

Abstract
Ambient particulate matter (PM), a component of air pollution, exacerbates airway inflammation and hyper-reactivity in asthmatic patients. Studies showed that PM possesses adjuvant-like properties that enhance the allergic inflammatory response; however, the mechanism(s) by which PM enhances the allergic response remains to be determined. The aim of this study was to assess how exposure to fine PM collected from Sacramento, CA, shapes the allergic airway immune response in BALB/c mice undergoing sensitization and challenge with ovalbumin (OVA). Eight-week old BALB/c male mice were sensitized/challenged with phosphate buffered saline (PBS/PBS; n = 6), PM/PBS (n = 6), OVA/OVA (n = 6), or OVA + PM/OVA (n = 6). Lung tissue, bronchoalveolar lavage fluid (BALF) and plasma were analyzed for cellular inflammation, cytokines, immunoglobulin E and heme oxygenase-1 (HO-1) expression. Mice in the OVA + PM/OVA group displayed significantly increased airway inflammation compared to OVA/OVA animals. Total cells, macrophages, and eosinophils recovered in BALF lavage were significantly elevated in the OVA + PM/OVA compared to OVA/OVA group. Histopathological grading indicated that OVA + PM/OVA treatment induced significant inflammation compared to OVA/OVA. Both IgE and TNFα levels were significantly increased in OVA/OVA and OVA + PM/OVA groups compared to PBS/PBS control. The number of HO-1 positive alveolar macrophages was significantly elevated in lungs of mice treated with OVA + PM/OVA compared to OVA/OVA. Our findings suggest that fine PM enhances allergic inflammatory response in pulmonary tissue through mechanisms involving increased oxidative stress.
Introduction
Allergic asthma is an immune disorder characterized by overactive IgE antibody responses to antigens (Bateman et al. 2008). There are two hallmark phenotypes of allergic asthma: (1) inflammatory, due to a T helper cell type 2 (Th2)-mediated immune response resulting in elevated inflammatory cells in the airways, and (2) airway hyper-reactivity, during which volume and flow of inhaled air decreases as a consequence of airway constriction. Numerous epidemiological studies demonstrated an association of increased asthma prevalence and exacerbation of symptoms with exposure to ambient air pollutants (McConnell et al. 2006; von Klot et al. 2002; Kim and Bernstein 2009; Jayawardene et al. 2013; Cheng et al 2014; Chen et al, 2016; Greenberg et al, 2016).
The City of Sacramento is situated in California’s Central Valley, a region that contains 6 of the 10 cities in the nation with the worst air pollution (State of the Air 2015). Sacramento also ranks as the 14th city in the nation to display the highest short-term particle air pollution. In 2012, Sacramento County’s lifetime asthma prevalence was estimated to be 10.6% for children (0-18 years old) and 16.4% for adults (18 years old and above), which is greater for both groups compared to the national rates of 9.5 and 8.2%, respectively (California Health Interview Survey 2012). Genetic factors cannot fully explain the higher incidence of asthma in Sacramento, and it is postulated that other factors, such as environmental exposures, may play an important role in promoting asthma susceptibility. Various studies demonstrated that particulate matter (PM) exposure is associated with enhanced sensitization to aeroallergens and worsening of asthmatic symptoms (Fuertes and Heinrich 2015; Bowatte et al. 2015; Fuertes et al. 2013; Mortimer et al. 2008). Particularly, living in close proximity to major highways has been strongly associated with a higher risk of childhood asthma (McConnell et al. 2006).
Although the immune system cannot become sensitized to PM, data suggest that PM possess immunological adjuvant activity that promotes allergic responses (Li et al. 2009; 2010); PM alone, in the absence of allergens, was found to modulate the inflammatory immune response towards an allergic/IgE-mediated response as previously reported by Diaz-Sanchez et al (1994; 1999). Oxidative stress was postulated as the primary mechanism underlying PM-mediated toxicity. However, given that PM contains hundreds to thousands of distinct constituents, it is likely that not all mechanisms by which PM exacerbates allergic responses or the key cells involved have been identified. Identifying the mechanisms underlying PM-mediated toxicity is an important step in understanding how air pollution might exacerbate asthma in those with disease and make individuals with no history of atopy more susceptible to asthma and other allergies given that the incidence of asthma and allergy has risen substantially in the past decades.
The aim of this study was to investigate the role ambient fine PM (PM2.5) collected from an urban sampling site located in downtown Sacramento plays in allergic sensitization of a mouse model of human asthma. BALB/c mice were sensitized to the allergen ovalbumin (OVA) via intranasal instillation on days 1, 3 and 5 followed by OVA challenge on days 12-14. PM was introduced, with or without OVA, only during allergic sensitization to assess its effects in modulating the developing allergic immune response.
Methods
Ambient PM Collection, Extraction, and Chemical Characterization
Field studies were conducted during the summer 2011 at an urban sampling site located on the rooftop of a two story building at the northeast corner of T St. and 13th St. in downtown Sacramento, CA. The sampling site is surrounded by a mixture of residential, commercial and industrial sources and within a quarter mile of a major freeway interchange. Samples were collected using PM2.5 high-volume sampler systems equipped with PM10 size-selective heads, operating at a flow rate of 40 cfm and loaded with aluminum foil substrates for collecting the coarse PM fraction (PM10-2.5 = 2.5 <Dp< 10 μm) and Teflon coated borosilicate glass microfiber filters for collecting the fine PM fraction (PM2.5 = Dp< 2.5 μm). Aluminum foil substrates were pre-baked at 500° C for 24 hr, and glass microfiber filters were pre-cleaned via successive sonication in milli-Q H2O, dichloromethane (DCM) and hexane (Hx). Field blanks were included for all investigations. The samplers were operated continuously for 1-2 week sampling intervals.
PM2.5 filter samples and field blanks were extracted using a multi-solvent filter extraction technique that combines sonication in multiple solvents, liquid-liquid extraction, microporous membrane filtration and detailed gravimetric analyses to (1) maximize extraction efficiency, (2) minimize compositional biases, (3) minimize extraction artifacts and (4) provide precise and accurate direct measurements of extracted PM mass. A comprehensive characterization of this method was published by Bein and Wexler (2014; 2015).
All PM and field blank extracts were subjected to an exhaustive suite of chemical analyses, including trace metals via inductively coupled plasma-mass spectrometry (ICP-MS), water soluble inorganic and organic ions via ion chromatography, automated colorimetry and atomic absorption spectrophotometry (AAS), molecular organic compounds via thermal desorption-gas chromatography mass spectrometry (GC-MS), and elemental and organic carbon via thermal optical reflectance. A detailed description of the application of these analytical techniques to extract PM samples have been described by Bein and Wexler (2015). Lipopolysaccharide (LPS) levels were quantitated by Lonza Kinetic Chromogenic LAL Endotoxin-Assay (Basel, Switzerland) and found to be below the limits of detection (LOD) >0.005 endotoxin units.
Animal Model: Allergen and Particulate Matter Administration
Eight week old male BALB/c mice (22-24 g body weight) were obtained from Harlan Laboratories (Livermore, CA). Animals were housed at the Center for Health and the Environment at the University of California, Davis. The university’s Institutional Animal Care and Use Committee Experiments reviewed and approved the experimental protocols associated with the study. Twenty-four mice were acclimated for 2 weeks and randomly divided into 4 exposure groups (each group n=6) that were sensitized/challenged with one of the following: (1) phosphate buffered saline (PBS)/PBS, (2) Sacramento PM2.5 (PM)/PBS, (3) OVA/OVA, or (4) OVA+PM/OVA.
The exposure protocol is depicted in Figure 1. Mice were sensitized on days 1, 3, and 5 and challenged on days 12-14. PM and OVA were administered in a total volume of 30 μl/day/mouse of PBS (delivery vehicle) via intranasal instillation, which was selected to ensure complete and accurate delivery of PM. Ovalbumin, which was depleted of endotoxin via Detoxi-Gel™ endotoxin removal columns (Thermo Fisher Scientific Inc., Rockford, IL), was administered at a dose of 10 μg/day/mouse. PM was administered at a concentration of 33.3 μg/day/mouse (100 μg total sensitization dose). PM was sonicated immediately before administration. PM/PBS and OVA+PM/OVA treatment groups only received PM during the sensitization period to assess its adjuvant-like effect on the adaptive response (challenge). In mice sensitized with OVA+PM, OVA and PM were dosed separately, approximately 15 min apart, to avoid particle-protein interactions. For detailed information regarding this experimental protocol, please refer to Castañeda and Pinkerton (2016). Animals were euthanized with pentobarbital solution (65 mg/ml) i.p. on day 15, 24-hr after the last intranasal challenge to assess pulmonary inflammation. Serology and histopathologic assessment of sentinel animals was conducted at the start and end of the experimental period, and results showed that mice were infection free.
Figure 1.
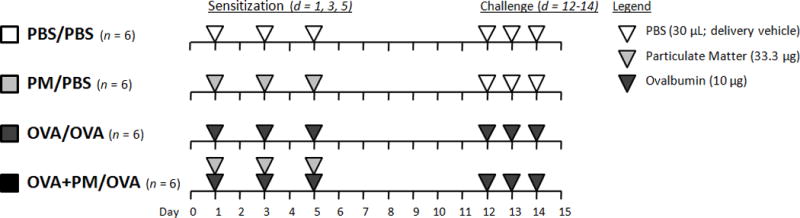
Allergic sensitization and challenge protocol. Mice were sensitized (day 1, 3, and 5) and challenged (day 12-14) intranasally with PBS (30 μl/day; delivery vehicle; white triangles), PM (33.3 μg/day, light grey triangles), OVA (10 μg/day, dark grey triangles) or OVA+PM. Mice were euthanized 24 hr after the final challenge (day 15) to assess pulmonary inflammation.
Bronchoalveolar Lavage Fluid (BALF) and Cellular Analysis
Mice were cannulated intratracheally and lungs lavaged with two volumes of 0.7ml sterile PBS (Sigma Aldrich, St. Louis, MO). The lavage fluid was centrifuged at 500 × g for 15 min at 4°C. The supernatant was removed, frozen for future analysis, and cells resuspended in 500 ul PBS. Cell numbers and viability were determined via hemocytometer using 0.4% trypan blue solution (Sigma-Aldrich). Cells were centrifuged onto slides (1.5 × 103 cells/slide) using a Shandon Cytospin (Thermo Shandon, Inc., Pittsburg, PA). Slides were stained with hematoxylin and eosin (H&E) or DippKwik stain (American MasterTech, Lodi, CA) for cellular differential analysis. A total of 500 cells were counted per slide to determine macrophage, neutrophil, eosinophil, and lymphocyte cell composition of BALF.
Histological Analysis and Immunohistochemistry
The right lung was removed from the thoracic cavity by midline dissection and inflation fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) at 30 cm water pressure for one hr. The cranial, caudal, middle, and accessory lobes of the fixed lung were separated, embedded in paraffin wax, and cut into 5 μm sections mounted onto slides. Slides were stained with hematoxylin and eosin (H&E) or used for immunohistochemistry (see below).
Hematoxylin and eosin stained sections were employed for overall pathological assessment, including identification of sites of inflammation, and cell types involved. Sections from each of the 4 right lung lobes were assessed for inflammation based on extent and severity of: (1) epithelial/bronchoalveolar inflammation, (2) endothelial/perivascular inflammation, and (3) alveolar/parenchymal inflammation. Extent and severity of inflammation was scored using a scale of 0 (no inflammation), 1 (mild), 2 (marked), 3 (extensive), and 4 (severe). Extent and severity scores were multiplied to determine the final score.
Immunohistochemistry
Staining for heme oxygenase-1 (HO-1) was performed by hydrating mounted tissue paraffin embedded sections in decreasing concentrations of ethanol (100, 95 and 75%), with a final wash in distilled water. Antigen retrieval was performed with ethylenediaminetetraacetic acid buffer (EDTA, Sigma Aldrich, St. Louis MO; 1mM, pH 8) at a temperature of 123˚C for 10 min at 18psi. Endogenous peroxidase was blocked with 3% hydrogen peroxide. Tissue sections were treated with protein block (Dako, Carpinteria, CA) to prevent non-specific protein binding. This was followed by incubation with primary IgG antibody HO-1 (Abcam, Cambridge, MA; ab13243, anti-mouse made in rabbit) at a dilution of 1:400 (2.5μg/ml) for 1 hr at room temperature. Tissue sections were treated with the EnVision System horseradish peroxidase (HRP)-labeled anti-rabbit polymer (Dako, Carpinteria, CA) for 30 min and then with the 3,3′-diaminobenzidine (DAB) substrate chromogen (Dako) for 5 min. Tissue sections were counterstained with hematoxylin (American MasterTech), dehydrated in increasing concentrations of ethanol (75, 95 and 100%), and coverslipped. A negative control consisted of non-immune IgG substituted for the primary HO-1 antibody that underwent identical methodology as HO-1 treated slides. HO-1 positive macrophages were counted in a total of 10 random high power fields (400x) beneath the epithelium of the central airway.
Blood Plasma Collection and Immunoglobulin Analysis
Blood was collected immediately following euthanasia via cardiac puncture in EDTA coated cryotubes (BD, Franklin Lakes, NJ) and centrifuged at 3,000 × g for 10 min at 4°C to collect plasma. Plasma was utilized to quantify total immunoglobulin E (IgE) via ELISA. The ELISA was performed by coating 96-well plates (Maxisorp, Rockford, IL) overnight at 4°C with rat-anti mouse IgE capture antibody, 50 μl/well at a concentration of 5μg/ml in a coating buffer of sodium carbonate (NaCO3, Sigma Aldrich) and sodium bicarbonate (NaHCO3, Sigma Aldrich) in distilled water (ddH2O). Plates were washed with wash buffer, PBS-0.05% Tween (Sigma Aldrich), and blocked with 50μl 1% bovine serum albumin (BSA; Sigma Aldrich) for 1 hr at room temperature. Following the washing step, 50μl standards (IgE Calibrator Serum, Bethyl Laboratories, Montgomery, TX) and samples (diluted 1:5 in 1%BSA) were added to the wells and incubated for 2 hr at room temperature. Wells were subsequently washed with wash buffer and incubated with 50μl/well of rat anti-mouse IgE-HRP detection antibody (1:1000 dilution; Southern Biotech, Birmingham, AL) for 1 hr at room temperature. Wells were washed and treated with 50μl/well of 3,3’,5,5’-tetramethybenzidine (TMB) substrate (Pierce-Thermo, Rockford, IL) for 15 min in the dark. The reaction was stopped by adding 50μl/well of 2N H2SO4. Plates were immediately read using a Spectramax Microplate spectrophotometer (Molecular Devices, Sunnyvale, CA) at a wavelength of 450nm.
Pulmonary Cytokine Analysis
The left lung was collected, flash frozen and stored for biochemical analysis. Lung lobes were homogenized with a cell lysis kit (Bio-Rad, Hercules, CA). Total protein concentration was assessed via Lowry protein assay (Bio-Rad). Lung homogenates were diluted 1:50 in 1% BSA (Sigma Aldrich). ELISA’s for IL-1β, IL-5, IL-6, IL-17A, IL-25, and TNF-α were performed using Biolegend ELISA kits (BioLegend, San Diego, CA) according to the manufacturer’s protocol. Cytokine levels (ng/ml) were standardized to total lung protein (mg/ml) and expressed as nanograms of cytokine per milligram of lung tissue (ng/mg).
Statistical Methods
Data are expressed as means ± standard error of the mean (SEM). All comparisons (PBS/PBS, PM/PBS, OVA/OVA, and OVA+PM/OVA) were assessed by one-way ANOVA followed by post hoc Tukey’s Multiple Comparison Test using GraphPad PRISM 5 software. A value of p<0.05 was considered statistically significant.
Results
Composition of PM
In brief, summertime PM2.5 in Sacramento was dominated by organic carbon (49% composition by mass), including polycyclic aromatic hydrocarbons(PAH) and non-aromatic hydrocarbons, and water soluble inorganic ions (21% composition by mass). Elemental carbon accounted for 1.4% of PM mass and various metals ranging from lithium to lead were detected at levels significantly above LOD.
Bronchoalveolar Lavage Fluid (BALF) and Immune Cell Differential Analysis
Immune cells (macrophage, neutrophil, eosinophil, and lymphocyte) collected in the BALF were differentially counted to assess the manner in which PM impacts the pulmonary allergic response (Figure 2). Mice sensitized/challenged with OVA+PM/OVA displayed significantly higher numbers of total cells, macrophages, and eosinophils than animals treated with OVA/OVA. These findings suggest that PM exacerbated the allergic inflammatory response, as the eosinophil is a key immune cell involved in Th2-mediated immune responses. Although both neutrophil and lymphocyte cells were significantly elevated in OVA/OVA and OVA+PM/OVA treatment groups compared to controls, there was no marked difference between these two exposed groups.
Figure 2.
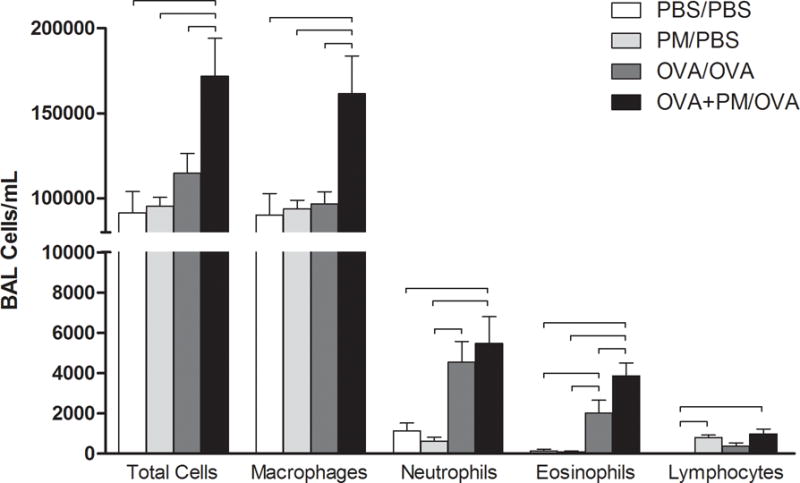
Cellular profiles of recovered bronchoalveolar lavage fluid (BALF) from mice sensitized/challenged with PBS/PBS (control; white), PM/PBS (light grey), OVA/OVA (dark grey), or OVA+PM/OVA (black). Total cellular influx, macrophages, neutrophils, eosinophils, and lymphocytes are shown for all 4 groups in number of cells/ml. PM enhanced OVA-induced allergic inflammation with respect to total cells, macrophages, and eosinophils. Results are presented as mean ± SEM (n=6 mice per group). Bars indicate a significant difference of p < 0.05 between groups.
Histopathology and Scoring
To further assess the localization of the immune cells in pulmonary tissue and degree of inflammation, blinded histopathological scoring of the left lung was performed to evaluate the extent and severity of inflammation. Micrographs of lung demonstrated a greater influx of macrophages, neutrophils, and eosinophils to the subepithelium of airways in addition to airway epithelial hyperplasia in mice treated with OVA+PM/OVA compared to animals administered OVA/OVA (Figure 3, D vs C). The overall histopathological score of lung tissue, which was based on additive scores from pulmonary bronchiolar, perivascular, and alveolar regions, supports these observations (Figure 3E, Total Lung). Predominant inflammation occurred in the parenchymal region (Figure 3E, Alveolar).
Figure 3.
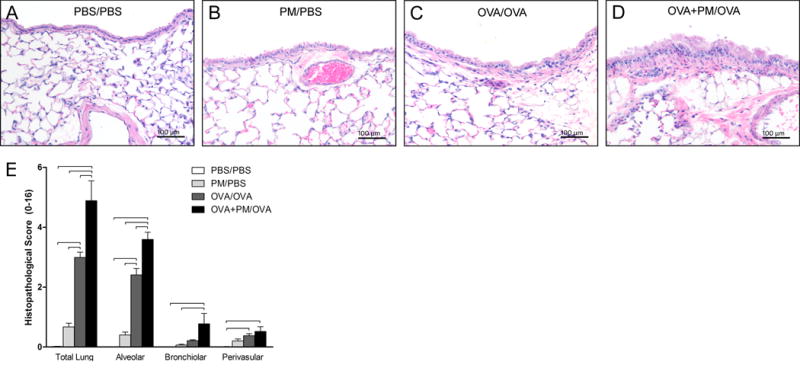
Histopathological analysis of total lung and specific lung compartments. (A-D) Micrographs of lung tissue (200x magnification) stained with hematoxylin & eosin (H&E). (E) Histopathological analysis of lung tissue stained with H&E. OVA/OVA treatment resulted in significant pulmonary inflammation compared with PBS/PBS control treatment. OVA+PM/OVA treatment significantly enhanced inflammation over OVA/OVA treatment. Data is presented as mean ± SEM (n=6 mice per group). Bars indicate a significant difference of p < 0.05 between groups. The scale bar represents a distance of 100 micrometers.
Plasma Immunoglobulin E (IgE)
To determine whether the allergic response was detectable systemically, total plasma IgE protein levels were quantified. Both OVA/OVA and OVA+PM/OVA treatment groups displayed significantly higher IgE plasma levels compared to controls (Figure 4), demonstrating OVA administration produced an IgE-mediated allergic response. Notably, PM alone numerically enhanced IgE levels, although not significantly different from control.
Figure 4.
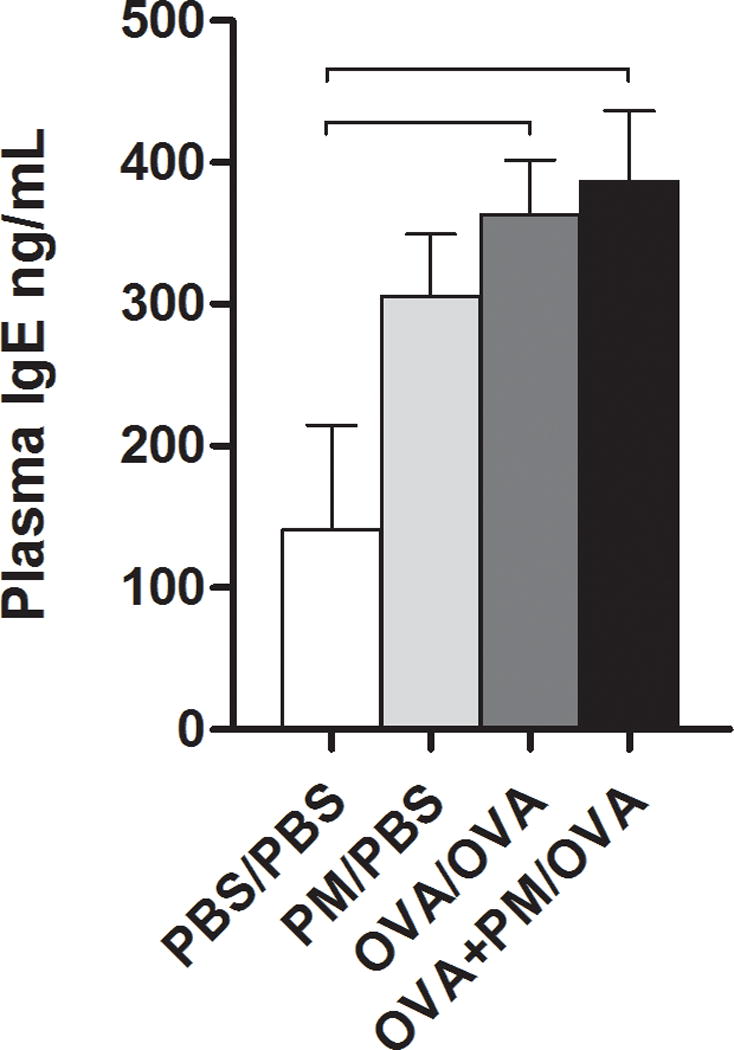
Immunoglobulin E (IgE) levels from blood plasma of mice sensitized/challenged with PBS/PBS, PM/PBS, OVA/OVA, or OVA+PM/OVA. Concentration is shown as nanograms (ng) of IgE per milliliter (ml) plasma. Data is presented as mean ± SEM (n = 3-5 mice per group). Bars indicate a significant difference of p < 0.05 between groups.
Pulmonary Cytokine Analysis
The enhancement of allergic inflammation by PM prompted investigation of cytokines (IL-1β, IL-6, IL-17A and TNFα) associated with PM-mediated pulmonary inflammation (Mitschik et al. 2008). In addition, levels of IL-5 and IL-25 cytokines, both eosinophil chemoattractants, were measured. Both IL-25 and TNFα protein levels were significantly elevated in both OVA/OVA and OVA+PM/OVA treatment groups compared to control (Table 1).
Table 1.
Pulmonary Cytokines (ng/mg)
| PBS/PBS | PM/PBS | OVA/OVA | OVA+PM/OVA | |
|---|---|---|---|---|
| IL-1β | 3.593 ± 1.268 | 3.453 ± 0.457 | 6.315 ± 1.157 | 5.336 ± 0.361 |
| IL-5 | 44.727 ± 6.609 | 40.041 ± 4.625 | 65.227 ± 10.096 | 68.002 ± 5.048 |
| IL-6 | 1.226 ± 0.155 | 1.105 ± 0.150 | 1.894 ± 0.302 | 1.769 ± 0.156 |
| IL-17A | 0.110 ± 0.010 | 0.121 ± 0.025 | 0.295 ± 0.049*† | 0.233 ± 0.029 |
| IL-25 | 1.299 ± 0.186 | 1.479 ± 0.269 | 2.508 ± 0.434* | 2.527 ± 0.211* |
| TNFα | 0.003 ± 0.001 | 0.013 ± 0.002 | 0.024 ± 0.005* | 0.026 ± 0.004* |
Groups are listed as sensitization/challenge treatment: PBS = phosphate buffer saline, PM = particulate matter, OVA = ovalbumin; Cytokines: IL = interleukin, TNFα = tumor necrosis factor alpha.
Cytokine levels were quantified via ELISA and were standardized to total lung protein and expressed as nanograms (ng) of cytokine per milligram (mg) of lung tissue. Data is presented as mean ± SEM (n = 5-6 mice per group),
p < 0.05 versus PBS/PBS,
p < 0.05 versus PM/PBS.
Pulmonary Heme Oxygenase-1 (HO-1) Expression
Because inflammatory effects of PM have been attributed to oxidative stress (Li et al. 2008; 2009; 2013), protein expression of HO-1 in lung tissue was determined. Epithelial cells, fibroblasts, and macrophages were positive for HO-1 expression (Figure 5, A-D), especially following OVA/OVA and OVA+PM/OVA treatment. Further, the number of HO-1 positive macrophages in the OVA+PM/OVA group was significantly higher than OVA/OVA (Figure 5E). HO-1 positive cells were seldom seen in control and PM/PBS groups.
Figure 5.
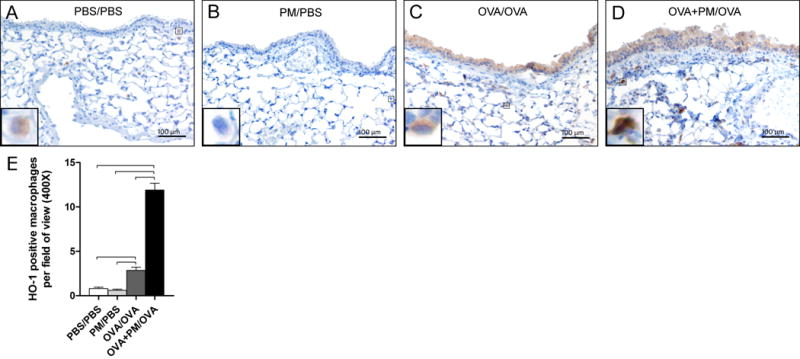
Hemeoxygenase-1 (HO-1) analysis. (A-D) Micrographs of paraffin embedded lung tissue sections immunohistochemically stained with HO-1 (brown stain, 200x magnification). The inserts show magnified macrophages within the respective image. The scale bar represents a distance of 100 micrometers. (E) The frequency of HO-1 positively stained alveolar macrophages in one total field of view (400x). Data is presented as mean ± SEM (n = 5-6 mice per group). Bars indicate a significant difference of p < 0.05 between groups.
Discussion
The purpose of this study was to assess whether exposure to PM2.5 during allergic sensitization in a BALB/c mouse model of asthma modulates inflammatory responses during allergic challenge in the absence of PM. PM used in this experiment was collected from the downtown area of the City of Sacramento near three major highways, and the dose used is equivalent of a one- to two-week exposure to high ambient PM levels for humans. The primary endpoint of this study was to evaluate airway inflammation based on recovered BALF cells. Secondary endpoints aimed to characterize the inflammatory cytokine profile to elucidate PM-mediated toxicological effects as well as examine lung HO-1 expression as an indicator of oxidative stress. The findings of our study demonstrate that PM2.5 enhanced allergic airway inflammation, as suggested by elevated influx of immune cells into the pulmonary compartment. These observations are supported by histopathological scoring analysis: compared to OVA/OVA treatment, OVA+PM/OVA administration resulted in epithelial perturbations, including greater immune cell migration to the subepithelial and alveolar regions. In addition, exposure to PM2.5 during allergic sensitization significantly elevated eosinophil pulmonary recruitment compared to allergen sensitization without PM. Taken together, evidence indicates that PM modifies and enhances the initial development of the allergic immune response in mice.
PM is primarily formed through (1) combustion processes, such as gas and diesel engines, cooking, power plants and wildfires, (2) mechanical abrasion processes, such as windblown dust, sea spray and various agricultural activities, and (3) photochemical processing, including nucleation and secondary organic aerosol (SOA) formation. Depending upon the source, PM may be composed of internal and/or external mixtures of inorganic compounds, metals, trace elements, elemental carbon (black carbon or soot) and an exhaustive list of organic compounds, including PAH (Ghio et al, 2012). PM were found to deplete endogenous antioxidants and induce the production of reactive oxygen species (ROS), promoting a state of cellular oxidative stress (Ayres et al. 2008; de Oliveira et al, 2014). Particle deposition in the lung primarily enhances oxidative stress in mucosal epithelium and alveolar macrophages, triggering redox-sensitive pathways such as NFκB that lead to the secretion of pro-inflammatory cytokines promoting tissue injury and inflammation (Brown et al. 2006). The effects of Sacramento air pollution were studied in a BALB/c mouse model in which oropharyngeal aspiration of fine PM (2.5 μm in size: PM2.5) collected in Sacramento induced pulmonary inflammation via neutrophil recruitment (Van Winkle et al. 2015).
Various cytokines (IL-1β, IL-5, IL-6, IL-17A, IL-25, and TNFα) were assessed to better understand the augmented inflammatory response exhibited in the OVA+PM/OVA group vs. OVA/OVA. However, no clear relationships between enhanced cellular pulmonary recruitment with PM treatment and these cytokines were found. The levels of IL-5, which serves as an important chemoattractant for eosinophils, and IL-25, a cytokine that promotes airway eosinophilia and Th2-mediated inflammation, were similar between these groups. It is possible that PM may mediate eosinophil migration to the lung via other eosinophil chemoattractants, such as CCL11, CCL24, and/or CCL26 chemokines. TNFα is an important cytokine involved in the extravasation of monocytes and neutrophils from the blood by upregulating endothelial selectins and integrins during inflammation. There was a trend for PM to elevate TNFα levels, but the results were not significant. The study was limited by a low sample size (n) and time between analysis (day 15) and PM exposure (days 1, 3, and 5), which may have masked any significant changes in cytokines. Notably, PM exposure alone (PM/PBS) elevated TNFα levels despite its final administration 10 days prior to analysis, lending support to the hypothesis that PM alone exerts a sustained pro-inflammatory effect. This may explain PM adjuvant-like effect when given in conjunction with the allergen.
Various investigations demonstrated that HO-1 serves as a marker of PM-mediated oxidative stress in in-vivo models (Ayres et al. 2008; Carosino et al. 2015; Kooter et al. 2006; Li et al. 2000; 2003). In this study, HO-1 levels were evaluated via immunochemical staining in pulmonary tissue. Allergic sensitization, with and without PM, led to distinct expression patterns of HO-1 in epithelial cells, macrophages, and fibroblasts that were less frequent in control sham and PM only. Exposure to PM during allergic sensitization to OVA led to a significant increase in HO-1 expressing macrophages compared to mice sensitized in the absence of PM. This observation may be explained by uptake of PM through alveolar macrophages, leading to cellular oxidative stress. The combination of OVA and PM exposure during sensitization may enhance macrophage cellular activation, compared to OVA-alone and augment secretion of pro-inflammatory mediators that may modulate early acute inflammatory response during sensitization, leading to enhanced inflammation upon allergen challenge.
Based upon the findings of this study, it is difficult to draw conclusions as to the mechanisms underlying PM-enhanced allergic responses. PM-mediated oxidative stress was previously found to enhance dendritic cell activation leading to greater Th2 lymphocyte responses in mice (Li et al. 2013). Our results lend support to these observations as both PM treated groups (PM/PBS and OVA+PM/OVA) displayed a significantly elevated number of lymphocytes in the recovered BALF, a feature not present in the OVA/OVA administered mice. Further, HO-1 expression was found to be markedly higher in macrophages of animals administered OVA+PM/OVA vs OVA/OVA, despite the fact that macrophages recovered through BALF showed no particle content. Therefore PM may induce prolonged oxidative stress when given in concert with allergen compared to PM alone. PM-mediated enhanced activation of antigen presenting cells such as macrophages and dendritic cells may ultimately augment activation of the adaptive immune system (versus OVA-alone treatment), promoting lymphocyte proliferation, recruitment, and ultimately more severe inflammation. This modulation of the immune response between OVA/OVA vs OVA+PM/OVA may not have been discernible in this investigation since cytokine analysis was performed 10 days (day 15) after the final PM dose (day 5), providing sufficient time for resolution of the acute inflammatory effects attributed to PM. Alternatively, various factors may influence cytokine responses such as PM source variability, concentration and method of administration.
The source of the PM used in this experiment was obtained from downtown Sacramento (T Street and 13th Street). Similar to other cities located in the Central Valley, Sacramento is susceptible to air flow stagnation periods where local PM emissions, such as from agriculture and regional background sources, might mix with urban emissions to form a highly complex airshed (Herner et al. 2005). The PM used in this study likely includes various PM sources, such as from vehicular, commercial, factory, residential, landscaping, construction, and agriculture. The collection site was less than 2000 feet from a major highway (Highway 50/80) and approximately 1 mile east and 1 mile west of two major California Highways (Highway 5 and 99, respectively), which likely resulted in a PM composition high in fossil fuel combustion emissions from gasoline and diesel engines. The collection site location was specifically selected because investigators demonstrated that PM collected near highways is able to exacerbate allergic conditions (Kelly and Fussell 2011; Peterson and Saxon 1996; McConnell et al. 2006). Our results are in agreement as data demonstrated that Sacramento PM2.5 exerts significant toxicity in an animal model of allergic airway inflammation. Specifically, exposure to Sacramento PM2.5 only during allergen sensitization, and not during allergen challenge, produced chronic inflammatory effects, exacerbating allergic inflammatory responses by augmented recruitment of immune cells into the pulmonary compartment. Our study, therefore, lends support to the postulation that PM in air pollution possesses adjuvant-like properties in modulating development of immune responses, likely enhancing adaptive immune consequences. Although a clear relationship between PM toxicity and cytokine levels was not established, the low level of HO-1 expression in PM-only treated animals suggests that PM modulates early innate immune responses during allergen sensitization, concurrently enhancing allergic inflammatory responses. This is partly supported by elevated lymphocyte levels seen in our model in both PM treated groups (PM/PBS and OVA+PM/OVA). Deiuliis et al (2012) showed that PM alone activate pulmonary T cells. However, data are lacking that demonstrate how PM in concert with allergens impact adaptive immune responses. Future studies needs to examine effects of PM on antigen presenting cells, such as macrophage and dendritic cells, and whether PM in the presence of allergens might amplify cell activation of these cells to produce greater B cell and T cell priming and subsequent adaptive responses.
In conclusion, our study demonstrated that exposure to PM2.5 from the City of Sacramento during allergen sensitization modulates the immune response, exacerbating allergic response via increased monocyte and eosinophil recruitment into the lung. HO-1 pulmonary tissue expression suggests oxidative stress is a possible explanation for the observed enhanced toxicity mediated by PM; however, no clear mechanistic relationship was established based upon various analyzed cytokines. These findings are novel in that this is the first report, to our knowledge, that demonstrates an urban/rural PM composition from the City of Sacramento enhances allergic airway inflammation in an animal model. Further study is needed to investigate the possible mechanisms through which PM mediates its immunotoxicological effects. The high rate of asthma in Sacramento cannot fully be explained by genetic predisposition, it is highly likely that environmental components such as air pollutants play an important role in enhancing asthma susceptibility in non-atopic populations (i.e., in individuals with no family history of asthma or allergy). Ultimately these observations highlight the need for more careful regulation of PM emissions that possess the potential to promote allergy by enhancing allergic sensitization, particularly in susceptible populations such as children.
Acknowledgments
We would like to acknowledge Dale Uyeminami, Imelda Espiritu, Janice Peake, Alexa Pham, Esther Patchin, Jocelyn Claude, and Katherine Johnson for their valuable help through this experiment.
Funding
This work was supported by the National Institute for Occupational Safety and Health funded Western Center for Agricultural Health and Safety (NIOSH OHO7550), the UC Davis Graduate Research Mentorship Fellowship, and the National Institute of Health Pharmacology T32 Training Grant (T32GM099608).
Footnotes
Author Contributions
ARC performed experiments, analyzed data, and interpreted the results. KEP provided overall experimental design and guidance. KJB collected, extracted, and analyzed particulate matter samples. SSJ provided extensive feedback and editing. The article was read, revised, and approved by all authors.
Competing Interests
The authors do not have competing interests.
Contributor Information
Alejandro R. Castañeda, Center for Health and the Environment, University of California, Davis, CA 95616 USA; Tel: 530-752-7751, Fax: 530-752-5300
Keith J. Bein, Center for Health and the Environment, Air Quality Research Center, University of California, Davis, CA 95616 USA; Tel: 530-754-6558, Fax: 530-752-5300
Suzette Smiley-Jewell, Center for Health and the Environment, University of California, Davis, CA 95616 USA; Tel: 530-752-2723, Fax: 530-752-5300.
Kent E. Pinkerton, Department of Pediatrics, Center for Health and the Environment, University of California, Davis, CA 95616 USA ; Tel: 530-752-8334: Fax: 530-752-5300
References
- Ayres JG, Borm P, Cassee FR, Castranova V, Donaldson K, Ghio A, Harrison RM, Hider R, Kelly F, Kooter IM, Marano F, Maynard RL, Mudway I, Nel A, Sioutas C, Smith S, Baeza-Squiban A, Cho A, Duggan S, Froines J. Evaluating the toxicity of airborne particulate matter and nanoparticles by measuring oxidative stress potential–a workshop report and consensus statement. Inhal Toxicol. 2008;20:75–99. doi: 10.1080/08958370701665517. [DOI] [PubMed] [Google Scholar]
- Bateman ED, Hurd SS, Barnes PJ, Bousquet J, Drazen JM, FitzGerald M, Gibson P, Ohta K, O’Byrne P, Pedersen SE, Pizzichini E, Sullivan SD, Wenzel SE, Zar HJ. Global strategy for asthma management and prevention: GINA executive summary. Eur Respir J. 2008;31:143–178. doi: 10.1183/09031936.00138707. [DOI] [PubMed] [Google Scholar]
- Bein K, Wexler A. A high-efficiency, low-bias method for extracting particulate matter from filter and impactor substrates. Atmos Environ. 2014;90:87–95. [Google Scholar]
- Bein K, Wexler A. Compositional variance in extracted particulate matter using different filter extraction techniques. Atmos Environ. 2015;107:24–34. [Google Scholar]
- Bowatte G, Lodge C, Lowe AJ, Erbas B, Perret J, Abramson MJ, Matheson M, Dharmage SC. The influence of childhood traffic-related air pollution exposure on asthma, allergy and sensitization: A systematic review and a meta-analysis of birth cohort studies. Allergy. 2015;70:245–256. doi: 10.1111/all.12561. [DOI] [PubMed] [Google Scholar]
- Brown D, Hutchison G, Stone V, Barlow P. In: Particle Toxicology. Donaldson K, Borm P, editors. Boca Raton, FL: Taylor & Francis Group; p. 2006. [Google Scholar]
- California Health Interview Survey. Los Angeles, CA: UCLA Center for Health Policy Research; 2012. [Google Scholar]
- Carosino CM, Bein KJ, Plummer LE, Castañeda AR, Zhao Y, Wexler AS, Pinkerton KE. Allergic airway inflammation is differentially exacerbated by daytime and nighttime ultrafine and submicron fine ambient particles: Heme oxygenase-1 as an indicator of PM-mediated allergic inflammation. J Toxicol Environ Health A. 2015;78:254–266. doi: 10.1080/15287394.2014.959627. [DOI] [PubMed] [Google Scholar]
- Castañeda AR, Pinkerton KE. Investigating the effects of particulate matter on house dust mite and ovalbumin allergic airway inflammation in mice. Curr Protoc Toxicol. 2016;68:18.18.1–18.18.17. doi: 10.1002/cptx.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Chiu HF, Yang CY. Air pollution exposure and daily clinical visits for allergic rhinitis in a subtropical city: Taipei, Taiwan. J Toxicol Environ Health A. 2016;79:494–501. doi: 10.1080/15287394.2016.1182002. [DOI] [PubMed] [Google Scholar]
- Cheng MH, Chen CC, Chiu HF, Yang CY. Fine particulate air pollution and hospital admissions for asthma: A case-crossover study in Taipei. J Toxicol Environ Health A. 2014;77:1075–1083. doi: 10.1080/15287394.2014.922387. [DOI] [PubMed] [Google Scholar]
- Deiuliis JA, Kampfrath T, Zhong J, Oghumu S, Maiseyeu A, Chen LC, Sun Q, Satoskar AR, Rajagopalan S. Pulmonary T cell activation in response to chronic particulate air pollution. Am J Physiol Lung Cell Mol Physiol. 2012;302:L399–L409. doi: 10.1152/ajplung.00261.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira BF, Chacra AP, Frauches TS, Vallochi A, Hacon S. A curated review of recent literature of biomarkers used for assessing air pollution exposures and effects in humans. J Toxicol Environ Health B. 2014;17:369–410. doi: 10.1080/10937404.2014.976893. [DOI] [PubMed] [Google Scholar]
- Diaz-Sanchez D, Dotson AR, Takenaka H, Saxon A. Diesel exhaust particles induce local IgE production in vivo and alter the pattern of IgE messenger RNA isoforms. J Clin Invest. 1994;94:1417–1425. doi: 10.1172/JCI117478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Sanchez D, Garcia MP, Wang M, Jyrala M, Saxon A. Nasal challenge with diesel exhaust particles can induce sensitization to a neoallergen in the human mucosa. J Allergy Clin Immunol. 1999;104:1183–1188. doi: 10.1016/s0091-6749(99)70011-4. [DOI] [PubMed] [Google Scholar]
- Fuertes E, Heinrich J. The influence of childhood traffic-related air pollution exposure on asthma, allergy and sensitization. Allergy. 2015;70:1350–1351. doi: 10.1111/all.12611. [DOI] [PubMed] [Google Scholar]
- Fuertes E, Standl M, Cyrys J, Berdel D, von Berg A, Bauer CP, Kramer U, Sugiri D, Lehmann I, Koletzko S, Carlsten C, Brauer M, Heinrich J. A longitudinal analysis of associations between traffic-related air pollution with asthma, allergies and sensitization in the GINIplus and LISAplus birth cohorts. PeerJ. 2013;1:e193. doi: 10.7717/peerj.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghio AJ, Carraway MS, Madden MC. Composition of air pollution particles and oxidative stress in cells, tissues and living systems. J Toxicol Environ Health B. 2012;15:1–21. doi: 10.1080/10937404.2012.632359. [DOI] [PubMed] [Google Scholar]
- Greenberg N, Carel RS, Derazne E, Bibi H, Shpriz M, Tzur D, Portnov BA. Different effects of long-term exposures to SO2 and NO2 air pollutants on asthma severity in young adults. J Toxicol Environ Health A. 2016;79:342–351. doi: 10.1080/15287394.2016.1153548. [DOI] [PubMed] [Google Scholar]
- Herner JD, Aw J, Gao O, Chang DP, Kleeman MJ. Size and composition distribution of airborne particulate matter in northern California: I–particulate mass, carbon, and water-soluble ions. J Air Waste Manage Assoc. 2005;55:30–51. doi: 10.1080/10473289.2005.10464600. [DOI] [PubMed] [Google Scholar]
- Jayawardene WP, Youssefagha AH, Lohrmann DK, El Afandi GS. Prediction of asthma exacerbations among children through integrating air pollution, upper atmosphere, and school health surveillances. Allergy Asthma Proc. 2013;34:e1–8. doi: 10.2500/aap.2013.34.3629. [DOI] [PubMed] [Google Scholar]
- Kelly FJ, Fussell JC. Air pollution and airway disease. Clin Exp Allergy. 2011;41:1059–1071. doi: 10.1111/j.1365-2222.2011.03776.x. [DOI] [PubMed] [Google Scholar]
- Kim H, Bernstein JA. Air pollution and allergic disease. Curr Allergy Asthma Rep. 2009;9:128–133. doi: 10.1007/s11882-009-0019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooter IM, Boere AJ, Fokkens PH, Leseman DL, Dormans JA, Cassee FR. Response of spontaneously hypertensive rats to inhalation of fine and ultrafine particles from traffic: experimental controlled study. Part Fibre Toxicol. 2006;3:7. doi: 10.1186/1743-8977-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Harkema JR, Lewandowski RP, Wang M, Bramble LA, Gookin GR, Ning Z, Kleinman MT, Sioutas C, Nel AE. Ambient ultrafine particles provide a strong adjuvant effect in the secondary immune response: Implication for traffic-related asthma flares. Am J Physiol Lung Cell Mol Physiol. 2010;299:L374–L383. doi: 10.1152/ajplung.00115.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, Wang M, Oberley T, Froines J, Nel A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Persp. 2003;111:455–460. doi: 10.1289/ehp.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Venkatesan MI, Miguel A, Kaplan R, Gujuluva C, Alam J, Nel A. Induction of heme oxygenase-1 expression in macrophages by diesel exhaust particle chemicals and quinones via the antioxidant-responsive element. J Immunol. 2000;165:3393–3401. doi: 10.4049/jimmunol.165.6.3393. [DOI] [PubMed] [Google Scholar]
- Li N, Wang M, Barajas B, Sioutas C, Williams MA, Nel AE. Nrf2 deficiency in dendritic cells enhances the adjuvant effect of ambient ultrafine particles on allergic sensitization. J Innate Immun. 2013;5:543–554. doi: 10.1159/000347060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wang M, Bramble LA, Schmitz DA, Schauer JJ, Sioutas C, Harkema JR, Nel AE. The adjuvant effect of ambient particulate matter is closely reflected by the particulate oxidant potential. Environ Health Persp. 2009;117:1116–1123. doi: 10.1289/ehp.0800319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Xia T, Nel AE. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic Biol Med. 2008;44:1689–1699. doi: 10.1016/j.freeradbiomed.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell R, Berhane K, Yao L, Jerrett M, Lurmann F, Gilliland F, Kunzli N, Gauderman J, Avol E, Thomas D, Peters J. Traffic, susceptibility, and childhood asthma. Environ Health Persp. 2006;114:766–772. doi: 10.1289/ehp.8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitschik S, Schierl R, Nowak D, Jorres RA. Effects of particulate matter on cytokine production in vitro: A comparative analysis of published studies. Inhal Toxicol. 2008;20:399–414. doi: 10.1080/08958370801903784. [DOI] [PubMed] [Google Scholar]
- Mortimer K, Neugebauer R, Lurmann F, Alcorn S, Balmes J, Tager I. Early-lifetime exposure to air pollution and allergic sensitization in children with asthma. J Asthma. 2008;45:874–881. doi: 10.1080/02770900802195722. [DOI] [PubMed] [Google Scholar]
- Peterson B, Saxon A. Global increases in allergic respiratory disease: The possible role of diesel exhaust particles. Ann Allergy Asthma Immunol. 1996;77:263–268. doi: 10.1016/S1081-1206(10)63318-2. quiz 269-270. [DOI] [PubMed] [Google Scholar]
- State of the Air. Nashville, TN: American Lung Association; 2015. [Google Scholar]
- Van Winkle LS, Bein K, Anderson D, Pinkerton KE, Tablin F, Wilson D, Wexler AS. Biological dose response to PM2.5: effect of particle extraction method on platelet and lung responses. Toxicol Sci. 2015;143:349–359. doi: 10.1093/toxsci/kfu230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Klot S, Wolke G, Tuch T, Heinrich J, Dockery DW, Schwartz J, Kreyling WG, Wichmann HE, Peters A. Increased asthma medication use in association with ambient fine and ultrafine particles. Eur Respir J. 2002;20:691–702. doi: 10.1183/09031936.02.01402001. [DOI] [PubMed] [Google Scholar]