

It is a privilege and honor to have the opportunity to present the Kober Lecture at the premier meeting for physician-scientists and to have the lecture published in The Journal of Clinical Investigation.
Like other members of the Association of American Physicians, I am often asked by younger physician-scientists, “What were the most important decisions of your career?” On reflection, there were three, which I will review briefly before presenting an overview of our recent work on fatty liver disease (FLD), a burgeoning health problem in the Western world.
First career decision.
In 1979, while an intern in medicine at Columbia Presbyterian Hospital, I made the first and most important decision of my career, and of my personal life. I decided to marry my late husband, Dennis Stone (Figure 1, left), who was then a senior resident. Dennis had attended the University of Texas (UT) Southwestern Medical Center, and wanted to return there for subspecialty training. So after completing my internship at Columbia, we moved to Dallas and I continued my clinical training at UT Southwestern.
Figure 1. My muses and mentors.

My career would not have survived the deletion test of any of these men: Dennis K. Stone, my late husband (left, picture taken in 1980 when we met as residents at Columbia-Presbyterian Hospital); Donald W. Seldin, Chief of Medicine, UT Southwestern (middle, picture taken in 1983 when I was chief resident of internal medicine, UT Southwestern); and Michael S. Brown and Joseph L. Goldstein, scientific mentors at UT Southwestern (right, picture taken in 1985 when I was a postdoctoral fellow in their laboratory).
In Dallas, I met three outstanding mentors who changed the trajectory of my life. The first was the late Dr. Donald Seldin, Chairman of Internal Medicine (Figure 1, middle). I was a proverbial “late bloomer.” Only after becoming chief resident of medicine did I decide to train in clinical endocrinology. Dr. Seldin thought my decision was a terrible one, and strongly advised me to enter a laboratory and train as a scientist. And he was specific about the laboratory to join, that of Drs. Michael Brown and Joseph Goldstein. I inquired, naively, as to why I should go to that laboratory, given that I had no special predilection for lipoprotein metabolism. His response was terse and clear: always go for the best. Get the best and most rigorous scientific training available, which at UT Southwestern would be in the Brown and Goldstein laboratory.
Why would I take Dr. Seldin’s advice, since I was enjoying patient care and had never entertained training in a research laboratory? I followed his advice because he really knew me. He knew my strengths and, more importantly to me at the time, my weaknesses, and I trusted his judgement.
So at age 30 I dutifully joined the Brown and Goldstein laboratory (Figure 1, right), never having held a pipetman. My transition from the wards to the bench proved extremely difficult and frustrating. (It was equally difficult and frustrating for Drs. Brown and Goldstein, I am certain!) I could write a treatise on what I learned in the four years I spent in their laboratory. Suffice it to say, I am sure that I was among the best-mentored physician-scientists of that time.
So the first, and most important decision in my career (and life) was to marry Dennis, a man whom my mother aptly stated wanted more for me than I wanted for myself. If I had not married Dennis, I would not have traveled to Texas and worked with Dr. Seldin. And if I had not worked with Dr. Seldin, I would never have trained with Drs. Brown and Goldstein. My scientific career would not have survived the so-called “deletion test” of any of these four men (Figure 1).
Second career decision: finding my scientific sweet spot.
I skip now to the midpoint of my career, when I made two decisions that put my research program on course. It is no coincidence that the first of these decisions was made soon after my younger son got his driver’s license, which afforded me the time and energy to think more deeply about my science. I had been working on a scavenger receptor called scavenger receptor class B member 1 (SR-B1, now called SCARB1). This cell surface receptor was discovered by Monty Krieger (Massachusetts Institute of Technology) and we had shown in mice that SCARB1 delivers cholesteryl esters from circulating HDL to hepatocytes and steroidogenic tissues (1, 2). I went on to probe the mechanism by which SCARB1 selectively transfers neutral lipids from lipoproteins to cells (3, 4). I wrote several papers on the topic, but made no mechanistic breakthroughs. In retrospect, it is not surprising that I made so little progress, given my limited knowledge of the physical chemistry of lipids.
While working on SCARB1, I seriously questioned my decision to pursue a scientific career, and even considered alternative career paths. I shared my existential doubts with Drs. Goldstein and Brown. They both advised me to focus on my laboratory and make a discovery. A discovery, they assured me, would dispel my doubts about being a scientist.
Around that time, I attended a scientific meeting in Europe, where the mapping and cloning of the gene defective in Tangier disease was reported (5). Although very rare, Tangier disease is well known in the lipoprotein field because of its association with very low plasma levels of HDL-cholesterol (HDL-C) and the accumulation of cholesteryl esters in scavenger cells in tissues, most famously in the oropharynx (yellow tonsils) (6). The defective gene encodes a member of the family of ATP-binding cassette (ABC) transporter proteins, ABCA1 (5, 7, 8). I recognized immediately the importance of the discovery. The identification of ABCA1 would provide new insights into a poorly characterized pathway in cholesterol transport. I also recognized the implications for my own work. Why had I not made that discovery? I had seen patients with Tangier disease and even had their genomic DNA in my freezer.
When I returned to Dallas, I immediately retooled my laboratory, collected families with recessive forms of hypercholesterolemia, and mapped and then subsequently cloned the defective genes. First we cloned the gene that is inactivated in autosomal recessive hypercholesterolemia, a disease that is common on Sardinia (9). The culprit gene (ARH or LDLRAP1) encodes an adaptor protein that links the LDL receptor to the endocytic machinery in hepatocytes (9–11). Absence of ARH results in delayed clearance of LDL from the circulation, resulting in hypercholesterolemia and premature coronary atherosclerosis (11–13). Next we cloned the genes defective in sitosterolemia, a disorder in which plant and animal sterols accumulate in blood and tissues. The defective genes in this disorder, ABCG5 and ABCG8, encode two ABC half-transporters that we found heterodimerize to form a major conduit for excretion of sterols from the body (14–16).I had finally identified my scientific sweet spot: a place where my clinical acumen, my proficiency in human genetics, and my scientific expertise in lipoprotein metabolism overlapped.
Third career decision: making a scientific partnership.
The third major decision of my career was made while designing the Dallas Heart Study, a multiethnic, population-based study in Dallas. I joined forces with Jonathan C. Cohen to write a grant based on the hypothesis that rare and low-frequency genetic variants contribute to complex diseases, such as coronary atherosclerosis (Figure 2 and ref. 17). The hypothesis was contrary to the prevailing view at the time, which held that common variants cumulatively cause common diseases (the common variant, common disease hypothesis) (18). We reasoned that if low-frequency variants with large effects were present in the population, their identification would expedite the translation of a genetic association into a therapeutic product.
Figure 2. A scientific partnership.

This picture was taken in 2006, six years after Jonathan Cohen and I merged our laboratories. We were attending a meeting in Rome where I presented our work on PCSK9 to the cardiovascular research community.
To achieve our goal, Jonathan and I established high-throughput sequencing in our laboratory (using the Sanger method, so low-throughput by current standards). We then sequenced candidate genes in individuals at the upper and lower extremes of the distribution for each trait, starting with plasma HDL-C levels. We compared the numbers of nonsynonymous variants in the two groups and found that rare, loss-of-function variants were significantly more abundant in those with low HDL-C than in those with high HDL-C levels (19). Subsequently, we used the same “sequencing the extremes” strategy to discover that loss-of-function mutations in PCSK9 reduce plasma LDL-C levels (20), and protect against heart disease (21). We also identified a relative of a Dallas Heart Study participant who did not express any PCSK9, and yet was healthy and fertile (22). These observations, together with those of our colleague, Jay Horton (23, 24), contributed to the rapid development of FDA-approved anti-PCSK9 therapeutic antibodies. These antibodies dramatically lower plasma LDL-C levels and protect against coronary atherosclerosis, just as our genetic studies had predicted (25, 26).
Thus, our first goal in the Dallas Heart Study was to use DNA sequencing to identify variants that have major effects on traits of medical interest and importance. Our other goal was to identify a new trait that was associated with a poorly understood disorder. We would then use genetics to get a molecular handle on key players in disease pathogenesis in humans. The trait we selected to study was hepatic triglyceride (TG) content (HTGC) (27).
FLD: nature and nurture.
The first step in both nonalcoholic and alcoholic FLD is the accumulation of TG in cytoplasmic lipid droplets within hepatocytes (Figure 3). FLD has burgeoned in frequency due to the increased prevalence of its two major risk factors: obesity and insulin resistance. Hepatic steatosis is often referred to as “bland” or “simple” since it is generally considered to be benign. But a subset of individuals with steatosis develops inflammation and fibrosis (steatohepatitis). In some, the disease progresses from steatohepatitis to cirrhosis and then to hepatocellular carcinoma (HCC) (Figure 3). In a smaller subset of individuals, hepatic steatosis and steatohepatitis progress directly to HCC (28, 29).
Figure 3. Fatty liver disease (FLD): a continuum of related disorders.

Both alcoholic and nonalcoholic FLD encompass a continuum of histological and pathological diagnoses. All start with steatosis and some progress to steatohepatitis, cirrhosis, and hepatocellular carcinoma. Reversibility between different states is shown with double-headed arrows. Irreversible progression is shown with single-headed arrows. All arrows are dotted since liver disease does not progress in all those at risk.
We measured HTGC in the Dallas Heart Study (n = 2,287) using proton magnetic resonance spectroscopy, the most accurate noninvasive assay of hepatic fat (30). This study provided the first quantitative data on the distribution of HTGC in the general population (31, 32). Approximately 31% of the cohort had hepatic steatosis (defined as a hepatic TG content greater than 5.5%) (32). More intriguing was the finding that Hispanics had a much higher prevalence of hepatic steatosis (45%) than did individuals of either European (33%) or African descent (24%) (31). These differences could be only partially explained by interethnic differences in body weight and insulin sensitivity. We hypothesized that the ethnic differences in HTGC were heritable, a sequela of differences in genetic ancestry. Accordingly, we performed a genome-wide association study (GWAS) (33). We restricted our survey to nonsynonymous variants to focus on those most likely to have large effects on HTGC, and to minimize the loss of statistical power incurred by correction for multiple testing. The analysis revealed a highly significant association with a single nucleotide polymorphism (SNP) in PNPLA3 (P < 7.0 × 10–14). This variant remains the most important genetic risk factor for FLD (33, 34). We subsequently identified a second risk allele for hepatic steatosis by performing a GWAS with a denser panel of exonic SNPs (35). The second variant was in a gene of unknown function that encodes transmembrane 6 superfamily member 2 (TM6SF2). Multiple investigators have subsequently shown that both the PNPLA3 and TM6SF2 variants are not only associated with increased hepatic fat, but also with the full spectrum of alcoholic as well as nonalcoholic FLD (for review, see refs. 34, 36, and Figure 3).
Two genes, two pathways contributing to FLD: implications for disease pathogenesis.
Both the PNPLA3 and TM6SF2 risk variants we identified are missense variants (Figure 4). In PNPLA3, methionine is substituted for isoleucine at position 148. In TM6SF2 the variant results in the substitution of lysine for glutamic acid at position 167. Homozygosity for either of the risk alleles is associated with an approximately 2-fold increase in median HTGC (33, 35). Heterozygotes have an intermediate HTGC.
Figure 4. Comparison of two major genetic risk factors for fatty liver disease: PNPLA3(148M) and TM6SF2(167K).

The major features of the two missense mutations in PNPLA3 and TM6SF2 that confer susceptibility to fatty liver disease are summarized.
The frequency of PNPLA3(148M) parallels the prevalence of hepatic steatosis among ethnicities (33). The PNPLA3 risk allele is very common in Hispanics (49%) and much less frequent among African-Americans (17%) than among non-Hispanic whites (23%). This variant alone explains 60% to 70% of the interethnic differences in the prevalence of hepatic steatosis. TM6SF2(167K) is much less frequent in all three ethnic groups (3%–7%) (35). Although neither variant is associated with body mass index or insulin sensitivity (33), the impact of the variants on expression of FLD is highly dependent on the presence of these two risk factors. Both adiposity and insulin resistance increase the penetrance of both risk alleles (37).
Despite having similar effects on HTGC, the two risk alleles are associated with different levels of circulating lipids. PNPLA3(148M) is associated with significantly lower TG levels, but only among the very obese (38), and it has no effect on plasma cholesterol levels. In contrast, individuals with the TM6SF2(167K) variant have lower plasma levels of both cholesterol and TG (35, 39). These differences in circulating lipid levels provided the first clue that the variants caused FLD by different mechanisms.
PNPLA3 is a member of the patatin-like phospholipase domain–containing family of proteins that shares a common fold with patatin, a plentiful plant protein that has nonspecific acyl hydrolase activity (40, 41). A structural model based on the crystal structure of patatin predicts that the risk variant (I148M) is located in a hydrophobic groove that forms part of the substrate-binding site (42, 43). The longer side chain of methionine is predicted to prevent access to the serine of the catalytic dyad.
PNPLA3 is expressed at highest levels in adipose tissue and liver, where it is expressed in predominantly in hepatocytes (44). Approximately 90% of the protein localizes to lipid droplets and the C-terminal half of the protein is required for this localization (42). In mouse liver, PNPLA3 is expressed at very low levels in the fasting state and is among the most upregulated transcripts with refeeding (44). The gene is a direct target of sterol regulatory element–binding protein 1c (44), an insulin-responsive transcription factor that also orchestrates the upregulation of fatty acid synthesis (45).
The mechanism by which the I148M variant confers susceptibility to FLD remains to be clearly defined. The enzyme has TG hydrolase activity in vitro, and the I148M substitution attenuates this activity (46, 47), but Pnpla3–/– mice do not have hepatic steatosis, thus ruling out a pure loss-of-function mechanism (48, 49). Hepatic overexpression of the mutant protein, but not the wild-type protein, causes fatty liver, which is consistent with the variant being a neomorph (50). Wild-type PNPLA3 is rapidly degraded, whereas PNPLA3(148M) has a much slower turnover due at least partially to reduced ubiquitylation and proteasomal degradation (51). The mutant protein accumulates on lipid droplets (52), where it may alter the composition and/or architecture of the droplet in such a manner that it interferes with the action of other lipases on the droplet, thus impairing TG mobilization.
In contrast to PNPLA3, TM6SF2 is a polytopic protein of the endoplasmic reticulum (ER) and Golgi complex (53, 54); the protein contains a Golgi retrieval signal sequence at the C-terminus (KKQH) and presumably cycles between these two compartments (55). TM6SF2 is expressed at highest levels in the intestine and liver, the two organs that synthesize ApoB-containing lipoproteins (35). Unlike PNPLA3, the expression of TM6SF2 is not altered significantly by dietary manipulation (53).
The E167K substitution destabilizes the protein and is therefore presumed to be a loss-of-function mutation (35). This notion is supported by our finding that Tm6sf2–/– mice recapitulate the phenotype observed in humans (53). These mice have reduced rates of secretion of TG from the liver. TG is secreted as a component of VLDL, which then matures into LDL as it circulates through peripheral tissues. The rate of secretion of VLDL-ApoB from the liver does not differ between wild-type and Tm6sf2–/– mice. Thus, TM6SF2 is involved in the lipidation of nascent VLDL particles. Absence of the protein results in the accumulation of TG in the liver, and reduced circulating levels of Apo-B–containing lipoproteins.
In conclusion, these two genetic risk variants confer susceptibility to hepatic steatosis by different pathways. PNPLA3(148M) is a lipid droplet protein that appears to disrupt TG mobilization from droplets, whereas TM6SF2(167K) is an ER/Golgi protein that limits VLDL-TG secretion.
Lipids, inflammation, and fibrosis.
Despite causing fatty liver by different mechanisms, both PNPLA3(148M) and TM6SF2(E167K) are associated with the full spectrum of FLD, both nonalcoholic and alcoholic (Figure 3). These findings suggest that hepatic steatosis may not be so bland, especially if it is established at an early age and maintained for many years.
The relationship between hepatic fat accumulation and liver disease progression resembles that observed with hypercholesterolemia and coronary atherosclerosis (Figure 5). Chronic exposure of the coronary arteries to excess LDL, irrespective of the molecular basis of the hypercholesterolemia, promotes inflammation and fibrosis. In a similar fashion, chronic exposure of hepatocytes to excess TG, irrespective of the molecular cause, promotes progression of liver disease. Lipid accumulation is a necessary first step in the development of the common forms of both diseases (Figure 3).
Figure 5. The primacy of lipid accumulation in fatty liver disease (FLD) and coronary atherosclerosis.
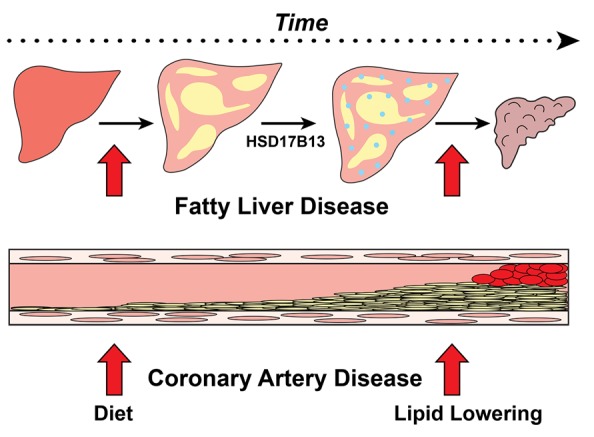
Neutral lipid accumulation in hepatocytes and in the intima of coronary arteries is the first step toward the development of FLD and coronary atherosclerosis, respectively. In both complex disorders, multiple factors confer susceptibility to disease progression with inflammation and fibrosis. HSD17B13 expression is one such susceptibility factor for FLD.
The optimal approach to preventing coronary atherosclerosis is to reduce plasma cholesterol levels from an early age. This can best be accomplished by reducing dietary cholesterol and saturated fat while maintaining an ideal body weight. These dietary interventions have their greatest impact when initiated early in life so that cumulative exposure of the coronary arteries to LDL is minimized. Individuals with loss-of-function mutations in PCSK9 enjoy relative protection from coronary heart disease because they have had lower cholesterol levels throughout their lives.
In a similar fashion, individuals who consume a prudent diet and maintain an ideal body weight are at low risk of developing hepatic steatosis, even if they inherit an FLD risk allele (37).
Currently, no FDA-approved agents to reduce hepatic TG are as effective as statins, ezetimibe, and PCSK9 antibodies for lowering plasma cholesterol levels. It is likely that reducing hepatic TG levels in FLD will have beneficial effects in the liver just as lipid-lowering therapy does for the prevention of heart disease.
Protective alleles for FLD.
Why does chronic exposure of hepatocytes to TG, which is sequestered in lipid droplets, promote inflammation, fibrosis, and cancer? We could not address this question in the Dallas Heart Study due to its small sample size. Therefore, we have established a cohort of patients with FLD. Our goal in these studies is to identify alleles that confer protection from FLD in a manner that is analogous to loss-of-function mutations in PCSK9 and protection from heart disease.
Together with Regeneron, we recently reported a variant that is not associated with hepatic steatosis in the Dallas Heart Study and yet protects against the progression of FLD (56). The variant, which is in a steroid dehydrogenase of unknown function, hydroxysteroid 17-β dehydrogenase (HSD17B13), is depleted in those with steatohepatitis or cirrhosis. This observation has the potential to lead to a new approach for preventing and treating this increasingly common and potentially devastating disease.
Science, serendipity, and the single degree.
For me, medicine was a portal to science. I would never have become a scientist without first becoming a physician. I have never regretted being “just an MD,” but I do regret not studying chemistry, biochemistry, and mathematics more deeply or having more laboratory experience before getting my clinical training.
Serendipity played a major role in my career. As outlined above, a series of unlikely events led me to become a physician-scientist. During my residency training, Tom H. Lee, a resident in training at Brigham and Women’s Hospital, told me, “If you get into a well-functioning organization, have talent, and work hard, the organization will pull you up.” UT Southwestern has been such a place for me and I am indebted to the institution. Finally, any success I have enjoyed is shared equally with Jonathan C. Cohen, my scientific partner of the last 18 years, and with the gifted students and postdoctoral fellows who have worked in our laboratory.
Acknowledgments
This work was supported by the Howard Hughes Medical Institute and the following grants from the NIH: R01 HL072304, R01 DK090066, P01 HL20948, and UL1 TR001105. I wish to thank Jonathan C. Cohen and Jay D. Horton (UT Southwestern), and Stephen G. Young (UCLA) for manuscript review.
Version 1. 10/01/2018
Print issue publication
Footnotes
Reference information: J Clin Invest. 2018;128(10):4218–4223. https://doi.org/10.1172/JCI124404.
This article is adapted from a presentation at the 2018 AAP/ASCI/APSA Joint Meeting, April 21, 2018, in Chicago, Illinois, USA.
References
- 1.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271(5248):518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 2.Landschulz KT, Pathak RK, Rigotti A, Krieger M, Hobbs HH. Regulation of scavenger receptor, class B, type I, a high density lipoprotein receptor, in liver and steroidogenic tissues of the rat. J Clin Invest. 1996;98(4):984–995. doi: 10.1172/JCI118883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stangl H, Cao G, Wyne KL, Hobbs HH. Scavenger receptor, class B, type I-dependent stimulation of cholesterol esterification by high density lipoproteins, low density lipoproteins, and nonlipoprotein cholesterol. J Biol Chem. 1998;273(47):31002–31008. doi: 10.1074/jbc.273.47.31002. [DOI] [PubMed] [Google Scholar]
- 4.Stangl H, Hyatt M, Hobbs HH. Transport of lipids from high and low density lipoproteins via scavenger receptor-BI. J Biol Chem. 1999;274(46):32692–32698. doi: 10.1074/jbc.274.46.32692. [DOI] [PubMed] [Google Scholar]
- 5.Rust S, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999;22(4):352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 6. Frederickson DS, Altrocchi PH, Avioli LV, Goodman DS, and Goodman HC. Tangier disease-combined clinical staff conference at the National Institutes of Health. Ann Intern Med. 1961;55(6):1016–1031. [Google Scholar]
- 7.Brooks-Wilson A, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22(4):336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 8.Bodzioch M, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22(4):347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 9.Arca M, et al. Autosomal recessive hypercholesterolaemia in Sardinia, Italy, and mutations in ARH: a clinical and molecular genetic analysis. Lancet. 2002;359(9309):841–847. doi: 10.1016/S0140-6736(02)07955-2. [DOI] [PubMed] [Google Scholar]
- 10.Garcia CK, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292(5520):1394–1398. doi: 10.1126/science.1060458. [DOI] [PubMed] [Google Scholar]
- 11.Wilund KR, et al. Molecular mechanisms of autosomal recessive hypercholesterolemia. Hum Mol Genet. 2002;11(24):3019–3030. doi: 10.1093/hmg/11.24.3019. [DOI] [PubMed] [Google Scholar]
- 12.Jones C, et al. Disruption of LDL but not VLDL clearance in autosomal recessive hypercholesterolemia. J Clin Invest. 2007;117(1):165–174. doi: 10.1172/JCI29415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones C, Hammer RE, Li WP, Cohen JC, Hobbs HH, Herz J. Normal sorting but defective endocytosis of the low density lipoprotein receptor in mice with autosomal recessive hypercholesterolemia. J Biol Chem. 2003;278(31):29024–29030. doi: 10.1074/jbc.M304855200. [DOI] [PubMed] [Google Scholar]
- 14.Berge KE, et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290(5497):1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 15.Yu L, et al. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci U S A. 2002;99(25):16237–16242. doi: 10.1073/pnas.252582399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu L, et al. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest. 2002;110(5):671–680. doi: 10.1172/JCI16001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Victor RG, et al. The Dallas Heart Study: a population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol. 2004;93(12):1473–1480. doi: 10.1016/j.amjcard.2004.02.058. [DOI] [PubMed] [Google Scholar]
- 18.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17(9):502–510. doi: 10.1016/S0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 19.Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305(5685):869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 20.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37(2):161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 21.Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Z, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79(3):514–523. doi: 10.1086/507488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem. 2007;282(29):20799–20803. doi: 10.1074/jbc.C700095200. [DOI] [PubMed] [Google Scholar]
- 24.Lagace TA, et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116(11):2995–3005. doi: 10.1172/JCI29383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stein EA, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380(9836):29–36. doi: 10.1016/S0140-6736(12)60771-5. [DOI] [PubMed] [Google Scholar]
- 26.Sabatine MS, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 27.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55(7):434–438. [PubMed] [Google Scholar]
- 28.Paradis V, et al. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology. 2009;49(3):851–859. doi: 10.1002/hep.22734. [DOI] [PubMed] [Google Scholar]
- 29.Piscaglia F, et al. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: A multicenter prospective study. Hepatology. 2016;63(3):827–838. doi: 10.1002/hep.28368. [DOI] [PubMed] [Google Scholar]
- 30.Longo R, et al. Proton MR spectroscopy in quantitative in vivo determination of fat content in human liver steatosis. J Magn Reson Imaging. 1995;5(3):281–285. doi: 10.1002/jmri.1880050311. [DOI] [PubMed] [Google Scholar]
- 31.Browning JD, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40(6):1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 32.Szczepaniak LS, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288(2):E462–E468. doi: 10.1152/ajpendo.00064.2004. [DOI] [PubMed] [Google Scholar]
- 33.Romeo S, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Speliotes EK, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7(3):e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kozlitina J, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46(4):352–356. doi: 10.1038/ng.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sookoian S, Pirola CJ. Genetic predisposition in nonalcoholic fatty liver disease. Clin Mol Hepatol. 2017;23(1):1–12. doi: 10.3350/cmh.2016.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stender S, Kozlitina J, Nordestgaard BG, Tybjærg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49(6):842–847. doi: 10.1038/ng.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romeo S, et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int J Obes (Lond) 2010;34(1):190–194. doi: 10.1038/ijo.2009.216. [DOI] [PubMed] [Google Scholar]
- 39.Holmen OL, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet. 2014;46(4):345–351. doi: 10.1038/ng.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Racusen D, Foote M. A major soluble glycoprotein of potato tubers. J Food Biochem. 1980;4(1):43–52. doi: 10.1111/j.1745-4514.1980.tb00876.x. [DOI] [Google Scholar]
- 41.Hirayama O, Matsuda H, Takeda H, Maenaka K, Takatsuka H. Purification and properties of a lipid acyl-hydrolase from potato tubers. Biochim Biophys Acta. 1975;384(1):127–137. doi: 10.1016/0005-2744(75)90102-3. [DOI] [PubMed] [Google Scholar]
- 42.He S, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285(9):6706–6715. doi: 10.1074/jbc.M109.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rydel TJ, et al. The crystal structure, mutagenesis, and activity studies reveal that patatin is a lipid acyl hydrolase with a Ser-Asp catalytic dyad. Biochemistry. 2003;42(22):6696–6708. doi: 10.1021/bi027156r. [DOI] [PubMed] [Google Scholar]
- 44.Huang Y, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci U S A. 2010;107(17):7892–7897. doi: 10.1073/pnas.1003585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem. 2011;286(43):37085–37093. doi: 10.1074/jbc.M111.290114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem. 2004;279(47):48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 48.Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology. 2010;52(3):1134–1142. doi: 10.1002/hep.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Basantani MK, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res. 2011;52(2):318–329. doi: 10.1194/jlr.M011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li JZ, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122(11):4130–4144. doi: 10.1172/JCI65179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. 2017;66(4):1111–1124. doi: 10.1002/hep.29273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smagris E, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61(1):108–118. doi: 10.1002/hep.27242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem. 2016;291(20):10659–10676. doi: 10.1074/jbc.M116.719955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mahdessian H, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A. 2014;111(24):8913–8918. doi: 10.1073/pnas.1323785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jackson MR, Nilsson T, Peterson PA. Retrieval of transmembrane proteins to the endoplasmic reticulum. J Cell Biol. 1993;121(2):317–333. doi: 10.1083/jcb.121.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abul-Husn NS, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med. 2018;378(12):1096–1106. doi: 10.1056/NEJMoa1712191. [DOI] [PMC free article] [PubMed] [Google Scholar]