

Abstract
An increased prevalence of familial neurodegenerative parkinsonism or cognitive deterioration was recently found in a small region of southeastern Moravia.
The aim of the study was to assess the genetic background of this familial disease.
Variants in the ADH1C, EIF4G1, FBXO7, GBA + GBAP1, GIGYF2, HTRA2, LRRK2, MAPT, PRKN, DJ-1, PINK1, PLA2G6, SNCA, UCHL1, VPS35 genes were examined in 12 clinically positive probands of the pedigree in which familial atypical neurodegenerative parkinsonism was identified in previous epidemiological studies. Libraries were sequenced by massive parallel sequencing (MPS) on the Personal Genome Machine (PGM; Ion Torrent). Data were analyzed using Torrent Suite and IonReporter software. All variants were then verified and confirmed by Sanger sequencing.
We identified 31 rare heterozygous variants: 11 missense variants, 3 synonymous variants, 8 variants in the UTR region, and 9 intronic variants. Six variants (rs1801334, rs33995883, rs35507033, rs781737269, rs779760087, and rs63750072) were evaluated as pathogenic by at least one in-silico predictor.
No single “founder” pathogenic variant associated with parkinsonism has been found in any of the probands from researched pedigree. It may rather be assumed that the familial occurrence of this disease is caused by the combined influence of several “small-effect” genetic variants that accumulate in the population with long-lasting inbreeding behavior.
Keywords: familial neurodegenerative parkinsonism, molecular-genetic background, population with long-lasting inbreeding behavior
1. Introduction
The results of epidemiological studies indicate that the prevalence of Parkinson disease (PD) and neurodegenerative parkinsonism in the general population is around 1.6% in Europe and approximately 1.5% in Asia and the Americas.[1–8] In the population over 65 years of age, the prevalence of these diseases ranges from 1.1% to 2.2%[1–11] and even higher prevalences have been repeatedly found in small, isolated populations.[3,4,6,9]
We recently described such a phenomenon in 10 villages of a remote, small, rural, and isolated Moravian region of Czech Republic called “Hornacko” [Upper Lands]. The last census, conducted in 2011, determined the presence of 8664 inhabitants, 2927 of which were over 50 years of age. “Hornacko” people have their own specific customs and traditions, which are reflected in the folk art, crafts, architecture, costumes, songs, and dances; also, the local dialect is very different from that of the surrounding areas (Fig. 1). This area is traditionally isolated from the neighboring Moravian regions and the people only rarely migrate permanently out of here.[12,13] In 2011 to 2015, we conducted an epidemiological study in this region which has found a significantly increased prevalence of neurodegenerative parkinsonism. The overall prevalence in the population over 50 years of age was 2.8% (95% confidence interval (CI), 2.2–3.4); the prevalence in the population between 50 and 64 years of age was 1.9% (95% CI, 1.2–2.5) and it was 4.06% (95% CI, 2.9–5.1) in the population over 65 years of age.[14,15] Three large pedigrees with a familial occurrence of parkinsonism were found in which clinical geneticists identified an autosomal-dominant inheritance pattern.[16] Two of these pedigrees eventually compiled one large family tree spanning several generations from 1840 to the present.[17] In this large pedigree, 12 probands with apparent clinical signs of parkinsonism were identified (Fig. 2). These probands were referred to a tertiary movement disorders center for detailed clinical and laboratory examinations including DNA sampling in order to obtain information about the clinical, biological, and genetic characteristics of this endemic and familial disorder.
Figure 1.
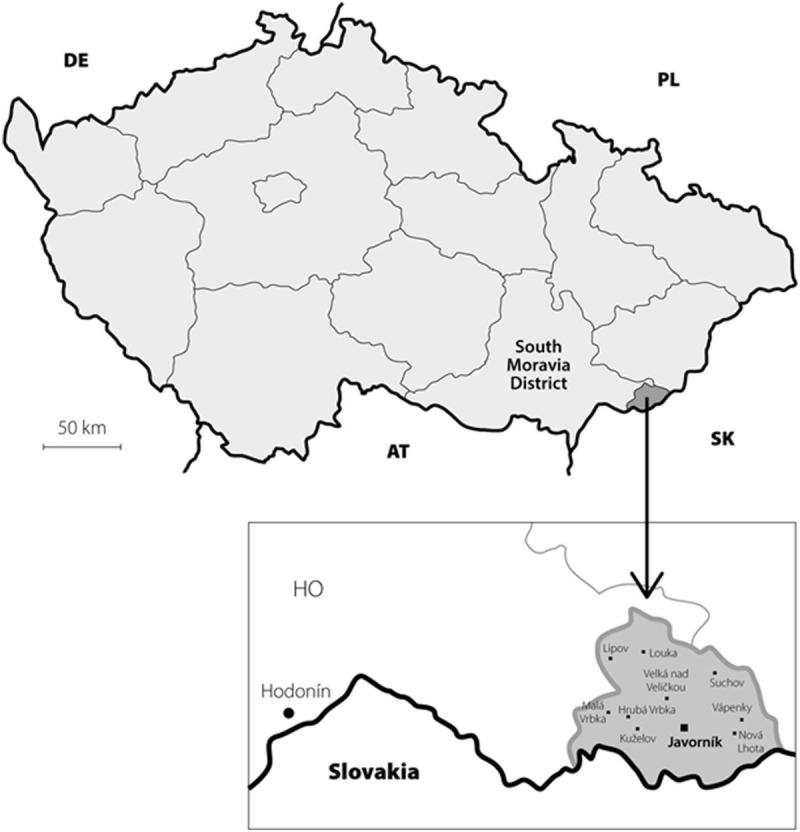
Map of Czech Republic with its district borders. The dark gray area in the lower right is the researched region with its 10 villages. The village where the researched pedigree originated (Javorník) is highlighted. DE: Germany, PL: Poland, AT: Austria, SK: Slovakia, HO: Hodonin county.
Figure 2.
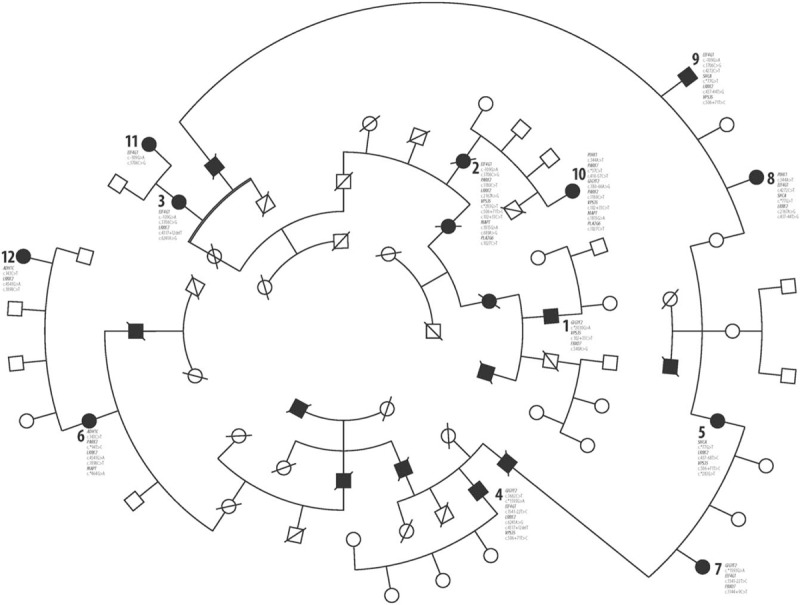
Large pedigree with familial parkinsonism and variants that have been identified in 12 symptomatic probands. The numbers of probands (1; 2; 3; etc.) in pedigree correspond with the numbers of probands in Table 1. The names of genes and their mutations which have been identified in individual probands are in the figure assigned to their numbers.
The village from which these families originate (Javorník nad Veličkou, PCN CZ-69674, elevation 412 m, population 720) is very specific even in this region: the population belongs almost entirely to the Lutheran denomination, unlike all neighboring villages, which are entirely Roman-Catholic. Marriages between the inhabitants of Javorník and residents of other villages were and still are quite exceptional, so the local population is practically isolated from a genetic point of view.[18]
2. Methods
The study was approved by the ethics committee of Palacký University Medical School, and all patients and probands gave their informed consent.
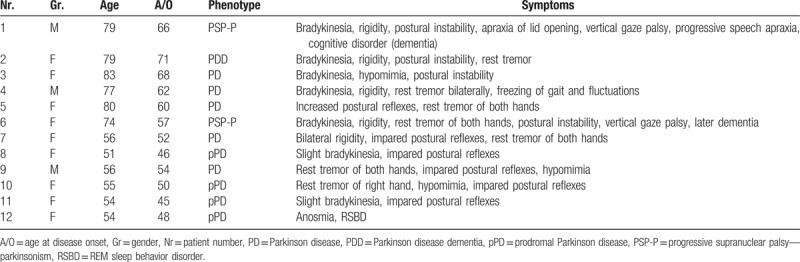
All 12 probands who were evaluated as positive for parkinsonian symptoms were examined by a movement disorders specialist. The United Kingdom Parkinson's Disease society Brain Bank clinical diagnostic criteria clinical diagnostic criteria were used for the diagnosis of parkinsonism; this was diagnosed when at least 2 of 4 Class I symptoms (resting tremor, rigidity, bradykinesia, and impaired postural reflexes) were present in a subject not receiving antiparkinsonian medication. Cognitive functions were assessed using the Mini Mental State Examination (MMSE) tool; cognitive deterioration was diagnosed when the MMSE score was ≤24. All positive probands were then examined in detail to exclude secondary forms of parkinsonism. The examination protocol included detailed neurological, psychological, and psychiatric examinations, examination of cerebrospinal fluid, magnetic resonance imaging of the brain; neurophysiological examinations were also performed (electroencephalographic recordings, electromyography, and assessments of visual, auditory, somatosensory, and motor evoked potentials).[16,17] The clinical details of all probands are presented in Table 1.
Table 1.
Clinical details of probands with clinical signs of parkinsonism.
DNA was isolated from peripheral blood by a DNA silica-gel membrane based isolation kit (QIAamp DNA Mini Kit - Qiagene; Strasse 1, 40724 Hilden, Germany). Each DNA sample was diluted to a final concentration of 10 ng/μL. The amplicon library was in silico designed by Ion AmpliSeq Designer (Thermo Fisher Scientific) for ADH1C, EIF4G1, FBXO7, GBA + GBAP1, GIGYF2, HTRA2, LRRK2, MAPT, PRKN, DJ-1, PINK1, PLA2G6, SNCA, UCHL1, and VPS35 genes. In total, 617 amplicons covered 92.59% of Coding Sequence (Coding Region of a Gene) including 100 bp in the introns and Untranslated Regions of mRNA of the regions. Libraries were prepared using Ion AmpliSeq Library Kit 2.0 (Ion Torrent; Termo Fisher Scientific, Waltham, Massachusetts, USA), emulsion PCR was done on the Ion OneTouch 2 Instrument (Ion Torrent) with Ion PGM Template OT2 200 Kit. Samples were barcoded to enable loading 8 samples on one Ion 316 Chip. The amplicons were sequenced by massive parallel sequencing (MPS) on the Personal Genome Machine (PGM; Ion Torrent) using Ion PGM Sequencing Kit (Ion Torrent) and Ion 316 Chip Kit v2. For the coverage control were the amplicons checked in Torrent Suite (Termo Fisher Scientific, Waltham, Massachusetts, USA) using plugin Coverage Analysis. Data analysis and variant searching (Mapping, Variant calling, Annotation) was done by Torrent Suite and IonReporter software (Termo Fisher Scientific, Waltham, Massachusetts, USA). Data from PGM were collected and converted to Fastq and BAM formats using Ion Torrent Suite software. The Bam/Bai files were reloaded to IonReporter and variant calling workflow was in the first level relaxed to minimal amplicon coverage of 4 and a minimal ratio of alternative/common variant of 0.1. Variants were then filtered using the parameter of minor allele frequency (MAF) <0.01. All variants were then verified and confirmed by Sanger sequencing. SIFT and PolyPhen-2 prediction software were used for missense variant evaluation. The PhyloP algorithm was used to assess the phylogenetical conservation.
3. Results
The variants identified in our 12 probands are shown in Table 2; 31 rare heterozygous variants were identified.
Table 2.
Genetic variants identified in probands with clinical signs of parkinsonism.
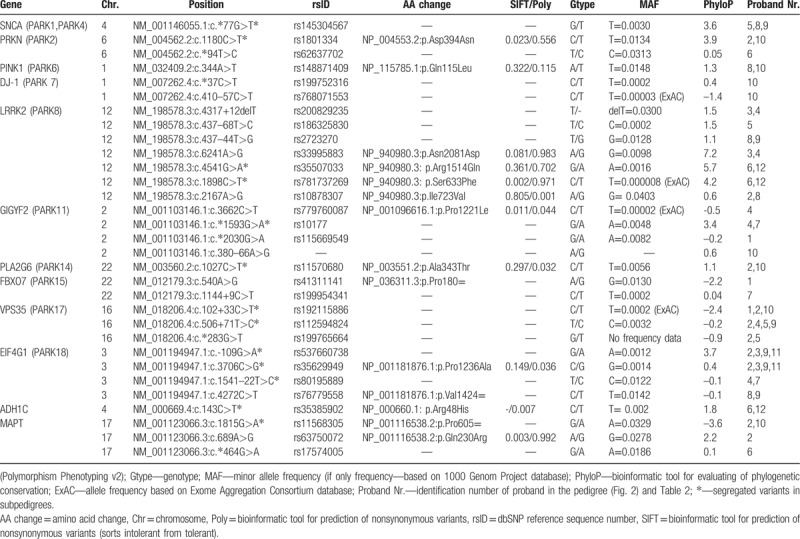
One variant was detected in the UTR-3 region in the SNCA (PARK1, PARK4) gene—c.∗77G>T (5,8,9). In the PRKN (PARK2) gene, we found 2 variants, 1 in the UTR-3 region—c.∗94T>C (6)—and 1 in exon—c.1180C>T (2,10). In the PINK1 (PARK6) gene was found 1 variant in the coding sequence—c.344A>T (8,10). In DJ-1 (PARK7) were found 2 variants in the noncoding region—c.∗37C>T (10) and c.410–57C>T (10). Three intronic—c.4317+12delT (3,4), c.437–68T>C (5), and c.437–44T>G (8,9)—and 4 exonic variants—c.2167A>G (2,8), c.6241A>G (3,4), c.4541G>A (6,12), and c.1898C>T (6,12) in the LRRK2 (PARK8) gene were detected. In the GIGYF2 (PARK11) gene were found 3 variants in the noncoding sequence—c.∗2030G>A (1), c.∗1593G>A (4,7), and c.380–66A>G (10) (this one has not been described before) and one in the coding sequence—c.3662C>T (4). In the PLA2G6 (PARK14) gene, one exonic variant—c.1027C>T (2,10) was found. In FBXO7(PARK15), 2 variants—c.540A>G (1) and c.1144+9C>T (7) were detected. In VPS35(PARK17), 3 variants in the noncoding sequence—c.∗283G>T (2,5), c.102+33C>T (1,2,10), and c.506+71T>C (2,4,5,9) were detected. Among 4 variants, 2 in the noncoding sequence—c.109G>A (2,3,9,11), c.1541–22T>C (4,7)—and 2 in the coding sequence—c.3706C>G (2,3,9,11), c.4272C>T (8,9)—were detected in the EIF4G1 (PARK18) gene. In the ADH1C gene was identified one variant located in the coding sequence—c.143C>T in 2 patients (6,12). Three variants, 1 in the noncoding sequence—c.∗464G>A (6)—and 2 in the coding sequence—c.1815G>A (2,10) and c.689A>G (2)—were detected in the MAPT gene. Segregated variants were seen only in particular subpedigrees and are signed as ∗ in the Table 2. The most “interesting” variants (PhyloP score ≥2 and at least 1 prediction tool evaluating it as pathogenic) are further emphasized in the Discussion section.
4. Discussion
Some past studies have suggested that people living in rural areas could be more exposed to putative environmental, mainly agricultural, influences and would be more likely to develop parkinsonism than those living in urban areas.[19] However, the area surveyed in our study is surrounded by a much larger rural area, in which the agricultural and environmental conditions are quite similar; to date, no signs of higher parkinsonism incidence in this (or any other) part of Central Europe have been reported. A genetic origin of familial parkinsonism thus seems to be the more probable explanation of presence in this population.
The molecular-genetic DNA analysis performed in 12 clinically positive probands in our pedigree has found several new variants in the loci associated with familial parkinsonism in the past:
4.1. NM_004562.2: c.1180 C>T PRKN (PARK2)
The missense substitution was evaluated as pathogenic using prediction programs (in SIFT pathogenic; in PolyPhen-2 probably pathogenic). It leads to an exchange of acidic for uncharged polar amino acid. Rare occurrence and also a higher degree of evolutionary conservation of the site has been reported. Moura et al[20] identified the variant in 9 (6.8%) of his patients; it was evaluated as a nonpathogenic polymorphism. Lücking et al[21] reported a statistically significant difference between patients and controls: the variant was found in 10/194 patients and in 12/125 controls. In another recent study, this variant was found in similar proportions in both patients and controls.[22]
4.2. NM_198578.3: c.6241A>G LRRK2(PARK8)
The substitution is located in an evolutionarily conserved region, which could, according to Polyphen-2, have a pathogenic character. However, SIFT evaluated this variant as benign. Its benign role is supported by the recent finding of Benitez et al,[23] who found this variant in 17 of 478 PD patients and 15 of 337 controls. In another study, Foo et al[24] found the variant in none of the patients and in 1 control (0.25%).
4.3. NM_198578.3: c.4541G>A LRRK2 (PARK8)
This missense variant in the coding region leads to the exchange of alkaline amino acid for polar uncharged in the phylogenetically conserved region. According to SIFT, it was evaluated as benign; according to PolyPhen-2, it was evaluated as probably pathogenic. Because of its rare occurrence in the human population and the high phylogenetic conservation of the region, it presumably influences the protein function. On the other hand, Nichols et al[25] described this variant as probably nonpathogenic. Toft et al[26] found the variant in 1 large kindred but they did not confirm its full segregation with parkinsonism. An analysis of the case-control series did not confirm an increased risk of parkinsonism with this variant, and no association with PD has been reported.
4.4. NM_198578.3: c.1898C>T LRRK2 (PARK8)
This is a missense variant in a coding conserved region, and it leads to the exchange of uncharged polar for nonpolar amino acid. It was evaluated as pathogenic (SIFT and PolyPhen-2). A pathogenic character can thus be expected, but no association with PD has been reported so far.
4.5. NM_001123066.3: c.689A>G MAPT (TAU)
This is a missense variant which leads to the substitution of uncharged polar amino acid for alkaline. The site is evolutionarily conserved. The variant was predicted as pathogenic. Jin et al[27] studied variants of MAPT associated with Alzheimer disease in the Spanish population. The variant was identified in 18 patients (10% probands) and 8 controls (5.8%). An association between the variant and the level of tau protein was described in the cerebrospinal fluid.[28] The variant has not yet been functionally analyzed.
The detailed study of the presence of mutual variants in the researched pedigree indicates that the genetic background of this endemic neurodegenerative disease has a clearly heterogenous character: 31 different variants were found in 12 genes. When we tracked the variants within the pedigree, we found a dominant trait in only 3 branches of the family and only for some variants. In patients 3 and 11, we found similar variants of the EIF4G1 gene. In contrast, LRRK2 variants were present in the maternal proband only. In patients 6 and 12, we found similar variants of ADH1C, LRRK2, and PRKN; MAPT variants were present in the maternal proband only. In patients 2 and 10, similar variants of PRKN, PLA2G6, VPS35, and MAPT were found. Variants of EIF4G1 and LRRK2 were present in the maternal proband only, while the variants of PINK1, DJ-1, and GIGYF2 were found only in her daughter. In other pedigree branches, we did not find any similarity in the variant presence in consanguineous relatives. Though many other genes could potentially be associated with PD; we focused on the most common candidate ones.
5. Conclusion
Our study has provided an evidence that the recently described endemic neurodegenerative parkinsonism, which can manifest in different phenotypes (see Table 1), has also different genetic background and apparently heterogenous traits. In this molecular–genetic study of the core pedigree, we have not found one “founder” pathogenic variant associated with parkinsonism in all or the most PD patients of this pedigree. Therefore, it could rather be assumed that the familial occurrence of this disease is caused by the combined influence of several “small-effect” genetic variants that accumulate in the population with long-lasting inbreeding behavior. Whether there is any link to the VPS35 positive familial parkinsonism, which has been discovered in the relatively near region of Upper Austria by Zimprich et al[29] and Struhal et al[30] remains open for further research. For the planned functional analyses, we have so far collected only one brain specimen from the research pedigreee.[31] Therefore, our future study will also utilize proper cell lines with combinations of targeted mutagenesis to assess the effects of particular rare variants.
Acknowledgments
The authors are grateful to Jan Pavlik, MD, native of Kuzelov and Hornacko researcher, for his substantial contribution to the manuscript by providing ethnographic data and references and to Jarmila Tomesova, the nurse of the general practitioner's office for her contribution to organizing research in the examined region.
Author contributions
Conceptualization: Katerina Mensikova, Radek Vodicka, Radek Vrtel, Marek Godava, Martin Bares, Vladimir Janout, Martin Prochazka, Petr Kanovsky.
Formal analysis: Kristyna Kolarikova, Radek Vodicka.
Investigation: Tereza Bartonikova, Katerina Mensikova, Kristyna Kolarikova, Radek Vodicka, Pavel Otruba, Michaela Kaiserova, Miroslav Vastik, Lenka Mikulicova, Josef Ovecka, Ludmila Sachova, Frantisek Dvorsky, Jiri Krsa, Petr Jugas, Marek Godava, Martin Bares.
Methodology: Katerina Mensikova, Kristyna Kolarikova, Radek Vodicka, Radek Vrtel, Marek Godava, Martin Bares, Vladimir Janout, Petr Kanovsky.
Supervision: Radek Vrtel, Martin Prochazka, Petr Kanovsky.
Writing – original draft: Tereza Bartonikova, Kristyna Kolarikova, Radek Vodicka.
Writing – review & editing: Katerina Mensikova, Petr Hlustik, Martin Prochazka, Petr Kanovsky.
Footnotes
Abbreviations: ADH1C = alcohol-dehydrogenase 1C gene, also known as PARK 2, also known as PARK7, ATP13A3 = ATPase 13A3gene, DJ-1 = gene which encodes a protein of the peptidase C56 family, EIF4G1 = eukaryotic translation initiation factor 4 gamma 1 gene, FBXO7 = F-box only protein 7 gene, GBA = glucosylceramidase beta gene, GBAP1 = glucosylceramidase beta pseudogene 1, GIGYF2 = GRB 10 interacting GYF protein 2, HTRA2 = HtrA serine peptidase 2 gene, LRRK2 = leucine rich- repeat kinase 2 gene, MAPT = microtubule associated protein tau gene, PARK1-PARK18 = Parkinson's disease protein 1-18, PD = Parkinson's disease, PDD = Parkinson disease dementia, PINK1= PTEN induced putative kinase 1 gene, PLA2G6 = phospholipase A2 group VI gene, pPD = prodromal Parkinson disease, PRKN = gene for making a protein called parkin, PSP-P = progressive supranuclear palsy-parkinsonism, RSBD = REM sleep behavior disorder, SNCA= synuclein alpha gene, UCHL1= ubiquitin C-terminal hydrolase L1 gene, VPS35 = vacuolar protein sorting 35 gene.
The study was supported by grant from the Ministry of Health of the Czech Republic, grant Nr. 15–32715A, by grant from the Palacky University Medical School Internal Grant Agency—IGA LF 2018–009, and by grant from the Ministry of Health, Czech Republic—conceptual development of research organization—MH CZ—DRO (FNOL, 00098892) 2017.
All other authors declare no financial or other conflicts of interest. All authors have approved the final article.
References
- [1].de Rijk MC, Tzourio C, Breteler MM, et al. Prevalence of parkinsonism and Parkinson's disease in Europe: EUROPARKINSON Collaborative Study. J Neurol Neurosurg Psychiatry 1997;62:10–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].de Rijk MC, Breteler MM, Graveland GA, et al. Prevalence of Parkinson's disease in the elderly: the Rotterdam Study. Neurology 1995;45:2143–6. [DOI] [PubMed] [Google Scholar]
- [3].Kis B, Schrag A, Ben-Shlomo Y, et al. Novel three-stage ascertainment method: prevalence of PD and parkinsonism in South Tyrol, Italy. Neurology 2002;58:1820–5. [DOI] [PubMed] [Google Scholar]
- [4].Benito-Leó J, Bermejo-Pareja F, Rodríguez J, et al. Prevalence of PD and other types of parkinsonism in three elderly populations of central Spain. Mov Disord 2003;18:267–74. [DOI] [PubMed] [Google Scholar]
- [5].Zhang ZX, Roman GC. Worldwide occurrence of Parkinson's disease: an updated Review. Neuroepidemiology 1993;12:195–208. [DOI] [PubMed] [Google Scholar]
- [6].Barbosa MT, Caramelli P, Maima DP, et al. Parkinsonism and Parkinson's disease in the elderly: a community based survey in Brazil (the Bambuí study). Mov Disord 2006;21:800–8. [DOI] [PubMed] [Google Scholar]
- [7].Das SK, Misra AK, Ray BK, et al. Epidemiology of Parkinson disease in the city of Kolkata, India. Neurology 2010;75:1362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Winkler AS, Tutuncu E, Trendafilova A, et al. Parkinsonism in a population of northern Tanzania: a community-based door-to-door study in combination with a prospective hospital-based evaluation. J Neurol 2010;257:799–805. [DOI] [PubMed] [Google Scholar]
- [9].Wermuth L, Joensen P, Bünger N, et al. High prevalence of Parkinson's disease in the Faroe Islands. Neurology 1997;49:426–32. [DOI] [PubMed] [Google Scholar]
- [10].Bergareche A, De la Puenta E, Lóez de Munain A, et al. Prevalence of Parkinson's disease and other types of Parkinsonism. A door- to-door survey in Bidasoa, Spain. J Neurol 2004;251:340–5. [DOI] [PubMed] [Google Scholar]
- [11].Clavería LE, Duarte J, Sevillano MD, et al. Prevalence of Parkinson's disease in Cantalejo, Spain: a door-to-door survey. Mov Disord 2002;17:242–9. [DOI] [PubMed] [Google Scholar]
- [12].Pavlicová M. Folk Culture and its Historic and Social Reflexes. Brno: Masaryk University Press; 2007. [Google Scholar]
- [13].Válka M. Social and Cultural Changes in the Village: Moravian Countryside at the Turn of Third Millennium. Brno: Masaryk University Press; 2011. [Google Scholar]
- [14].Mensikova K, Kanovsky P, Kaiserova M, et al. Prevalence of neurodegenerative parkinsonism in an isolated population in south-eastern Moravia, Czech Republic. Eur J Epidemiol 2013;28:833–6. [DOI] [PubMed] [Google Scholar]
- [15].CSU (Office) Report. Available at: http://czso.cz/csu/2011edicniplan.nsf/publ/0001-11-2010. [Google Scholar]
- [16].Mensikova K, Kanovsky P, Otruba P, et al. Epidemiological study of neurodegenerative parkinsonism in “Hornacko”, a specific region of the south-eastern Moravia, Czech Republic. Cesk Slov Neurol N 2014;77:714–20. [Google Scholar]
- [17].Mensikova K, Godava M, Kanovsky P, et al. Familial autosomal-dominant neurodegenerative parkinsonism with cognitive deterioration spanning five generations in a genetically isolated population of south-eastern Moravia, Czech Republic. Biomed Pap 2016;160:158–60. [DOI] [PubMed] [Google Scholar]
- [18].Frolec V, Holý D, Jeřábek R. Horňácko. The Life and Culture of the People from Moravian-Slovakian borderlands (In Czech). Blok, Brno, 1996, ISBN 80-86720-40-1.11. [Google Scholar]
- [19].Koller W, Vetere-Overfield B, Gray C, et al. Environmental risk factors in Parkinson's disease. Neurology 1990;40:1218–21. [DOI] [PubMed] [Google Scholar]
- [20].Moura KC, Campos Junior M, DeRosso AL, et al. Genetic analysis of PARK2 and PINK1 genes in Brazilian patients with early-onset Parkinson's disease. Dis Markers 2013;35:181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lücking CB, Chesneau V, Lohmann E, et al. Coding polymorphisms in the parkin gene and susceptibility to Parkinson disease. Arch Neurol 2003;60:1253–6. [DOI] [PubMed] [Google Scholar]
- [22].Fiala O, Zahorakova D, Pospisilova L, et al. Parkin (PARK 2) mutations are rare in Czech patients with early-onset Parkinson's disease. PLoS One 2014;9:e107585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Benitez BA, Davis AA, Jin SC, et al. Resequencing analysis of five Mendelian genes and the top genes from genome-wide association studies in Parkinson's Disease. Mol Neurodegener 2016;11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Foo JN, Tan LC, Liany H, et al. Analysis of non-synonymous-coding variants of Parkinson's disease-related pathogenic and susceptibility genes in East Asian populations. Hum Mol Gen 2014;23:3891–7. [DOI] [PubMed] [Google Scholar]
- [25].Nichols WC, Marek DK, Pauciulo MW, et al. R1514Q substitution in Lrrk2 is not a pathogenic Parkinson's disease mutation. Mov Disord 2007;22:254–7. [DOI] [PubMed] [Google Scholar]
- [26].Toft M, Mata IF, Ross OA, et al. Pathogenicity of the Lrrk2 R1514Q substitution in Parkinson's disease. Mov Disord 2007;22:389–92. [DOI] [PubMed] [Google Scholar]
- [27].Jin SC, Pastor P, Cooper B, et al. Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer's disease Ibero-American cohort. Alzheimers Res Ther 2012;4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kauwe JS, Cruchada C, Mayo K, et al. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci USA 2008;105:8050–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zimprich A, Benet-Pagès A, Struhal W, et al. A mutation on VPS35, encoding a subunit of the retromer complex, causes late onset Parkinson disease. Am J Hum Genet 2011;89:168–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Struhal W, Presslauer S, Spielberger S, et al. VPS35 Parkinson's disease phenotype resembles the sporadic disease. J Neural Transm 2014;21:755–9. [DOI] [PubMed] [Google Scholar]
- [31].Bartonikova T, Mensíkova K, Mikulicova L, et al. Familial atypical parkinsonism with rare variant in VPS35 and FBXO7 genes. Medicine (Baltimore) 2016;95:e5398. [DOI] [PMC free article] [PubMed] [Google Scholar]