

Abstract
Preparation of degradable materials using reversible deactivation radical polymerizations (RDRP) is of particular interest for biomedical applications. In this paper we report preparation of degradable copolymers of 2-methylene-4-phenyl-1, 3-dioxolane (MPDL), monomer which undergoes ring-opening reaction and forms ester bond upon radical polymerization, with hydrophobic and hydrophilic methacrylate monomers using atom transfer radical polymerization (ATRP). Copolymers composition and degradation were evaluated upon varied temperature and monomer type.
Keywords: degradable materials, reversible deactivation radical polymerizations, copolymers of 2-methylene-4-phenyl-1, 3-dioxolane, atom transfer radical polymerization
Degradability is one of the most important requirements for materials targeting biomedical applications [1–7], including degradable sutures, drug delivery systems, hydrogels, wound dressings and cell growing platforms [1–3, 8–11]. Indeed, designed degradable polymers have become the material of choice for drug/biomolecule delivery due to their initially large hydrodynamic size, solubility, stealth properties, and stimuli responsiveness [5–7, 12–15]. These degradable materials can be applied for delivery of hydrophobic drugs, which have very limited solubility in aqueous environment [16– 19] or biomolecules which would degrade or cause an immune response if added to a living entity on their own [13, 20–23]. A larger hydrodynamic radius provides longer circulation time, and also helps targeting cancer cells due to enhanced permeability and retention effect [20, 21, 23, 24]. However, robust drug delivery systems can accumulate in organs, such as liver and kidneys, during their circulation, and without timely excretion can cause immune response and inflammation [1, 4, 25]. Thus for the drug delivery applications, where the delivery material is targeted to circulate inside a human body, polymer degradability is especially important. This is why degradable synthetic polymers such as polycaprolactone, poly(lactic acid) or natural polymers such as chitosan are often utilized in this field [3, 4, 9, 26].
Reversible deactivation radical polymerization (RDRP) methods allow incorporation of various functionalities during the synthesis of polymers with diverse compositions and architectures [27]. However, if only vinyl monomers are incorporated into the polymers, the resulting materials consist solely of carbon-carbon bonds that have very limited degradability under physiological conditions [4]. Consequently, generating polymers by RDRP methods with appropriate degradation profiles remains a subject of high interest. There are several degradable linkages that are commonly utilized in synthetic delivery systems such as esters, acetals and disulfide bonds [2, 4, 10, 28, 29]. Acetals and esters can be hydrolytically degraded, while disulfide bonds are redox sensitive [2, 4, 28]. There are several approaches to incorporate degradable functionalities into copolymers synthesized by atom transfer radical polymerization (ATRP) [30]. Linear polymers can be grown from a degradable dual functional initiator, which would allow splitting polymer in half upon degradation [31–34]. For a star polymer synthesis one can either use multifunctional degradable initiators, or star cores prepared with a degradable crosslinker to dissociate the star copolymer into its arms [35–37]. Degradable crosslinkers or inimers can also be utilized in the synthesis of degradable hydrogels and nanogels [10, 38]. It is also possible to prepare degradable polymers containing heteroatoms by other techniques (ring opening, polycondensation) and extend them by ATRP [39–49]. However, some of these approaches can result in preparation of materials, which degrade into chains with broad molecular weight distributions (MWDs), and one has to consider the upper limits for molecular weight (MW) of the degraded components.
In order to incorporate several degradable groups along a polymer chains made from (meth)acrylates or (meth)acrylamides (comprised of only C-C bonds in a backbone) one can use cyclic comonomers with double bonds and incorporated degradable units such as cyclic ketene acetals (CKA), which will undergo ring opening once reacted with a radical, and the degradable moiety will be subsequently incorporated into the backbone of the copolymer [50–54]. Once such monomeric units undergo radical ring-opening polymerization (RROP) and are incorporated into the main C-C chain, the final product would contain ester bonds distributed along the backbone, which would provide desirable degradable properties under physiological conditions.
To date several CKAs have been examined as comonomers for RDRP procedures [Formulas (I)–(IV)].
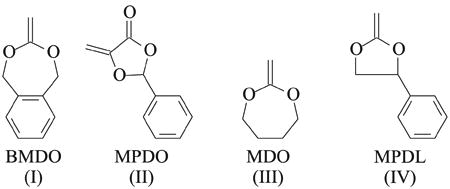
Copolymers with both water-soluble and hydrophobic monomers and CKA monomers, such as 5,6-benzo-2-methylene-1,3-dioxepane (BMDO), were synthesized by reversible addition–fragmentation chain transfer (RAFT), atom transfer radical polymerization (ATRP), and nitroxide-mediated radical polymerization (NMP) [29, 50, 51, 55–63]. Polymerizations were characterized by controlled/“living” behavior, yielding degradable copolymers. Among other CKAs polymerizable by RDRP were 5-methylene-2-phenyl-1,3-dioxolan-4-one (MPDO) [64, 65], 2-methylene-1,3-dioxepane (MDO), and 2-methylene-4-phenyl-1,3-dioxolane (MPDL) [29, 50, 61, 66]. Recently it was reported that NMP copolymerization of MPDL and a water-soluble methacrylate yielded polymers with the higher level of the incorporated CKA comonomer, compared to other tested CKAs like MDO and BMDO [29, 50, 61]. There was one report on homopolymerization of MPDL by ATRP [67], but copolymerization was not investigated. Therefore, it was of interest to investigate copolymerization of MPDL with various types of monomers, typically polymerizable by ATRP, for degradable polymers for potential biomedical applications.
This paper reports the results of a series of studies on the synthesis of copolymers of MPDL with hydrophobic and hydrophilic monomers. n-Butyl acrylate was chosen as a hydrophobic monomer. Methacrylates with either oligo(ethylene oxide) (8–9 units) or poly(ethylene oxide) (45 units) as a side chain were chosen as hydrophilic monomers. This type of water-soluble monomers form biocompatible polymers with comb structures due to their longer side chains. They are commonly used in biomaterials preparation, and it would be beneficial to develop their hydrolytically degradable equivalents. The level of MPDL incorporation, ring-opening efficiency and degradation behavior of the synthesized copolymers were studied.
Experimental Part
Materials
– Butyl acrylate (BA, 99 %, Sigma Aldrich), oligo(ethylene oxide) methyl ether acrylate (OEOA480, 99 %, number average molecular weight M̄n = 480, Sigma Aldrich), oligo(ethylene oxide) methyl ether methacrylate (OEOMA500, 99 %, M̄n = 475, Aldrich) were passed over a column of basic alumina (Fisher Scientific) prior to use.
– Poly(ethylene oxide) methyl ether acrylate (PEOMA2k′ 50 % aqueous solution, M̄n = 2000, Sigma Aldrich) was extracted by dichloromethane and precipitated into hexane prior to use.
– Copper(II) bromide (99.999 %, Sigma Aldrich), N,N-dimethylformamide (DMF, ACS grade, Fisher Scientific), dichloromethane (DCM, HPLC grade, Fisher Scientific), ethyl ether (ACS grade, Fisher Scientific), chloroform-d (Cambridge Isotope Laboratories), acetonitrile-d3 (Cambridge Isotope Laboratories), tris[2-(dimethylamino)ethyl]amine (Me6TREN, 97 %, Sigma Aldrich), ethyl-2-bromo-2-methylpropionate (EBiB, 98 %, Sigma Aldrich), were used as received.
– Radical thermal initiators: 2,2′-azobis(2-methylpropionitrile) (AIBN, Sigma Aldrich), 1,1′-azobis(cyclohexanecarbonitrile) (V40, Sigma Aldrich), 2,2′-azobis(N-butyl-2-methylpropionamide) (Vam110, Wako) were used as received.
– Chloroacetaldehyde dimethyl acetal (97 %), styrene glycol (97 %), Dowex 50WX8 hydrogen form and potassium tert-butoxide (KO-tert-Bu, 98 %) were purchased from Acros.
– 2-methylene-4-phenyl-1,3-dioxolane (MPDL) was synthesized according to previous procedure [69].
Methods of testing
1H NMR (300 and 500 MHz) spectra were recorded on a Bruker Avance 300/500 spectrometer. The conversion of acrylates and methacrylates were determined using near infrared spectroscopy. Molecular weights and distributions were determined by THF, DMF and aqueous GPC. The THF GPC system was based on Polymer Standards Services (PSS) columns (Styrogel 102, 103, 105 Å) with, respectively, tetrahydrofuran (THF) as the eluent at a flow rate of 1 cm3/min at 35°C. DMF GPC utilized dimethylformamide (DMF) containing 50 mM LiBr as the eluent at a flow rate of 1 cm3/min at 50°C. The differential refractive index (RI) detector (Waters, 2414) and multi-angle laser light scattering detector (MALLS) (Wyatt TREOS) were used. The apparent molecular weights and dispersity (M̄w/M̄n) were determined with a calibration based on linear poly(methyl methacrylate) standards using for THF GPC. The aqueous GPC system (model Alliance 2695) was based on an Ultrahydrogel linear column (7.8 – –300 mm, Waters) with phosphate buffered saline (PBS) as the eluent at a flow rate of 1 cm3/min at room temperature and differential RI detector (Waters, 2414). The apparent molecular weights and dispersity (M̄w/M̄n) were determined with a calibration based on linear PEG standards.
Synthesis of the copolymers with incorporated ester groups by radical ring-opening polymerization using atom transfer radical polymerization (ICAR ATRP)
ICAR ATRP of BA with MPDL
BA (2.4 g, 18.7 mmol), MPDL (1.5 g, 9.4 mmol) were mixed with 0.375 cm3 of radical initiator stock solution (25 mM), 0.375 cm3 of CuBr2/Me6TREN stock solution (1/2, 7.5 mM of CuBr2), 0.375 cm3 of EBiB stock solution (250 mM). Reaction mixture was placed in Schlenk flask, sealed and purged with nitrogen for 30 min. Polymerization was started by immersing reaction mixture in a heated oil bath set at either 65 °C, 90 °C, or 120 °C.
ICAR ATRP of OEOA480 with MPDL
OEOA480 (2.4 g, 5 mmol), MPDL (0.4 g, 2.5 mmol) were mixed with 0.1 cm3 of radical initiator stock solution (25 mM), 0.1 cm3 of CuBr2/Me6TREN stock solution (1/2, 7.5 mM of CuBr2), 0.1 cm3 of EBiB stock solution (250 mM), and 2.2 cm3 of DMF. Reaction mixture was placed in Schlenk flask, sealed and purged with nitrogen for 30 min. Polymerization was started by immersing reaction mixture in a heated oil bath set at 90 °C.
ICAR ATRP of OEOMA500 with MPDL
OEOMA500 (2.5 g, 5 mmol), MPDL (0.4 g, 2.5 mmol) were mixed with 0.05 cm3 of radical initiator V40 stock solution (25 mM), 0.1 cm3 of CuBr2/Me6TREN stock solution (1/2, 7.5 mM of CuBr2), 0.1 cm3 of EBiB stock solution (50 mM), and 2.2 cm3 of DMF. Reaction mixture was placed in Schlenk flask, sealed and purged with nitrogen for 30 min. Polymerization was started by immersing reaction mixture in a heated oil bath set at 90 °C.
ICAR ATRP of PEOMA2k with MPDL
PEOMA2k (3 g, 1.5 mmol) was dissolved in 4.5 cm3 of DMF. After that MPDL (0.4 g, 2.5 mmol) were mixed with 0.04 cm3 of radical initiator V40 stock solution (25 mM), 0.04 cm3 of CuBr2/Me6TREN stock solution (1/2, 7.5 mM of CuBr2), 0.1 cm3 of EBiB stock solution (50 mM) and added to the dissolved PEOMA2k. Reaction mixture was placed in Schlenk flask, sealed and purged with nitrogen for 30 min. Polymerization was started by immersing reaction mixture in a heated oil bath set at 90 °C.
Hydrolytic degradation
Poly(BA)-r-poly(MPDL) copolymers were degraded in 5 % KOH solution in mixture of THF/MeOH with a ratio 1/1. Degradation products were neutralized with HCl and precipitated into hexane prior to analysis. Water-soluble polymers were degraded in aqueous 5 % KOH. Samples were dissolved in PBS prior to analysis. Polymers were typically dissolved at 10 mg/cm3 concentration.
Results and Discussion
There are several factors which can influence ring-opening efficiency during RROP. It was reported that the presence of high ring strain in the monomer, the formation of a thermodynamically stable functional group, presence of a radical stabilizing group, and elevated temperatures, all favor a ring-opening reaction during a radical polymerization [69]. It was also reported that MPDL can be copolymerized by free radical polymerization (FRP) with 100% ring-opening at temperatures between 60 °C–120 °C [Scheme A, reaction (1)] [36, 37]. However, in the ATRP homopolymerization of MPDL the efficiency of the ring-opening reaction strongly depended on temperature. The ring-opening became prevalent over vinyl-addition [Scheme A, reaction (2)] only at higher temperatures, above 120 °C [67].
Scheme A.

Therefore, the first set of experiments was designed to investigate ring-opening efficiency during copolymerization of MPDL with BA at different temperatures and monomer concentrations (Scheme B, Table 1).
Scheme B.

Table 1. Copolymerization of BA with MPDL by ICAR ATRP.
| Entry | M1/M2/I/CuBr2/L/RI | T, °C | Conv., % | Time, h | Mnth | M̄n | M̄w/M̄n | fMPDL, % | RO, % |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| 1 | 100/50/1/0.015/0.03/0.1 | 65 | 55 | 9.7 | 10 320 | 5 400 | 1.35 | 29.9 | 35 |
| 2 | 100/50/1/0.015/0.03/0.1 | 88 | 52 | 6 | 11 030 | 4 820 | 1.39 | 23.4 | 46 |
| 3 | 100/50/1/0.015/0.03/0.1 | 110 | 46 | 2 | 9 800 | 4 360 | 1.41 | 26.4 | 55 |
[M1] = [BA] = 1 M, [M2] = [MPDL] = 0.5 M, [I] = [EBiB] = 10 mM, 10 ml total; L – Me6TREN; reaction solvent – DMF; RI – radical initiator: entry 1 – AIBN (Tt1/2=10h = 65 °C), entry 2– V40 (Tt1/2=10h = 88°C), entry 3– Vam110 (Tt1/2=10h = 110 °C); RO – % of MPDL monomer in ring-opened form to ring-closed form; Mnth – theoretical mass, fMPDL – fraction of MPDL incorporated into the p(BA) backbone, monomer conversion was measured by 1H NMR; M̄n and M̄w was obtained by THF GPC with PMMA calibration standards.
Polymerization analysis of the initial reaction conducted at 65 °C (Table 1, entry 1) indicated a well-controlled polymerization (Fig. 1), according to kinetic studies.
Fig. 1. Copolymerization of BA with MPDL by ICAR ATRP: a) first-order kinetic plots, b) evolution of M̄n and M̄w/M̄n with conversion, c) GPC traces for ATRP of p(BA)-r-p(MPDL).

Copolymerization conditions: [BA]: [MPDL]: [EBiB]: [CuBr2]: [Me6TREN]: [AIBN] = 100:50:1:0.015:0.03:0.1, reaction solvent – DMF, 65 °C, [BA] = 1 M, [MPDL] = 0.5 M. MW and GPC traces were obtained by THF GPC with PMMA calibration standards. Linear first-order kinetics plots were obtained for both comonomers, with MPDL being incorporated into the copolymer at a rate a little faster than BA, at the given monomer feed ratio, BA/MPDL = 2/1. At low monomer conversions, MW increased linearly with conversion, but started to deviate toward lower MW when conversion increased to > 20 % (Fig. 1b). M̄w/M̄n values also increased with conversion. According to GPC traces, last two samples were characterized by shift towards higher MW, but low MW tailing was detected (Fig. 1b). Such results suggested some loss of chain-end functionality. Nevertheless, the final copolymer still had a relatively low M̄w/M̄n′ and thus it was isolated and further characterized to determine its composition.
The purified copolymer was further characterized by 1H NMR to determine the mode of incorporation of MPDL, i.e., determine what fraction of incorporated monomer exhibited ring-opening vs. vinyl addition. The composition of the p(BA)-r-p(MPDL) copolymer was determined from the ratio of aromatic protons (P1-3) present in MPDL to the protons from butyl acrylate side chain (B1) (Fig. 2).
Fig. 2. 1H NMR of purified copolymer p(BA)-r-p(MPDL) synthesized at 65°C (300 MHz, CD3CN).
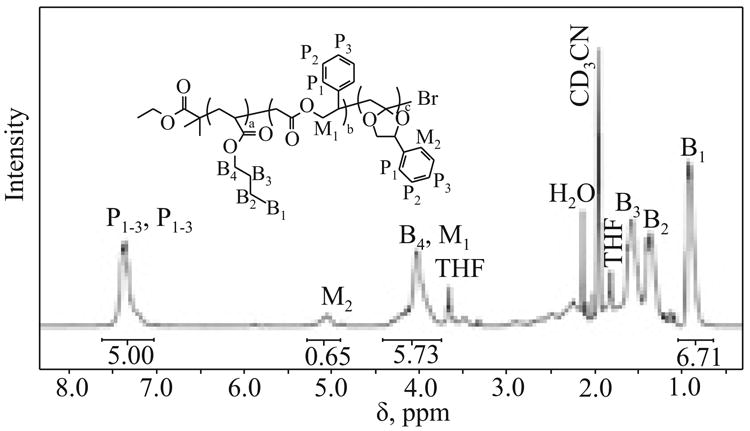
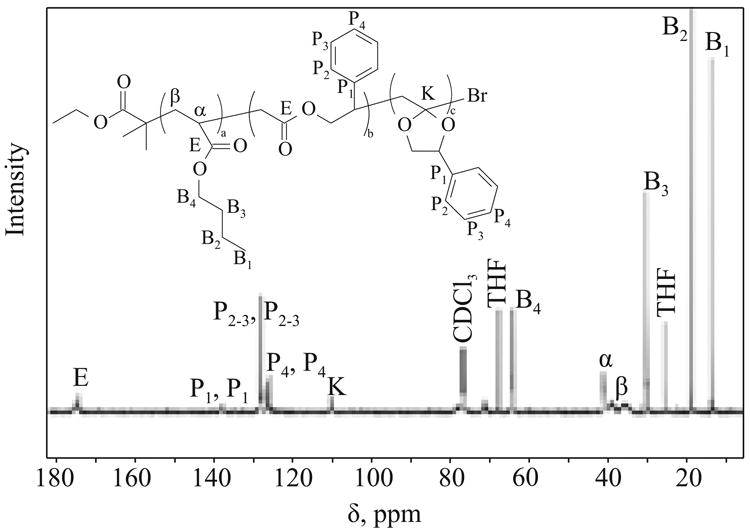
According to this calculation, MPDL incorporation was 29.9 %. The ring-opening efficiency was calculated from 1H NMR spectra, where the signal at ∼5.05 ppm corresponded to the methine proton (M2) on the carbon between the acetal oxygen and the phenyl group (Fig. 2). The difference between the integration of methine proton and phenyl proton provided a value of the percentage of MPDL which underwent the ring-opening reaction. According to the values calculated for copolymerization of BA with MPDL at 65 °C 35 % of incorporated MPDL was in its ring-opened form. 13C NMR was also used to confirm the presence of an acetal carbon (Fig. 3), detected at δ = 110 ppm.
Fig. 3. 13C NMR spectra of purified copolymer p(BA)-r-p(MPDL) synthesized at 65°C (500 MHz, CDCl3).
The next two copolymerizations of BA with MPDL were performed at higher temperatures (Table 1, entries 2 –3). Different free radical initiators were selected for each reaction: the initially used radical initiator (RI) AIBN was replaced by RIs with higher decomposition temperatures, V40 Tt1/2=10h = 88 °C (where t1/2=10h is the 10 h half lifetime of the initiator), and Vam110 with Tt1/2=10h = 110°C for the highest temperature reaction. Polymerizations at 90°C and 110°C were characterized by faster rate, but were also less controlled, yielding polymers with higher M̄w/M̄n. However, the final copolymers were characterized by higher percentage of incorporated MPDL, which underwent ring-opening instead of vinyl addition. According to 1H NMR analysis the peak due to the methine proton present in MPDL (M2), which represents incorporated MPDL that underwent vinyl addition, decreased for the polymers synthesized at the elevated temperatures (Fig. 4).
Fig. 4. 1H NMR of purified copolymers p(BA)-r-p(MPDL) synthesized at different temperatures (300 MHz, CD3CN); spectra were normalized to phenyl protons in each sample; signal at 5.05 ppm corresponds to methine proton (M2) in the polymer unit structure.
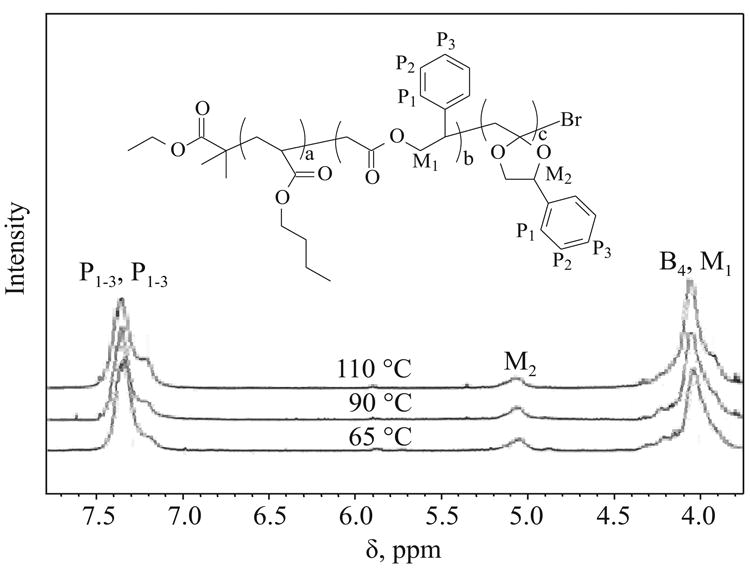
1H NMR spectra obtained for the polymers synthesized at different temperatures were normalized to aromatic protons, and their compositions were calculated based on integration values presented in the Table 2.
Table 2. Calculations of p(BA)-r-p(MPDL) compositions during copolymerization at different temperatures.
| Entry | T, °C | Integration values for the following protons in p(BA)-r-p(MPDL) | Composition a:b:c (mol %)* | ||
|---|---|---|---|---|---|
|
| |||||
| B1 | P1-3 | M2 | |||
|
| |||||
| 1 | 65 | 6.71 | 5 | 0.65 | 70:11:19 |
| 2 | 90 | 9.8 | 5 | 0.54 | 77:11:12 |
| 3 | 110 | 8.35 | 5 | 0.45 | 74:15:11 |
composition a:b:c is for the copolymer structure depicted in Fig. 4.
Increasing the temperature from 65 °C to 90 °C resulted in 30 % increase of incorporated MPDL via ring-opening process. A further increase from 90 °C to 110 °C resulted in another 20 % increase in the content of ring-opened MPDL in the copolymer. Therefore, while ring-opening efficiency could be improved by increase in temperature, the most significant improvement was detected for the first increase from 65 °C to 90 °C. A further 20 °C increase in temperature resulted in marginally higher ring-opening efficiency.
Furthermore, higher molecular weight p(BA)-r--p(MPDL) copolymers were synthesized at varied temperatures to evaluate their degradation behavior based on the ring-opening efficiency. As in a previous set of experiments, ring-opening efficiency increased at higher temperature (Fig. 5). Copolymerizations were conducted at higher monomer concentrations to facilitate higher yield of the targeted copolymers.
Fig. 5. GPC traces of copolymers p(BA)-r-p(MPDL) prepared at different temperatures before and after degradation.

Copolymerization conditions: [BA]: [MPDL]: [EBiB]: [CuBr2]: [Me6TREN]: [RI] = 200:100:1:0.03:0.06:0.1, reaction solvent - DMF, 65 °C–110 °C, [BA] = 3.4 M, [MPDL] = 1.7 M. Each sample was incubated for 45 h in 5 % KOH in THF/ MeOH (1/1), and the polymer was precipitated after acidification with 1 M HCl, dissolved in THF and analyzed by THF GPC. Such difference could potentially be relevant to the difference in the incorporation of the ring-opened form of MPDL for the sample prepared at the lowest temperature. The copolymers were incubated under basic conditions to determine their degradation properties and GPC was used to determine decrease in MW resulting from the degradation reactions (Fig. 5, Table 3).
Table 3. Studies of copolymers p(BA)-r-p(MPDL) prepared at different temperatures before and after degradation.
| Entry | T, °C | M̄n | M̄w/M̄n | fMPDL, % | RO, % |
|---|---|---|---|---|---|
|
| |||||
| 1 | 65 | 13 800 | 1.16 | 24 | 16 |
| 1 degraded | 4 850 | 1.32 | |||
| 2 | 90 | 9 500 | 1.43 | 27 | 45 |
| 2 degraded | 2 690 | 1.28 | |||
| 3 | 110 | 7 590 | 1.55 | 28 | 55 |
| 3 degraded | 1 180 | 1.27 | |||
As expected, according to this analysis, the p(BA)-r--p(MPDL) copolymer with highest MPDL content in the ring-opened form was characterized by the largest decrease in MW. Since the total incorporation of MPDL in these copolymers varied insignificantly, it is likely that drastic difference in the amount of ring-opened MPDL vs. MPDL incorporated via vinyl addition is responsible for more efficient degradation of copolymers prepared at 90 °C and 110 °C compared to the copolymer prepared at 65 °C.
In the next set of experiments, MPDL was copolymerized with water-soluble monomers, such as OEOA480, OEOMA500 and PEOMA2k (Table 4).
Table 4. Copolymerization of MPDL with hydrophilic monomers by ICAR ATRP.
| Entry | M1/M2/I/CuBr2/L/RI | M1 | Time, h | M̄n | M̄w/M̄n | fMPDL, % | RO, % |
|---|---|---|---|---|---|---|---|
|
| |||||||
| 1 | 200/100/1/0.03/0.06/0.1 | OEOA480 | 10.7 | 31 | 1.07 | 20.5 | 32 |
| 2 | 1000/500/1/0.15/0.3/0.5 | OEOA480 | 10 | 50 | 1.37 | 9.2 | 62 |
| 3 | 1000/500/1/0.15/0.3/0.25 | OEOMA500 | 10 | 147 | 1.73 | 6.1 | 82 |
| 4 | 1000/500/1/0.75/1.5/0.25 | OEOMA500 | 6 | 125 | 1.49 | 5.9 | 74 |
| 5 | 150/150/1/0.03/0.06/0.1 | PEOMA2k | 13 | 52 | 1.08 | 6.0 | 96 |
Volume – 5 ml total; reaction solvent – DMF; T = 90 °C; M1 – BA, M2 – MPDL; L – Me6TREN; [I] = [EBiB] = 5 mM; RI – radical initiator: V40 (Tt1/2=10h = 88 °C); entries 1–2: [M1] = 1 M, [M2] = 0.5 M; entry 5: [M1] = 0.3 M, [M2] = 0.3 M; RO – % of MPDL monomer in ring-opened form to ring-closed form; fMPDL – fraction of MPDL incorporated into the polyether backbone, final M̄n was measured by DMF GPC with MALLS detector.
The initial polymerization reaction for OEOA480 was conducted at 65 °C with the ratio of reagents identical to BA/MPDL copolymerization (Table 4, entry 1). According to the analysis, the final copolymer contained around 20 % of MPDL, and 32 % of this MPDL underwent ring-opening (Fig. 6). This was consistent with the results obtained for BA/MPDL copolymerization.
Fig. 6. 1H NMR of purified copolymer p(OEOA480)-r-p(MPDL) synthesized at 65 °C (300 MHz, CD3CN).
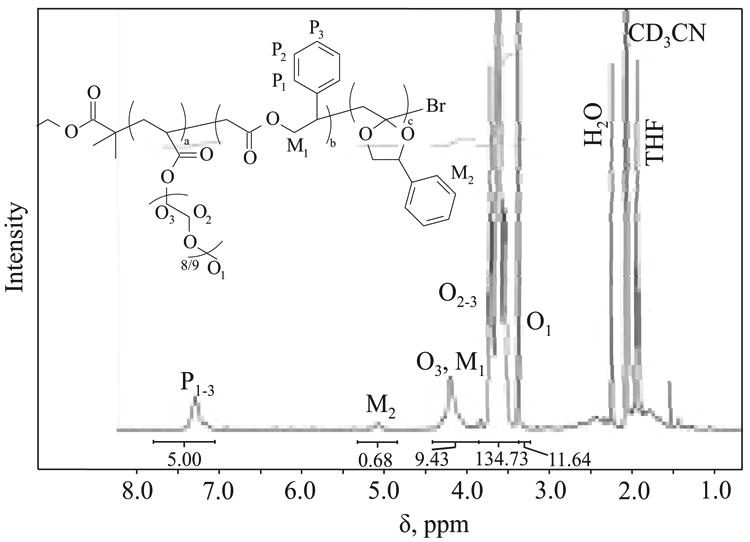
In the next experiment, OEOA480/MPDL copolymerization was conducted at 90 °C to improve percentage of MPDL incorporated into the copolymer in its ring-opened form. Additionally, the targeted degree of polymerization (DP) was increased to 1500. To date, most of synthesized copolymers with CKA were characterized by rather low MW (10 000–20 000), with some systems reaching ∼50 000 [55]. However for certain biological application the preparation of degradable high MW polymers would be especially beneficial for the reasons stated earlier and because lower MW polymers could be removed from a physiological circulation without need for their degradation. In a similar manner to copolymerization with BA, copolymerization of OEOA480 with MPDL at 90 °C resulted in the formation of a copolymer with a higher percentage of MPDL with ring-opened structure (Table 4, entry 2). The percentage of incorporated MPDL which underwent ring-opening during this copolymerization reached 62 %. The fraction of MPDL incorporated into the pOEOA480 backbone was, however, less than 10 %.
When MPDL was copolymerized with OEOA methacrylate analogue, OEOMA500, the overall incorporation of MPDL was lower (Table 4, entry 3–4). Polymerization resulted in high MW copolymer of almost 150 000, but its M̄w/M̄n value was relatively high indicating limited control over polymerization. In the presence of a higher concentration of catalyst, control over polymerization improved and resulted in formation of copolymers with lower M̄w/M̄n, 1.49 vs. 1.73 with 6 % of incorporated MPDL. Even though copolymers of MPDL with OEOMA500 were characterized by higher M̄w/M̄n compared to copolymerization with acrylate OEOA480, it was possible to obtain polymers with MW > 120 000 with M̄w/M̄n ∼1.5 (Table 4, entry 4).
The degradability of both p(OEOA480)-r-p(MPDL) and p(OEOMA500)-r-p(MPDL) was evaluated by incubating the copolymers in 5 % aqueous KOH. Hydrolytic degradation results were analyzed by aqueous GPC to determine the decrease in MW with time (Fig. 7, Table 5).
Fig. 7. Degradation studies of hydrophilic polymers; all samples were neutralized by 1 M HCl and analyzed by water GPC in PBS at pH = 7 (calibrated with linear PEO standards).

Table 5. Degradation studies of hydrophilic copolymers (hydrolysis in 5 % aq. KOH).
| Sample | Time, h | M̄n | M̄w/M̄n |
|---|---|---|---|
|
| |||
| p(OEOA480)-r-p(MPDL) | 0 | 15 500 | 2.27 |
| 20 | 4 620 | 1.40 | |
| 48 | 4 610 | 1.34 | |
|
| |||
| p(OEOMA500)-r-p(MPDL) | 0 | 35 200 | 2.75 |
| 20 | 8 540 | 1.86 | |
| 48 | 7 780 | 1.97 | |
After 20 h, the molecular weight of both the water-soluble polyacrylate and polymethacrylate copolymers decreased by a factor of 3–4, and did not change over the next 28 h, indicating a full degradation had occurred. Final degradation products were characterized by M̄n <10 000, according to calibration with PEO standards. However, it is important to point out that even though apparent M̄n (based on linear PEO standards) of degradable copolymers were only 15 500 for p(OEOA)-r-p(MPDL) and 35 200 for p(OEOMA)-r-p(MPDL), MW of copolymers as measured by MALLS detector was more than 100 000. Degradation of this higher MW fraction of copolymers resulted in formation of degraded products with MW significantly below their initial values.
The final copolymerization in this series of experiments was the copolymerization of MPDL with a PEOMA2k macromonomer. This was evaluated to determine if this procedure would form a degradable brush copolymer by the “grafting through” method (Table 4, entry 5). The synthesized polymer was characterized by incorporation of a similar fraction of MPDL (∼6%) as the lower MW OEOMA500 monomer, however, according to proton NMR analysis, 96% of the MPDL units had undergone ring-opening during the copolymerization (Fig. 8).
Fig. 8. 1H NMR of purified copolymers p(PEOMA2k)-r-p(MPDL) (500 MHz, CD3CN) with insert with zoomed in region 4.8–8 ppm.
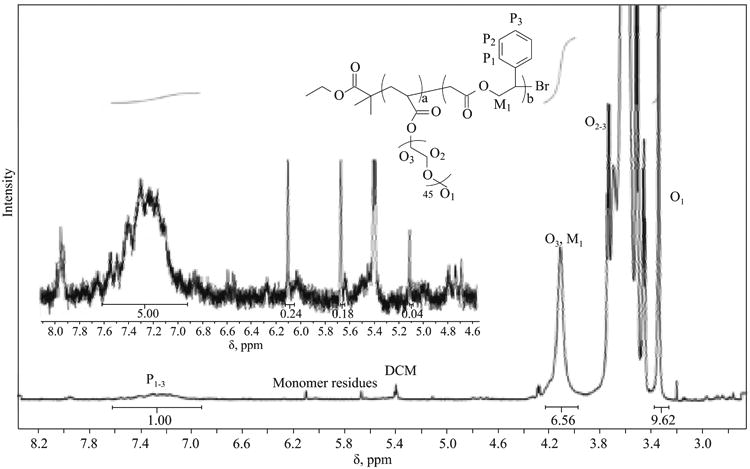
Besides structural difference of this type of macromonomer from other utilized monomers, the copolymerization was performed at very low comonomers concentrations (0.3 M) resulting in a relatively slow rate of polymerization (30 % monomer conversion in 13 h). This result indicated that it would be important to investigate further if copolymerization under dilute conditions and at a slower rate of polymerization could increase the prevalence of ring-opening of MPDL over vinyl-addition [70].
Conclusions
Degradable functional copolymers were synthesized by ATRP via copolymerization of methacrylates with MPDL as an exemplary CKA monomer. The efficiency of ring-opening of MPDL during copolymerization, which is required for formation of the degradable units in the backbone of the copolymer, increased at higher temperatures. MPDL was successfully copolymerized with both acrylates and methacrylates, and copolymers with acrylates were characterized by higher levels of incorporation of MPDL into the copolymers (∼2 to 3 times), compared to copolymers with methacrylates. High MW copolymers, MW > 100 000, were synthesized and successfully degraded forming fragmented chains below the renal threshold limit.
The final copolymers were characterized by relatively high dispersities, and the measured MWs were lower than theoretically predicted. The ring-opening efficiency of MPDL incorporation varied with different comonomers, which could be explained by several differences in reaction conditions including monomer concentration, deactivation efficiency, or (cross)propagation rate coefficients. Thus, additional detailed studies have to be performed to identify all side reactions and establish conditions for more effective ring-opening with specific comonomers despite temperature effects, and also to determine how to control MW, M̄w/M̄n and produce well-defined copolymers of complex architectures.
Acknowledgments
The support from NSF (DMR 1501324) and from NIH (R01DE020843) is gratefully acknowledged.
This paper is dedicated to memory of an outstanding scientist and a great friend and collaborator Prof. Andrzej Duda – on the occasion of his premature passing away.
References
- 1.Nair LS, Laurencin CT. Progress in Polymer Science. 2007;32:762. doi: 10.1016/j.progpolymsci.2007.05.017. [DOI] [Google Scholar]
- 2.Meng F, Hennink WE, Zhong Z. Biomaterials. 2009;30:2180. doi: 10.1016/j.biomaterials.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 3.Pillai CKS, Paul W, Sharma CP. Progress in Polymer Science. 2009;34:641. doi: 10.1016/j.progpolymsci.2009.04.001. [DOI] [Google Scholar]
- 4.Tian H, Tang Z, Zhuang X, et al. Progress in Polymer Science. 2012;37:237. doi: 10.1016/j.progpolymsci.2011.06.004. [DOI] [Google Scholar]
- 5.Tang Z, He C, Tian H, et al. Progress in Polymer Science. 2016;60:86. doi: 10.1016/j.progpolymsci.2016.05.005. [DOI] [Google Scholar]
- 6.Li Y, Maciel D, Rodrigues J, et al. Chemical Reviews. 2015;115:8564. doi: 10.1021/cr500131f. [DOI] [PubMed] [Google Scholar]
- 7.Dong R, Zhou Y, Huang X, et al. Advanced Materials. 2015;27:498. doi: 10.1002/adma.201402975. [DOI] [PubMed] [Google Scholar]
- 8.Ulery BD, Nair LS, Laurencin CT. Journal of Polymer Science Part B: Polymer Physics. 2011;49:832. doi: 10.1002/polb.22259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malafaya PB, Silva GA, Reis RL. Advanced Drug Delivery Reviews. 2007;59:207. doi: 10.1016/j.addr.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Oh JK, Siegwart DJ, Lee Hi, et al. Journal of the American Chemical Society. 2007;129:5939. doi: 10.1021/ja069150l. [DOI] [PubMed] [Google Scholar]
- 11.Kabanov AV, Vinogradov SV. Angewandte Chemie International Edition. 2009;48:5418. doi: 10.1002/anie.200900441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knop K, Hoogenboom R, Fischer D, Schubert US. Angewandte Chemie International Edition. 2010;49:6288. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- 13.Pack DW, Hoffman AS, Pun S, Stayton PS. Nature Reviews Drug Discovery. 2005;4:581. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 14.Schmaljohann D. Advanced Drug Delivery Reviews. 2006;58:1655. doi: 10.1016/j.addr.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 15.Lee KY, Yuk SH. Progress in Polymer Science. 2007;32:669. doi: 10.1016/j.progpolymsci.2007.04.001. [DOI] [Google Scholar]
- 16.Galaev IY, Mattiasson B. Trends in Biotechnology. 1999;17:335. doi: 10.1016/S0167-7799(99)01345-1. [DOI] [PubMed] [Google Scholar]
- 17.Savic R, Eisenberg A, Maysinger DJ. Journal of Drug Targeting. 2006;14:343. doi: 10.1080/10611860600874538. [DOI] [PubMed] [Google Scholar]
- 18.He C, Kim SW, Lee DS. Journal of Controlled Release. 2008;127:189. doi: 10.1016/j.jconrel.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 19.Liechty WB, Kryscio DR, Slaughter BV, Peppas NA. Annual Review of Chemical and Biomolecular Engineering. 2010;1:149. doi: 10.1146/annurev-chembioeng073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duncan R. Nature Reviews Drug Discovery. 2003;2:347. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 21.Langer R, Peppas NA. AICHE Journal. 2003;49:2990. doi: 10.1002/aic.690491202. [DOI] [Google Scholar]
- 22.George M, Abraham TE. Journal of Controlled Release. 2006;114:1. doi: 10.1016/j.jconrel.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 23.Haag R, Kratz F. Angewandte Chemie International Edition. 2006;45:1198. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 24.Maeda H, Wu J, Sawa T, et al. Journal of Controlled Release. 2000;65:271. doi: 10.1016/S0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 25.Vert M. Progress in Polymer Science. 2007;32:755. doi: 10.1016/j.progpolymsci.2007.05.006. [DOI] [Google Scholar]
- 26.Danhier F, Ansorena E, Silva JM, et al. Journal of Controlled Release. 2012;161:505. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 27.Matyjaszewski K, Tsarevsky NV. Journal of the American Chemical Society. 2014;136:6513. doi: 10.1021/ja408069v. [DOI] [PubMed] [Google Scholar]
- 28.Gillies ER, Goodwin AP, Frechet JMJ. Bioconjugate Chemistry. 2004;15:1254. doi: 10.1021/bc049853x. [DOI] [PubMed] [Google Scholar]
- 29.Delplace V, Tardy A, Harrisson S, et al. Biomacromolecules. 2013;14:3769. doi: 10.1021/bm401157g. [DOI] [PubMed] [Google Scholar]
- 30.Delplace V, Nicolas J. Nature Chemistry. 2015;7:771. doi: 10.1038/NCHEM.2343. [DOI] [PubMed] [Google Scholar]
- 31.Tsarevsky NV, Matyjaszewski K. Macromolecules. 2005;38:3087. doi: 10.1021/ma050020r. [DOI] [Google Scholar]
- 32.Johnson JA, Lewis DR, Díaz DD, et al. Journal of the American Chemical Society. 2006;128:6564. doi: 10.1021/ja0612910. [DOI] [PubMed] [Google Scholar]
- 33.de Graaf AJ, Mastrobattista E, Vermonden T, et al. Macromolecules. 2012;45:842. doi: 10.1021/ma2024667. [DOI] [Google Scholar]
- 34.Li Y, Nese A, Lebedeva NV, et al. Journal of the American Chemical Society. 2011;133:17 479. doi: 10.1021/ja207491r. [DOI] [PubMed] [Google Scholar]
- 35.Gao H, Tsarevsky NV, Matyjaszewski K. Macromolecules. 2005;38:5995. doi: 10.1021/ma0503099. [DOI] [Google Scholar]
- 36.Johnson JA, Finn MG, Koberstein JT, Turro NJ. Macromolecules. 2007;40:3589. doi: 10.1021/ma062862b. [DOI] [Google Scholar]
- 37.Cho HY, Srinivasan A, Hong J, et al. Biomacromolecules. 2011;12:3478. doi: 10.1021/bm2006455. [DOI] [PubMed] [Google Scholar]
- 38.Tsarevsky NV, Huang J, Matyjaszewski K. Journal of Polymer Science Part A: Polymer Chemistry. 2009;47:6839. doi: 10.1002/pola.23723. [DOI] [Google Scholar]
- 39.Wolf FF, Friedemann N, Frey H. Macromolecules. 2009;42:5622. doi: 10.1021/ma900894d. [DOI] [Google Scholar]
- 40.Coca S, Matyjaszewski K. Macromolecules. 1997;30:2808. doi: 10.1021/ma970073b. [DOI] [Google Scholar]
- 41.Coca S, Paik Hj, Matyjaszewski K. Macromolecules. 1997;30:6513. doi: 10.1021/ma970637b. [DOI] [Google Scholar]
- 42.Kajiwara A, Matyjaszewski K. Macromolecules. 1998;31:3489. doi: 10.1021/ma971445j. [DOI] [Google Scholar]
- 43.Ziegler MJ, Matyjaszewski K. Macromolecules. 2001;34:415. doi: 10.1021/ma001182k. [DOI] [Google Scholar]
- 44.Arehart SV, Matyjaszewski K. Macromolecules. 1999;32:2221. doi: 10.1021/ma981693v. [DOI] [Google Scholar]
- 45.Roos SG, Mueller AHE, Matyjaszewski K. Macromolecules. 1999;32:8331. doi: 10.1021/ma9819337. [DOI] [Google Scholar]
- 46.Matyjaszewski K. Journal of Physical Organic Chemistry. 1995;8:197. doi: 10.1002/poc.610080403. [DOI] [Google Scholar]
- 47.Matyjaszewski K, Gaynor S, Greszta D, et al. Journal of Physical Organic Chemistry. 1995;8:306. doi: 10.1002/poc.610080414. [DOI] [Google Scholar]
- 48.Matyjaszewski K, Nakagawa Y, Gaynor SG. Macromolecular Rapid Communications. 1997;18:1057. doi: 10.1002/marc.1997.030181209. [DOI] [Google Scholar]
- 49.Gaynor SG, Matyjaszewski K. Macromolecules. 1997;30:4241. doi: 10.1021/ma960720j. [DOI] [Google Scholar]
- 50.Tardy A, Delplace V, Siri D, et al. Polymer Chemistry. 2013;4:4776. doi: 10.1039/C3PY00719G. [DOI] [Google Scholar]
- 51.Agarwal S. Polymer Chemistry. 2010;1:953. doi: 10.1039/C0PY00040J. [DOI] [Google Scholar]
- 52.Bailey WJ. Polymer Journal. 1985;17:85. doi: 10.1295/polymj.17.85. [DOI] [Google Scholar]
- 53.Ding D, Pan X, Zhang Z, et al. Polymer Chemistry. 2016;7:5258. doi: 10.1039/C6PY01075J. [DOI] [Google Scholar]
- 54.Paulusse JMJ, Amir RJ, Evans RA, Hawker CJ. Journal of the American Chemical Society. 2009;131:9805. doi: 10.1021/ja903245p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siegwart DJ, Bencherif SA, Srinivasan A, et al. Journal of Biomedical Materials Research Part A. 2008;87A:345. doi: 10.1002/jbma.31708. [DOI] [PubMed] [Google Scholar]
- 56.Huang J, Gil R, Matyjaszewski K. Polymer. 2005;46:11 698. doi: 10.1016/j.polymer.2005.09.048. [DOI] [Google Scholar]
- 57.Lutz JF, Andrieu J, Üzgün S, et al. Macromolecules. 2007;40:8540. doi: 10.1021/ma070356w. [DOI] [Google Scholar]
- 58.Riachi C, Schüwer N, Klok HA. Macromolecules. 2009;42:8076. doi: 10.1021/ma901537x. [DOI] [Google Scholar]
- 59.Zhang Y, Chu D, Zheng M, et al. Polymer Chemistry. 2012;3:2752. doi: 10.1039/C2PY20403G. [DOI] [Google Scholar]
- 60.D'Ayala GG, Malinconico M, Laurienzo P, et al. Journal of Polymer Science Part A: Polymer Chemistry. 2014;52:104. doi: 10.1002/pola.26976. [DOI] [Google Scholar]
- 61.Delplace V, Harrisson S, Tardy A, et al. Macromolecular Rapid Communications. 2014;35:484. doi: 10.1002/marc.201300809. [DOI] [PubMed] [Google Scholar]
- 62.Decker CG, Maynard HD. European Polymer Journal. 2015;65:305. doi: 10.1016/j.eurpolymj.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hedir GG, Bell CA, O'Reilly RK, Dove AP. Biomacromolecules. 2015;16:2049. doi: 10.1021/acs.biomac.5b00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chung IS, Matyjaszewski K. Macromolecules. 2003;36:2995. doi: 10.1021/ma034029. + [DOI] [Google Scholar]
- 65.Smith Q, Huang J, Matyjaszewski K, Loo YL. Macromolecules. 2005;38:5581. doi: 10.1021/ma961327g. [DOI] [Google Scholar]
- 66.Delplace V, Guegain E, Harrisson S, et al. Chemical Communications. 2015;51:12 847. doi: 10.1039/C5CC04610F. [DOI] [PubMed] [Google Scholar]
- 67.Pan CY, Lou XD. Macromolecular Chemistry and Physics. 2000;201:1115. doi: 10.1002/1521-3935(20000701)201:11<1115::AID-MACP1115>3.0.CO;2-D. [DOI] [Google Scholar]
- 68.Bailey WJ, Wu SR, Ni Z. Die Makromolekulare Chemie. 1982;183:1913. doi: 10.1002/macp.1982.021830811. [DOI] [Google Scholar]
- 69.Sanda F, Endo T. Journal of Polymer Science Part A: Polymer Chemistry. 2001;39:265. doi: 10.1002/1099-0518(20010115)39:2<265::AID-POLA20>3.0.CO;2-D. [DOI] [Google Scholar]
- 70.Cho HY, Krys P, Szcześniak K, et al. Macromolecules. 2015;48:6385. doi: 10.1021/acs.macromol.5b01592. [DOI] [Google Scholar]