 70]. Even substitution of PheB24 by its near-isostere Tyr impairs IR binding by at least tenfold [70, 71]. SerB24 is a clinical mutation, known as insulin Los Angeles [72].
The native activity of GlyB24-insulin may reflect a “register shift” in which PheB25-TyrB26 take the place of PheB24-PheB25 with extrusion of a five-residue non-canonical turn (GERGG; residues B20-B24) [39]. An analogous shift may rationalize the enhanced affinity of non-standard insulin analogs containing D-amino-acid substitutions at B24 [38, 39, 73]. In such models the role of TyrB26 is taken by ThrB27 in general accordance with divergent rodent sequences (Figure 5 above).
Position B25.
The B25 side chain is less well ordered in solution; its aromatic resonances exhibit motional narrowing and fewer inter-residue nuclear Overhauser enhancements (NOEs) contacts relative to those of B24 or B26. Studies by the late H.S. Tager and his then-MD-PhD student Raghu Mirmira (now Director of the Diabetes Center and Professor of Medicine at the Indiana University School of Medicine) established that a broad range of aromatic residues are tolerated by the IR, including β-naphthyl-alanine. A general requirement was established for a trigonal γ-carbon, a shared feature of active aromatic substituents. LeuB25, found as a clinical mutation known as insulin Chicago [74] and containing a tetrahedral γ-carbon, impairs receptor binding by at least 50-fold [75, 76].
Position B26.
TyrB26 lies in part within a crevice between the A- and B chains, contacting the conserved side chains of ValB12, IleA2 and ValA3. This “closed” conformation thus conceals key aspects of the receptor-binding surface as inferred from mutagenesis and visualized in Section 3 through crystallographic studies of model hormone-receptor complexes. Although stabilization of the closed conformation by TyrB26 is not required for biological activity, it protects the free hormone from physical degradation [67].
Residue-specific photo-cross-linking studies, employing para-azido-Phe (Pap) as a photo-activatable probe, implied that residues B24-B26 each pack at the hormone-receptor interface. Domain mapping of photo-products demonstrated that PapB24 and PapB26 photo-cross-linked to the L1 domain of the IR α-subunit [77] whereas PapB25 contacted the C-terminal fragment of the α-subunit [78]. To our knowledge, the use of Pap as a short and rigid residue-specific photo-probe in insulin was first demonstrated by Steiner in collaboration with Katsoyannis [78]. Rigorous peptide mapping of PapB25-photo-products by Kurose et al. [78] (exploiting mass-spectrometric methods) provided the first identification of the conserved αCT motif in the IR ectodomain as a critical hormone-binding element; its central structural role is highlighted in Section 3. Although incompletely visualized in electron-density maps, physical evidence that αCT functions in trans within the IR (αβ)2 dimer was provided by bifunctional Pap derivatives of insulin containing photo-probes at opposite surfaces [41].
Modifications at A2 and A3—sites partially concealed by the C-terminal segment of the B chain in the closed state—are also associated with markedly impaired activities [79, 80]. Even such subtle aliphatic substitutions as IleA2→allo-Ile (i.e., inversion of β-carbon chirality within the A2 side chain) [81] or substitution ValA3→Leu (insulin Wakayama [44]) cause profound defects in IR binding. In the following section we describe how detachment of the aromatic triplet at the receptor surface unmasks these A-chain residues. The latter are critical to receptor engagement via conserved αCT contacts.
3 |. INTERACTION OF THE AROMATIC TRIPLET WITH THE RECEPTOR’S PRIMARY INSULIN-BINDING SITE.
3.1 |. STRUCTURE OF THE INSULIN RECEPTOR ECTODOMAIN
In order to understand the remarkable role played by the aromatic triplet of insulin in mediating both hormone storage and receptor activation, it is appropriate to provide an overview of the structural biology of the insulin receptor itself. The receptor is a member of the modular receptor tyrosine kinase (RTK) family [82, 83]; two other members belong to the same sub-family of RTKs, namely, the type 1 insulin like growth factor receptor (IGF-1R) [82] and the IR-related receptor (IRR) [84]. These three receptors are distinct from other RTKs in that they are disulfide-linked homodimers, whereas the others are monomers. Conserved modules in the IR are shown in Figure 8.
Figure 8.
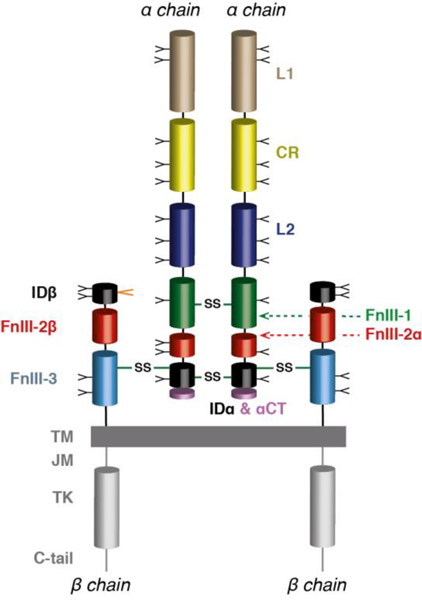
Primary structure of the insulin receptor (αβ)2 homodimer. Ll: first leucine-rich repeat domain; CR: cysteine-rich region; L2:second leucine-rich repeat domain; Fnlll-1 , Fnlll-2 and Fnlll-3: first, second and third fibronectin type III domains; ID: insert domain (IDα: α-chain component; IDβ: β-chain component); αCT: C-terminal segment of the receptor α chain, TM: trans-membrane segment; JM: juxta-membrane segment; TK: tyrosine kinase domain; C-tail: C-terminal tail of the receptor β chain. Inter-chain disulfide bonds are indicated by solid green lines, individual N-linked glycosylation sites by “Y” symbols and the location of the O-linked glycosylation segment by an orange “V” symbol.
The sequence of the human IR was determined in 1985 [82, 83]. The mature form of the receptor is a disulfide-linked (αβ)2 homodimer, formed by furin processing of disulfide-linked pro-receptor polypeptides into α- and β chains. Within the insulin receptor homodimer, a single disulfide bond links each α chain to its β chain partner and at least two disulfide bonds cross-link the α chains to each other [85].
The receptor is heavily glycosylated: thirteen glycans are N-linked to each α chain and four to each β chain [86], and six glycans are O-linked to each β chain [87]. The human insulin receptor occurs as two splice variants, termed IR-A and IR-B, respectively, with IR-A isoform lacking (and IR-B isoform retaining) the twelve amino-acid protein product of exon 11 [88]. IR-B is the predominant isoform in peripheral tissue and is responsible for metabolic signaling [89]. IR-A is the predominant isoform within fetal tissue and also within the adult brain. IR-A shows enhanced binding to insulin-like growth factors compared to IR-B and is upregulated in certain cancers [89].
The 3D structure of the IR extracellular region (its “ectodomain”) was derived from X-ray analysis of a crystal of the isolated ectodomain in complex with four Fab fragments; the latter were attached to the ectodomain fragment in order to overcome the hindrance posed to crystallization by the extensive N-linked glycosylation [90]. Within this structure, the extra-cellular component of each αβ chain pair was revealed to have the following domain organization [48] (describing from the N terminus of the α chain through to the C terminus of the β chain): a leucine-rich repeat domain (L1), a cysteine-rich region (CR), a second leucine-rich repeat domain (L2), and finally three fibronectin type III domains (FnIII-1, FnIII-2 and FnIII-3; see Figure 8).
Within the holo receptor, the FnIII-3 domain is followed by a transmembrane helical segment (TM), a juxtamembrane region, the tyrosine kinase domain (TK) and a C-terminal tail segment [90]. Domain FnIII-2 contains within its canonical CC’ loop [48] a segment of approximately 110 residues termed the insert domain (ID) that in turn contains the α/β furin processing site (Figure 8). The three-dimensional crystal structure of the ectodomain has a Λ shape, with each “leg” of the Λ containing the L1-CR-L2 module of one receptor monomer juxtaposed in an anti-parallel fashion against the (FnIII-1)-(FnIII-2)-(FnIII-3) module of the opposing receptor monomer [48, 90], placing the sites of membrane entry at ends of the respective “legs” of the assembly (Figure 9).
Figure 9.
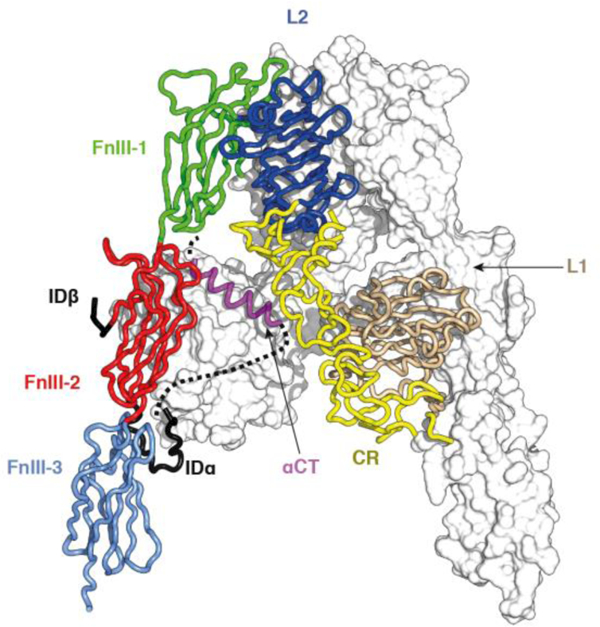
Crystal structure of the insulin receptor (αβ)2 ectodomain. One αβ monomer is shown in ribbon representation (foreground), the other in molecular surface representation (background). The coloring of domains is as in Panel A. Part of IDα is indicated as a dashed line (foreground monomer only), as it was disordered within the crystal. The schematic is based on PDB entry 4ZXB [48].
The “head” of the Λ-shaped assembly contains of the [L2-(FnIII-1)]2 module, the two FnIII-1 domain components of which are disulfide bonded to each other at residue Cys524. The ID domains are mostly disordered and lie within the interior of the Λ-shaped structure, except for a segment (termed αCT, located near the C terminus of the α chain) that assembles as an α helix on the central β sheet (termed L1-β2) of the L1 domain of the opposing receptor monomer [48, 91]. A single inter-α-chain disulfide bond connects residues Cys524 (within the CC’ loop of domain FnIII-1) [90, 91], and at least one inter-α-chain disulfide bond occurs within the ID at the triplet Cys682-Cys683-(Ser684)-Cys685 [48, 85]. Each α chain is linked to its β-chain counterpart by a disulfide bond between residues Cys647 (within the α-chain segment of the ID) and Cys860 (within domain FnIII-3; IR-A numbering) [85]. All remaining disulfide bonds are intra-chain and there is a single free cysteine residue within FnIII-3 (Cys872; IR-A numbering) [85]. It should be noted that the crystal contained an (IR-A)-based construct termed IRΔβ, which was engineered to remove the heavily glycosylated segment near the N terminus of the receptor β chain (again to reduce hindrance to crystallization).
The inter-domain interfaces (both intra-monomer and inter-monomer) within the above structure are sparsely packed, with the only exception being that between the L1 domain and the N-terminal region of its immediately downstream CR domain [48, 90]. These loosely-packed interfaces are reminiscent of crystal packing interfaces, suggesting that all or some of them may disassemble upon insulin binding. However, this raises the question as to whether reported apo ectodomain structure has been modulated by Fab attachment and/or crystal lattice embedding. Two lines of evidence support the view that the Λ-shaped structure represents that adopted by the ectodomain in the context of a membrane-embedded holo receptor. First, recent negative-stain electron microscopy imaging of holo insulin receptor embedded in lipid nanodiscs show, at least qualitatively, that the Λ-shaped assembly concurs with that observed in the nanodisc context [92] (Figure 10). Second, crystallographic analysis of the isolated and growth factor-free IGF-1R ectodomain reveals it to have a similar shape and domain assembly to that of the insulin receptor, albeit that the IGF-1R construct employed included the attachment of a pair of antibody Fv modules [93].
Figure 10.
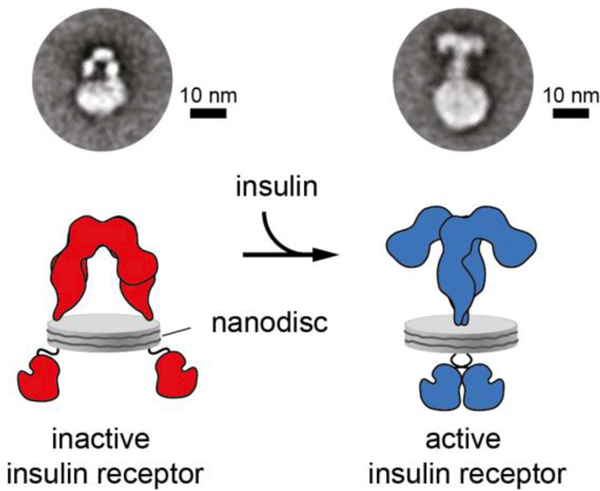
Negative-stain transmission electron microscopy images of holo insulin receptor embedded in nanodiscs [92]. The left-hand panel is an image of the receptor in an insulin-free state, the right-hand panel is an image of the receptor in an insulin-bound state, with the panels underneath each offering an interpretation of the respective images above. The correspondence of the Λ-shaped particle in the left-hand panel to those of that of the crystal structure of the isolated ectodomain is apparent. The right-hand panels show the closing of the receptor legs upon addition of insulin and a likely folding out of the L1-CR-L2 modules. Republished with permission of the Rockefellar University Press, from “Journal of Cell Biology, Gutmann et al., v217, p1643 (2018)”; permission conveyed through Copyright Clearance Center, Inc. [92].
3.2 |. STRUCTURE OF MODEL HORMONE-RECEPTOR COMPLEXES
Insulin binding to the IR displays complex kinetics, characterized by curvilinear Scatchard plots and negative cooperativity. The latter was demonstrated by accelerated dissociation of labeled insulin upon dilution with cold insulin [94–97]. Further, the accelerated dissociation of insulin is reduced at high cold-insulin concentration (> 1 μM). These data can be explained by a model wherein insulin binding involves the formation of a cross-link between two distinct sites (designated site 1 and site 2’) on the receptor, where ‘ is used here and throughout to denote—within the context being described—entities from the alternate αβ monomer. Site 1 is taken to be the higher-affinity (“primary”) binding site and site 2’ the lower-affinity (“secondary”) binding site. Insulin binding to one site 1 / site 2’ pair is then proposed to reduce the affinity of insulin for the alternate site 1’ / site 2 pair (negative cooperativity). The reduction in accelerated dissociation at high insulin concentrations is explained within this model by postulating that, under such conditions, three insulin molecules can bind to the homodimeric receptor: one cross-linking the site 1 / site 2’ pair with high affinity, one binding to site 1’ and one to site 2. Insulin binding independently to the latter two sites then abrogates both the formation of a high-affinity cross-link between sites 1’ and 2 and the reduction in affinity of the site 1 / site 2’ cross-link.
The complexities of the hormone receptor-binding kinetics taken together with the structural complexities of the receptor itself have confounded attempts to crystallize the ectodomain in complex with insulin. Partial success has thus come via another route, namely, by analysis of insulin bound to fragments of the ectodomain [40, 41]. The smallest receptor fragment know to bind insulin is the isolated two-domain L1-CR construct in the presence of an exogenous peptide segment spanning the receptor αCT helix [98], which together display nanomolar affinity for insulin and arguably reconstitute site 1. This tandem receptor element (L1-CR + αCT) has been termed the insulin “micro-receptor” (μIR) [41], in the same vein of nomenclature as the earlier terms “mini-receptor” (the receptor L1-CR-L2 module plus an exogenous αCT peptide [99, 100]) and “midi-receptor” (the receptor [L1-CR-L2-(FnIII-1)]2 fragment wherein each constituent FnIII-1 domain is extended at its C terminus by the α-chain component of the ID domain [98, 101].
An X-ray crystallographic structure of a μIR complex consisting of (i) receptor residues 1–310 (an L1-CR fragment), (ii) an αCT peptide fragment spanning residues 704–719 of IR-A and (iii) human insulin, has been determined to a resolution of 3.5 Å [40, 41] (Figure 11).
Figure 11.
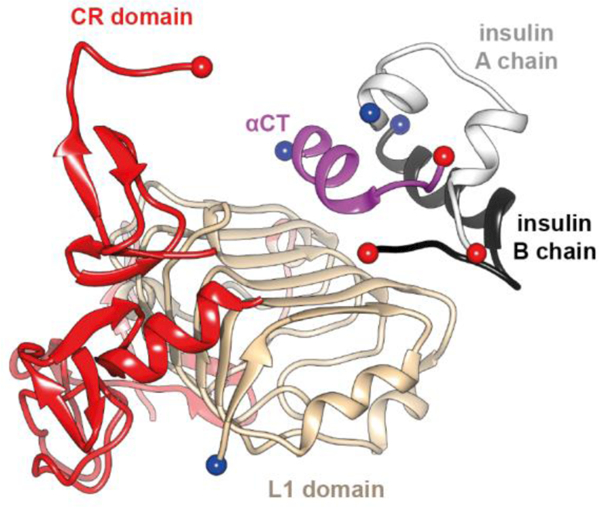
Interaction of insulin with the receptor’s primary binding site. Schematic shows overall organization of the insulin microreceptor (μIR, the L1-CR module + an αCT peptide) in complex with insulin [40, 41]. The schematic is based on PDB entry 4OGA [41].
This structure reveals conformational change both in insulin and in the αCT segment upon their engagement and dissection of the atomic details has led to significant structural insight into the insulin’s classical structure-activity relationships. The details of the insulin / insulin micro-receptor complex are as follow [40, 41].(i) The αCT helix, compared to its conformation within the apo ectodomain structure, has undergone restructuring upon the L1-β2 surface (Figure 12). In particular, its axis is rotated ~30° to lie approximately at right angles to the direction of the β strands. In addition, it has unwound at its N terminus and wound on at its C terminus; the resultant helix spans residues 705–715 rather than residues 694–710.
Figure 12.
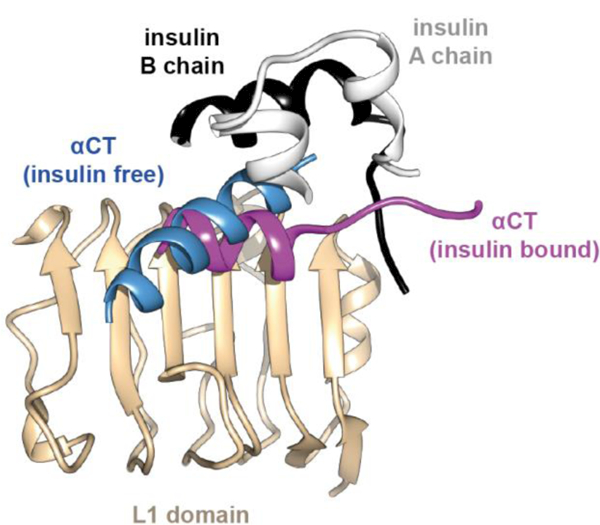
Conformational change in the primary binding site of the IR upon insulin binding. Schematic shows rotation and re-organization of αCT on the surface of domain L1 upon insulin binding. The schematic is based on PDB entries 4ZXB [48] and 4OGA [41].
(ii) The insulin B-chain helix (observed here to span residues B9-B20) lies on the C-terminal edge of the L1-β2 surface, packing adjacent and anti-parallel to the αCT helix. To allow such accommodation, insulin B-chain residues B24 to B27 fold away from the core of the hormone, with the direction of the polypeptide segment B24-B27 now being approximately perpendicular to the axes of the receptor αCT helix and insulin B-chain helix [41] (Figure 13).
Figure 13.
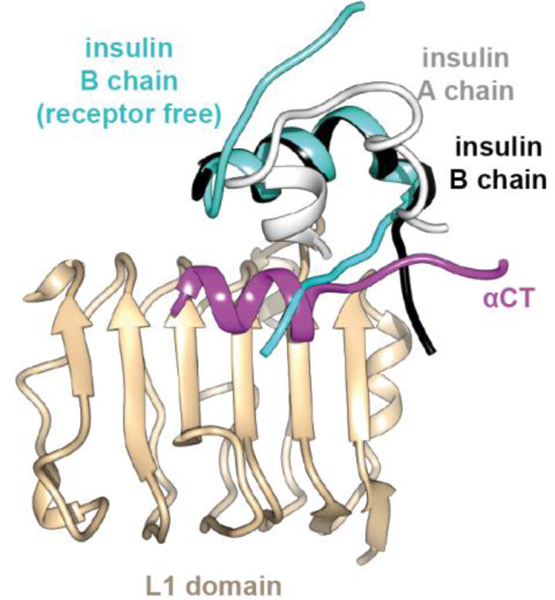
Folding away from the hormone core of insulin residues B24-B30 upon engagement with μIR. The receptor-bound insulin B chain is in black and A chain in white, overlaid with the B chain of the free hormone in cyan. The schematic is based on PDB entries 4INS [6] and 4OGA [41].
(iii) No crystallographic electron density is apparent in this structure for insulin B-chain residues beyond residue B27, suggesting a lack of specific interaction of these elements with the receptor. (iv) No indication is apparent in this structure that the N-terminal helical extension of the insulin B-chain helix is formed upon microreceptor engagement. (v) The side chains of the αCT residues His 710 and Phe714 engage the exposed hormone core directly, with that of Phe714 effectively occupying the position of the side chain of TyrB26 in receptor-free insulin (Figure 14).
Figure 14.
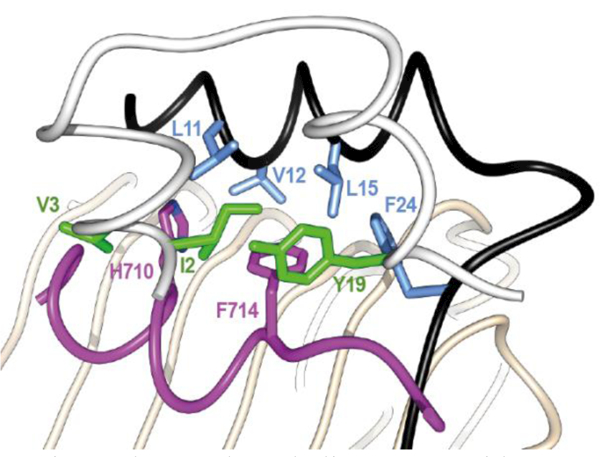
Details of the interaction of αCT residue His710 and Phe714 with the hormone core. The insulin A chain is in white with green side chains, the B chain is in black with light blue side chains. The receptor L1 domain is in tan and the receptor αCT helix is in magenta. The schematic is based on PDB entry 4OGA [41].
3.3 |. PACKING OF THE AROMATIC TRIPLET AT HORMONE-RECEPTOR INTERFACE
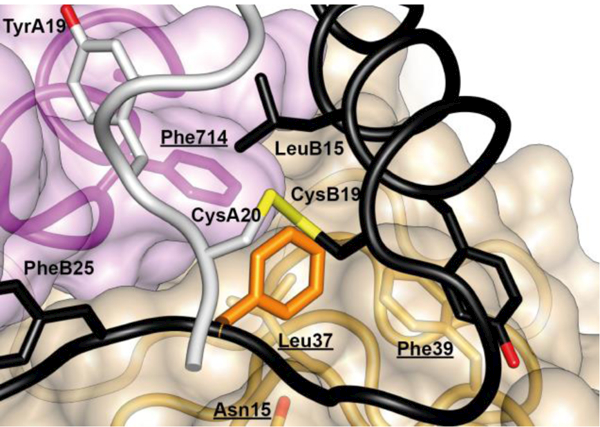
Upon displacement of the B-chain C terminus from the hormone core, the side chain of insulin residue PheB24 undergoes a rotameric re-configuration leaving the phenyl ring in effectively the same relative place within the hormone core that it occupies in receptor-free insulin (Figure 15). Such positioning buries insulin residue PheB24 within a large hydrophobic cavity formed by insulin residues TyrA19, CysA20, LeuB15, receptor αCT residue Phe714 and receptor L1 domain residues An15, Leu37 and Phe39 (Figure 16).
Figure 15.
Re-positioning of insulin residues B24-B27 upon engagement with μIR. The receptor-bound insulin B chain is in black and A chain in white, overlaid with the B chain of the free hormone in cyan. The schematic is based on PDB entries 4INS [6] and 4OGA [41].
Figure 16.
Atomic environment of PheB24 within the Site 1 μIR complex. Schematic shows all side chains of all residues within 4 Å of that of insulin PheB24 (unlabeled, orange). Insulin A chain is in white, insulin B chain is in black, L1 domain of the insulin receptor is in tan, αCT segment of the insulin receptor is in magenta. Receptor residues have underlined labels whereas insulin residues are in normal font. The schematic is based on PDB entry 4OGA [41].
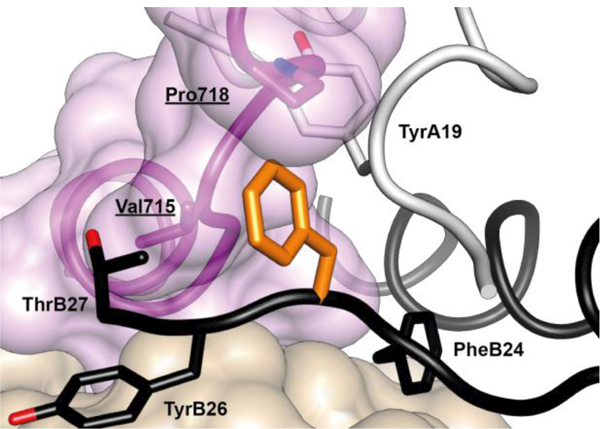
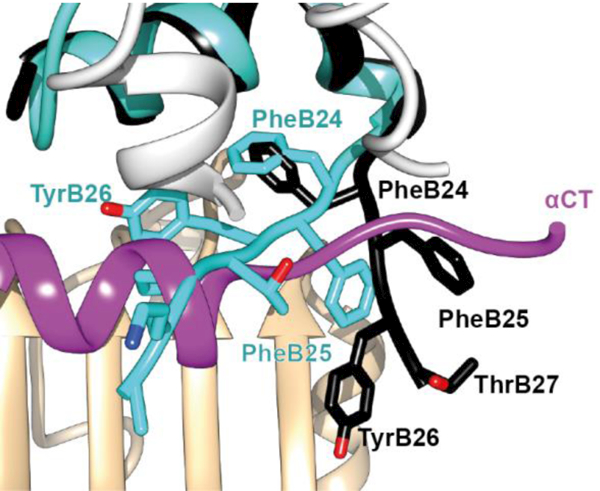
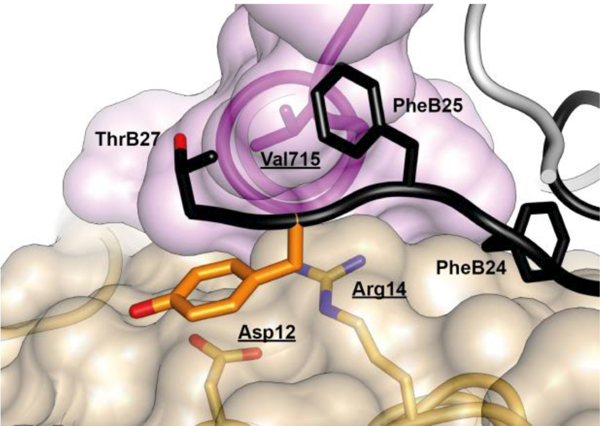
In contrast to the positioning of the side chain of PheB24, the side chains of residues PheB25 and TyrB26 are largely exposed within the μIR complex. The side chain of residue PheB25 engages the αCT residues Val715 and Pro718 and is otherwise positioned adjacent to that of insulin residue ThrB27 (Figure 17). The side chain of residue TyrB26 is rotated away from the hormone core, stacking parallel to the side chains of the receptor L1 domain residues Asp12 and Arg14 (Figure 18).
Figure 17.
Atomic environment of PheB25 within the Site 1 μIR complex. Schematic shows all side chains of all residues within 4 Å of that of insulin PheB25 (unlabeled, orange). Color and labeling schemes are otherwise as in Figure 12. (q.v.). The schematic is based on PDB entry 4OGA [41].
Figure 18.
Atomic environment of TyrB26 within the Site 1 μIR complex. Schematic shows all side chains of all residues within 4 Å of that of insulin TyrB26 (unlabeled, orange). Color- and labelling schemes are otherwise as in Fig. 12. (q.v.). The schematic is based on PDB entry 4OGA [41].
Residue ThrB27 appears weakly ordered in the μIR complexes, with B chain residues beyond B27 almost completely disordered. The latter observation is consistent with the ability of these residues to tolerate significant mutation, chemical modification or deletion [6, 102].
4 |. FUTURE PERSPECTIVES
A major challenge is posed by extension of present structures of model hormone-receptor complexes [47, 48] to the holo-receptor. Such progress is likely to require a series of structures, each representing a step in the complex mechanism of conformational change leading to signal propagation. A significant advance was recently obtained through single-particle cryo-electron microscopy analysis of the insulin-ectodomain complex [49]. Because the isolated ectodomain lacks a single high-affinity site and because the present analysis encompassed only ca. 10% of the images, it is not yet clear how the rearrangement of fibronectin-homology domains observed in the structure [49] relates to the structure of the free ectodomain [47, 48] or μIR complexes [40, 41]. Of particular interest will be understanding the connection between Site 2 as inferred from the cryo-EM structure [49] and Site 2 as defined by kinetic analysis of insulin analogs [96, 97]. We speculate that the aromatic triplet plays a critical role to initiate a series of conformational changes associated with the modular reorganization of the ectodomain on hormone binding.
The aromatic triplet of vertebrate insulins and insulin-like growth factors may not be essential for insulin-like ligands. The recent structure of a venom insulin-like protein from an invertebrate (Conus geographus) provides an example of a “miniature” insulin lacking the C-terminal β-strand of the B chain [103].It is possible that such minimal insulin scaffolds could provide templates for design of novel therapeutic insulins. The C. geographus ligand contains multiple post-translational modifications whose contributions to stability and activity will require characterization.
A key constraint in the pharmacology of insulin analogs is posed by chemical- and physical degradation [7, 18]. The receptor-bound open conformation of the hormone may be particularly susceptible to such degradation, including formation of amyloid (insulin fibrillation). A promising structure-based route to the engineering of ultra-stable analogs is provided by foreshortened C domains [104, 105]. In such single-chain insulins (SCIs) the aromatic triplet resides in a closed conformation—tethered by a foreshortened connecting peptide—and yet sufficient “play” is provided to enable its detachment on receptor binding. It would be of future interest to investigate crystal- or cryo-EM structures of SCI-receptor complexes as probes of the closed-open transition. In the context of this issue, dedicated to the late Prof. Steiner, it would be fitting if the B-C-A structure of proinsulin [22, 23] should find application in next-generation therapeutic insulin analogs.
We envisage that continuing characterization of an insulin-related superfamily, spanning 500 million years in the natural history of both vertebrates and invertebrates, will provide an increasingly fertile model for studies of protein evolution. How and why particular amino acids are conserved are questions that cross from structural biology to cell biology and physiology. Whereas the structural origins of some evolutionary constraints (such as co-conservation of critical hormone-receptor contacts) may be apparent in structures (e.g., of complexes at higher resolution), deciphering other constraints may require study of off-pathway events, such as toxic protein misfolding [18–21]. Other residues may contribute to on-pathway folding efficiency and yet be dispensable once the native state is achieved.
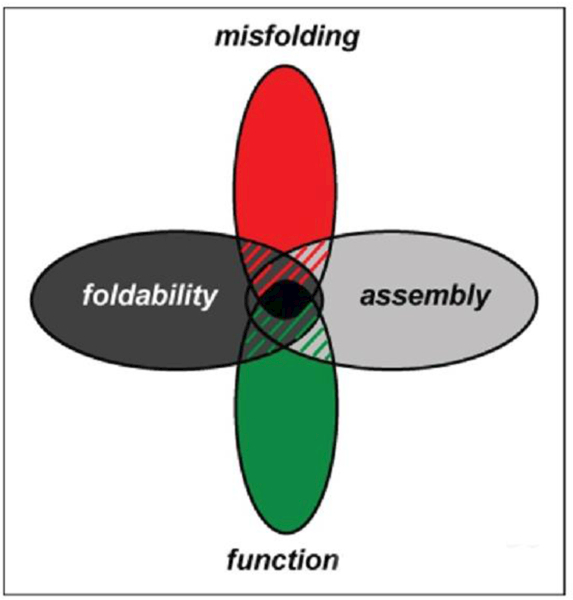
Extant insulin sequences thus lie at the intersection of multiple explicit and implicit constraints (Figure 19). Many—and indeed perhaps most—residues in insulin are likely to play different roles at distinct stages in the hormone’s complex “life cycle”: dissecting these roles will require high-resolution analysis of conformational change in this remarkable protein from biosynthesis to function and degradation.
Figure 19.
Evolutionary constraints and insulin fibrillation: Venn diagram showing intersection of multiple constraints: function, foldability, misfolding and assembly.
Acknowledgements
We dedicate this review to our late colleagues G.G. Dodson, M. Kochoyan, N.M. McKern, and C.W. Ward with special recognition to the late D.F. Steiner, whose multifaceted contributions inspired this conference. We thank J.G. Menting, B. Smith for helpful discussion and N. Rege and N. Wickramasinghe for assistance with the manuscript. MAW thanks R. Mirmia and S.E. Shoelson for sharing their recollections of insulin research at the University of Chicago in the period 1982–1992, during which studies of the aromatic triplet were of central interest to the laboratories of Professors Steiner and Tager in the Department of Biochemistry & Molecular Biology and to the laboratories of Professors Rubenstein and Polonsky in the Department of Medicine.
Funding Information:
M.A.W. is supported by the National Institutes of Health (R01 DK040949 and DK079233) and by grants from the JDRF and the Leona M. and Harry B. Helmsley Charitable Trust.
M.C.L. is supported by grants from the Australian National Health and Medical Research Council Project Grant APP1058233 (to M. C. L.), by Victorian State Government Operational Infrastructure Support, and by the Australian NHMRC Independent Research Institutes Infrastructure Support Scheme (the latter two to his institution).
Footnotes
Disclosures: M.A.W. has equity in Thermalin Diabetes, LLC (Cleveland, OH USA), where he serves as Chief Innovation Officer. Part of M.C.L.’s research is funded by Sanofi (Germany). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies cited.
Author Contributions. The two authors contributed equally to this article.
REFERENCES
- [1].Bliss M ed. The discovery of insulin. Chicago, Illinois: University of Chicago Press, 1982. [Google Scholar]
- [2].Sanger F, Thompson EO.The amino-acid sequence in the glycyl chain of insulin. Biochem J. 1952; 52: iii. [PubMed] [Google Scholar]
- [3].Ryle AP, Sanger F, Smith LF, Kitai R.The disulphide bonds of insulin. Biochem J. 1955; 60: 541–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sanger F Chemistry of insulin; determination of the structure of insulin opens the way to greater understanding of life processes. Science. 1959; 129: 1340–1344 [DOI] [PubMed] [Google Scholar]
- [5].Abel JJ.Crystalline Insulin. Proc Natl Acad Sci U S A. 1926; 12: 132–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baker EN, Blundell TL, Cutfield JF, et al. The structure of 2Zn pig insulin crystals at 1.5 Å resolution. Philos Trans R Soc Lond B Biol Sci. 1988; 319: 369–456 [DOI] [PubMed] [Google Scholar]
- [7].Brange J ed. Galenics of Insulin: The Physico-chemical and Pharmaceutical Aspects of Insulin and Insulin Preparations. Berlin: Springer Berlin Heidelberg, 1987. [Google Scholar]
- [8].Ferry G.Dorothy Hodgkin: A Life 1edn: Cold Spring Harbor Laboratory Press, 2000 [Google Scholar]
- [9].Adams MJ, Blundell TL, Dodson EJ, et al. Structure of rhombohedral 2 zinc insulin crystals. Nature. 1969; 224: 491–495 [Google Scholar]
- [10].Blundell TL, Cutfield JF, Cutfield SM, et al. Atomic positions in rhombohedral 2-zinc insulin crystals. Nature. 1971; 231: 506–511 [DOI] [PubMed] [Google Scholar]
- [11].Polonsky KS, Rubenstein AH Donald F Steiner MD, 1930–2014: pioneering diabetes researcher. Diabetologia. 2015; 58: 419–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Weiss MA, Chan SJ Remembering Donald F. Steiner. Front Endocrinol (Lausanne). 2015; 6: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Katsoyannis PG. Synthesis of insulin. Science. 1966; 154: 1509–1514 [DOI] [PubMed] [Google Scholar]
- [14].Katsoyannis PG. Synthetic studies on the A and B chains of insulin. Vox Sang. 1964; 9: 227–248 [PubMed] [Google Scholar]
- [15].Zahn H Chemical synthesis of proteins. Naturwissenschaften. 1967; 54: 396–402 [DOI] [PubMed] [Google Scholar]
- [16].Kung YT, Du YC, Huang WT, Chen CC, Ke LT. Total synthesis of crystalline bovine insulin. Sci Sin. 1965; 14: 1710–1716 [PubMed] [Google Scholar]
- [17].Wang CC, Tsou CL. The insulin A and B chains contain sufficient structural information to form the native molecule. Trends Biochem Sci. 1991; 16: 279–281 [DOI] [PubMed] [Google Scholar]
- [18].Brange J, Andersen L, Laursen ED, Meyn G, Rasmussen E. Toward understanding insulin fibrillation. J Pharm Sci. 1997; 86: 517–525 [DOI] [PubMed] [Google Scholar]
- [19].Hua QX, Weiss MA. Mechanism of insulin fibrillation: the structure of insulin under amyloidogenic conditions resembles a protein-folding intermediate. J Biol Chem. 2004; 279: 21449–21460 [DOI] [PubMed] [Google Scholar]
- [20].Ahmad A, Uversky VN, Hong D, Fink AL. Early events in the fibrillation of monomeric insulin. J Biol Chem. 2005; 280: 42669–42675 [DOI] [PubMed] [Google Scholar]
- [21].Devlin GL, Knowles TP, Squires A, et al. The component polypeptide chains of bovine insulin nucleate or inhibit aggregation of the parent protein in a conformation-dependent manner. J Mol Biol. 2006; 360: 497–509 [DOI] [PubMed] [Google Scholar]
- [22].Steiner DF, Oyer PE. The biosynthesis of insulin and a probable precursor of insulin by a human islet cell adenoma. Proc Natl Acad Sci U S A. 1967; 57: 473–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Steiner DF, Cunningham D, Spigelman L, Aten B. Insulin biosynthesis: evidence for a precursor. Science. 1967; 157: 697–700 [DOI] [PubMed] [Google Scholar]
- [24].Steiner DF, Clark JL, Nolan C, et al. Proinsulin and the biosynthesis of insulin. Recent Prog Horm Res. 1969; 25: 207–282 [DOI] [PubMed] [Google Scholar]
- [25].Sun J, Cui J, He Q, Chen Z, Arvan P, Liu M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes☆. Molecular aspects of medicine. 2015; 42: 105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu M, Weiss MA, Arunagiri A, et al. Biosynthesis, structure, and folding of the insulin precursor protein Dibetes, Obesity Metab. 2018; in press: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bentley G, Dodson E, Dodson G, Hodgkin D, Mercola D. Structure of insulin in 4-zinc insulin. Nature. 1976; 261: 166–168 [DOI] [PubMed] [Google Scholar]
- [28].Derewenda U, Derewenda Z, Dodson EJ, et al. Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer. Nature. 1989; 338: 594–596 [DOI] [PubMed] [Google Scholar]
- [29].Badger J, Harris MR, Reynolds CD, et al. Structure of the pig insulin dimer in the cubic crystal. Acta Crystallogr B Struct Sci. 1991; 47: 127–136 [DOI] [PubMed] [Google Scholar]
- [30].Ciszak E, Smith GD. Crystallographic evidence for dual coordination around zinc in the T3R3 human insulin hexamer. Biochemistry. 1994; 33: 1512–1517 [DOI] [PubMed] [Google Scholar]
- [31].Smith GD, Pangborn WA, Blessing RH. The structure of T6 human insulin at 1.0 A resolution. Acta Crystallogr D Biol Crystallogr. 2003; 59: 474–482 [DOI] [PubMed] [Google Scholar]
- [32].Boelens R, Ganadu ML, Verheyden P, Kaptein R. Two-dimensional NMR studies on des-pentapeptide-insulin. Proton resonance assignments and secondary structure analysis. Eur J Biochem. 1990; 191: 147–153 [DOI] [PubMed] [Google Scholar]
- [33].Knegtel RM, Boelens R, Ganadu ML, Kaptein R. The solution structure of a monomeric insulin. A two-dimensional 1H-NMR study of des-(B26-B30)-insulin in combination with distance geometry and restrained molecular dynamics. Eur J Biochem. 1991; 202: 447–458 [DOI] [PubMed] [Google Scholar]
- [34].Hua QX, Shoelson SE, Kochoyan M, Weiss MA. Receptor binding redefined by a structural switch in a mutant human insulin. Nature. 1991; 354: 238–241 [DOI] [PubMed] [Google Scholar]
- [35].Jørgensen AM, Kristensen SM, Led JJ, Balschmidt P. Three-dimensional solution structure of an insulin dimer. A study of the B9(Asp) mutant of human insulin using nuclear magnetic resonance, distance geometry and restrained molecular dynamics. J Mol Biol. 1992; 227: 1146–1163 [DOI] [PubMed] [Google Scholar]
- [36].Hua QX, Hu SQ, Frank BH, et al. Mapping the functional surface of insulin by design: structure and function of a novel A-chain analogue. J Mol Biol. 1996; 264: 390–403 [DOI] [PubMed] [Google Scholar]
- [37].Olsen HB, Ludvigsen S, Kaarsholm NC. Solution structure of an engineered insulin monomer at neutral pH. Biochemistry. 1996; 35: 8836–8845 [DOI] [PubMed] [Google Scholar]
- [38].Hua QX, Xu B, Huang K, et al. Enhancing the activity of insulin by stereospecific unfolding. Conformational life cycle of insulin and its evolutionary origins. J Biol Chem. 2009; 284: 14586–14596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Žáková L, Kletvíková E, Veverka V, et al. Structural integrity of the B24 site in human insulin is important for hormone functionality. J Biol Chem. 2013; 288: 10230–10240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Menting JG, Whittaker J, Margetts MB, et al. How insulin engages its primary binding site on the insulin receptor. Nature. 2013; 493: 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Menting JG, Yang Y, Chan SJ, et al. A structural hinge in insulin enables its receptor engagement. Proc Natl Acad Sci USA. 2014; 111: 1–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ismail-Beigi F, Yang Y, Carr K, et al. High-Resolution Structure and Biological Function of an Ultra-Stable Single-Chain Insulin Analog (in preparation). Biochemistry. 2017: [Google Scholar]
- [43].Shoelson S, Haneda M, Blix P, et al. Three mutant insulins in man. Nature. 1983; 302: 540–543 [DOI] [PubMed] [Google Scholar]
- [44].Nanjo K, Sanke T, Miyano M, et al. Diabetes due to secretion of a structurally abnormal insulin (insulin Wakayama). Clinical and functional characteristics of [LeuA3] insulin. J Clin Invest. 1986; 77: 514–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chan SJ, Seino S, Gruppuso PA, Schwartz R, Steiner DF. A mutation in the B chain coding region is associated with impaired proinsulin conversion in a family with hyperproinsulinemia. Proc Natl Acad Sci U S A. 1987; 84: 2194–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Steiner DF, Tager HS, Chan SJ, Nanjo K, Sanke T, Rubenstein AH. Lessons learned from molecular biology of insulin-gene mutations. Diabetes Care. 1990; 13: 600–609 [DOI] [PubMed] [Google Scholar]
- [47].Ward CW, Menting JG, Lawrence MC. The insulin receptor changes conformation in unforeseen ways on ligand binding: Sharpening the picture of insulin receptor activation. Bioessays. 2013; 35: 945–954 [DOI] [PubMed] [Google Scholar]
- [48].Croll TI, Smith BJ, Margetts MB, et al. Higher-Resolution Structure of the Human Insulin Receptor Ectodomain: Multi-Modal Inclusion of the Insert Domain. Structure. 2016; 24: 469–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Scapin G, Dandey VP, Zhang Z, et al. Structure of the Insulin Receptor-Insulin Complex by Single Particle CryoEM analysis. Nature. 2018; 556(7699):122–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brader ML, Dunn MF. Insulin hexamers: new conformations and applications. Trends Biochem Sci. 1991; 16: 341–345 [DOI] [PubMed] [Google Scholar]
- [51].Blundell TL, Dodson GG, Hodgkin DC, Mercola DA. Insulin: the structure in the crystal and its reflection in chemistry and biology. Adv Protein Chem. 1972; 26: 279–402 [Google Scholar]
- [52].Dodson G, Steiner D. The role of assembly in insulin’s biosynthesis. Curr Opin Struct Biol. 1998; 8: 189–194 [DOI] [PubMed] [Google Scholar]
- [53].Chimienti F, Favier A, Seve M. ZnT-8, a pancreatic β-cell-specific zinc transporter. Biometals. 2005; 18: 313–317 [DOI] [PubMed] [Google Scholar]
- [54].Zinc Rungby J., zinc transporters and diabetes. Diabetologia. 2010; 53: 1549–1551 [DOI] [PubMed] [Google Scholar]
- [55].Gursky O, Badger J, Li Y, Caspar DL. Conformational changes in cubic insulin crystals in the pH range 7–11. Biophys J. 1992; 63: 1210–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chothia C, Lesk AM, Dodson GG, Hodgkin DC. Transmission of conformational change in insulin. Nature. 1983; 302: 500–505 [DOI] [PubMed] [Google Scholar]
- [57].Brems DN, Alter LA, Beckage MJ, et al. Altering the association properties of insulin by amino acid replacement. Protein Eng. 1992; 5: 527–533 [DOI] [PubMed] [Google Scholar]
- [58].Schwartz GP, Burke GT, Katsoyannis PG. A superactive insulin: [B10-aspartic acid]insulin(human). Proc Natl Acad Sci U S A. 1987; 84: 6408–6411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Weiss MA, Hua QX, Lynch CS, Frank BH, Shoelson SE. Heteronuclear 2D NMR studies of an engineered insulin monomer: assignment and characterization of the receptor-binding surface by selective 2H and 13C labeling with application to protein design. Biochemistry. 1991; 30: 7373–7389 [DOI] [PubMed] [Google Scholar]
- [60].Nakagawa SH, Zhao M, Hua QX, et al. Chiral mutagenesis of insulin. Foldability and function are inversely regulated by a stereospecific switch in the B chain. Biochemistry. 2005; 44: 4984–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Engels M, Jacoby E, Kurger P, Schlitter J, Wollmer A. The T↔R structural transition of insulin: pathways suggested by targeted energy minimization. J Mol Biol. 1992; 238: 405–414 [DOI] [PubMed] [Google Scholar]
- [62].Kruger P, Hahnen J, Wollmer A. Comparative studies on the dynamics of crosslinked insulin. Eur Biophys J. 1994; 23: 177–187 [DOI] [PubMed] [Google Scholar]
- [63].Tidor B, Karplus M. The contribution of vibrational entropy to molecular association. The dimerization of insulin. J Mol Biol. 1994; 238: 405–414 [DOI] [PubMed] [Google Scholar]
- [64].Zoete V, Meuwly M, Karplus M. Study of the insulin dimerization: Binding free energy calculations and per-residue free energy decomposition. Proteins. 2005; 61: 79–93 [DOI] [PubMed] [Google Scholar]
- [65].Hellman U, Wernstedt C, Westermark P, O’Brien TD, Rathbun WB, Johnson KH. Amino acid sequence from degu islet amyloid-derived insulin shows unique sequence characteristics. Biochem Biophys Res Commun. 1990; 169: 571–577 [DOI] [PubMed] [Google Scholar]
- [66].Nishi M, Steiner DF. Cloning of complementary DNAs encoding islet amyloid polypeptide, insulin, and glucagon precursors from a New World rodent, the degu, Octodon degus. Mol Endocrinol. 1990; 4: 1192–1198 [DOI] [PubMed] [Google Scholar]
- [67].Pandyarajan V, Phillips NB, Rege NK, Lawrence MC, Whittaker J, Weiss MA. Contribution of TyrB26 to the Function and Stability of Insulin. Structure-activity relationships at a conserved hormone-receptor interface. J Biol Chem. 2016; 291: 12978–12990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Nakagawa SH, Hua QX, Hu SQ, et al. Chiral mutagenesis of insulin. Contribution of the B20-B23 β-turn to activity and stability. J Biol Chem. 2006; 281: 22386–22396 [DOI] [PubMed] [Google Scholar]
- [69].Mirmira RG, Tager HS. Role of the phenylalanine B24 side chain in directing insulin interaction with its receptor: Importance of main chain conformation. J Biol Chem. 1989; 264: 6349–6354 [PubMed] [Google Scholar]
- [70].Pandyarajan V, Smith BJ, Phillips NB, et al. Aromatic Anchor at an Invariant Hormone-Receptor Interface Function of Insulin Residue B24 with Aplication to Protein Design. J Biol Chem. 2014; 289: 34709–34727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mirmira RG, Nakagawa SH, Tager HS. Importance of the character and configuration of residues B24, B25, and B26 in insulin-receptor interactions. J Biol Chem. 1991; 266: 1428–1436 [PubMed] [Google Scholar]
- [72].Shoelson SE, Polonsky KS, Zeidler A, Rubenstein AH, Tager HS. Human insulin B24 (Phe →Ser), secretion and metabolic clearance of the abnormal insulin in man and in a dog model. J Clin Invest. 1984; 73: 1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kobayashi M, Ohgaku S, Iwasaki M, Maegawa H, Shigeta Y, Inouye K. Supernormal insulin: [D-PheB24]-insulin with increased affinity for insulin receptors. Biochem Biophys Res Commun. 1982; 107: 329–336 [DOI] [PubMed] [Google Scholar]
- [74].Kwok SC, Steiner DF, Rubenstein AH, Tager HS. Identification of a point mutation in the human insulin gene giving rise to a structurally abnormal insulin (insulin Chicago). Diabetes. 1983; 32: 872–875 [DOI] [PubMed] [Google Scholar]
- [75].Nakagawa SH, Tager HS. Role of the phenylalanine B25 side chain in directing insulin interaction with its receptor. Steric and conformational effects. J Biol Chem. 1986; 261: 7332–7341 [PubMed] [Google Scholar]
- [76].Mirmira RG, Tager HS. Disposition of the phenylalanine B25 side chain during insulin-receptor and insulin-insulin interactions. Biochemistry. 1991; 30: 8222–8229 [DOI] [PubMed] [Google Scholar]
- [77].Xu B, Huang K, Chu YC, et al. Decoding the cryptic active conformation of a protein by synthetic photoscanning: Insulin inserts a detachable arm between receptor domains. J Biol Chem. 2009; 284: 14597–14608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kurose T, Pashmforoush M, Yoshimasa Y, et al. Cross-linking of a B25 azidophenylalanine insulin derivative to the carboxyl-terminal region of the a-subunit of the insulin receptor. Identification of a new insulin-binding domain in the insulin receptor. J Biol Chem. 1994; 269: 29190–29197 [PubMed] [Google Scholar]
- [79].Kitagawa K, Ogawa H, Burke GT, Chanley JD, Katsoyannis PG. Critical role of the A2 amino acid residue in the biological activity of insulin: [2-glycine-A]- and [2-alanine-A]insulins. Biochemistry. 1984; 23: 1405–1413 [DOI] [PubMed] [Google Scholar]
- [80].Nakagawa SH, Tager HS. Importance of aliphatic side-chain structure at positions 2 and 3 of the insulin A chain in insulin-receptor interactions. Biochemistry. 1992; 31: 3204–3214 [DOI] [PubMed] [Google Scholar]
- [81].Xu B, Hua QX, Nakagawa SH, et al. Chiral mutagensis of insulin’s hidden receptor-binding surface: structure of an allo-isoleucineA2 analogue. J Mol Biol. 2002; 316: 435–441 [DOI] [PubMed] [Google Scholar]
- [82].Ullrich A, Bell JR, Chen EY, et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature. 1985; 313: 756–761 [DOI] [PubMed] [Google Scholar]
- [83].Ebina Y, Ellis L, Jarnagin K, et al. The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signalling. Cell. 1985; 40: 747–758 [DOI] [PubMed] [Google Scholar]
- [84].Shier P, Watt VM. Primary structure of a putative receptor for a ligand of the insulin family. J Biol Chem. 1989; 264: 14605–14608 [PubMed] [Google Scholar]
- [85].Sparrow LG, McKern NM, Gorman JJ, et al. The disulfide bonds in the C-terminal domains of the human insulin receptor ectodomain. J Biol Chem. 1997; 272: 29460–29467 [DOI] [PubMed] [Google Scholar]
- [86].Sparrow LG, Lawrence MC, Gorman JJ, et al. N-linked glycans of the human insulin receptor and their distribution over the crystal structure. Proteins. 2008; 71: 426–439 [DOI] [PubMed] [Google Scholar]
- [87].Sparrow LG, Gorman JJ, Strike PM, et al. The location and characterisation of the O-linked glycans of the human insulin receptor. Proteins. 2007; 66: 261–265 [DOI] [PubMed] [Google Scholar]
- [88].Seino S, Bell GI. Alternative splicing of human insulin receptor messenger RNA. Biochem Biophys Res Commun. 1989; 159: 312–316 [DOI] [PubMed] [Google Scholar]
- [89].Belfiore A, Malaguarnera R, Vella V, et al. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr Rev. 2017; 38: 379–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].McKern NM, Lawrence MC, Streltsov VA, et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature. 2006; 443: 218–221 [DOI] [PubMed] [Google Scholar]
- [91].Schäffer L, Ljungqvist L. Identification of a disulfide bridge connecting the α-subunits of the extracellular domain of the insulin receptor. Biochem Biophys Res Commun. 1992; 189: 650–653 [DOI] [PubMed] [Google Scholar]
- [92].Gutmann T, Kim KH, Grzybek M, Walz T, Coskun U. Visualization of ligand-induced transmembrane signaling in the full-length human insulin receptor. J Cell Biol. 2018; 17(5):1643–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Xu Y, Kong GK, Menting JG, et al. How ligand binds to the type 1 insulin-like growth factor receptor. Nat Commun. 2018; 9: 821–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].De Meyts P, Roth J, Neville J, Gavin JR, Lesniak MA. Insulin ineractions with its receptors: experimental evidence for negative cooperativity. Biochem Biophys Res Commun. 1973; 55: 154–161 [DOI] [PubMed] [Google Scholar]
- [95].De Meyts P, van Obberghen E, Roth J. Mapping of the residues responsible for the negative cooperativity of the receptor-binding region of insulin. Nature. 1978; 273: 504–509 [DOI] [PubMed] [Google Scholar]
- [96].De Meyts P The structural basis of insulin and insulin-like growth factor-I receptor binding and negative co-operativity, and its relevance to mitogenic versus metabolic signaling. Diabetologia. 1994; 37: S135–S148 [DOI] [PubMed] [Google Scholar]
- [97].Schäffer L A model for insulin binding to the insulin receptor. Eur J Biochem. 1994; 221: 1127–1132 [DOI] [PubMed] [Google Scholar]
- [98].Brandt J, Andersen AS, Kristensen C. Dimeric fragment of the insulin receptor alpha-subunit binds insulin with full holoreceptor affinity. J Biol Chem. 2001; 276: 12378–12384 [DOI] [PubMed] [Google Scholar]
- [99].Kristensen C, Wiberg FC, Andersen AS. Specificity of insulin and insulin-like growth factor I receptors investigated using chimeric mini-receptors. Role of C-terminal of receptor α subunit. J Biol Chem. 1999; 274: 37351–37356 [DOI] [PubMed] [Google Scholar]
- [100].Kristensen C, Andersen AS, Østergaard S, Hansen PH, Brandt J. Functional reconstitution of insulin receptor binding site from non-binding receptor fragments. J Biol Chem. 2002; 277: 18340–18345 [DOI] [PubMed] [Google Scholar]
- [101].Chan SJ, Nakagawa S, Steiner DF. Complementation analysis demonstrates that insulin cross-links both a subunits in a truncated insulin receptor dimer. J Biol Chem. 2007; 282: 13754–13758 [DOI] [PubMed] [Google Scholar]
- [102].Cosmatos A, Ferderigos N, Katsoyannis PG. Chemical synthesis of [des(tetrapeptide B27−30), Tyr(NH2)26-B] and [des(pentapeptide B26−30), Phe(NH2)25-B] bovine insulins. Int J Protein Res. 1979; 14: 457–471 [DOI] [PubMed] [Google Scholar]
- [103].Menting JG, Gajewiak J, MacRaild CA, et al. A minimized human insulin-receptor-binding motif revealed in a Conus geographus venom insulin. Nat Struc Mol Biol. 2016; 23: 916–920 [DOI] [PubMed] [Google Scholar]
- [104].Glidden MD, Yang Y, Smith NA, et al. Solution structure of an ultra-stable single-chain insulin analog connects protein dynamics to a novel mechanism of receptor binding. J Biol Chem. 2018; 293: 69–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Glidden MD, Aldabbagh K, Phillips NB, et al. An ultra-stable single-chain insulin analog resists thermal inactivation and exhibits biological signaling duration equivalent to the native protein. J Biol Chem. 2018; 293: 47–68 [DOI] [PMC free article] [PubMed] [Google Scholar]