

Abstract
Limb-girdle muscular dystrophy 2L (LGMD2L) is mainly characterized by late adult onset, atrophy of proximal muscles, chronic progressive and asymmetric weakness, accompanied by increased creatine kinase (CK) levels, dystrophic pathological changes and electromyography showing myogenic damage. To date, familial LGMD2L was reported in European countries and had not been reported in China.
A careful investigation of the clinical manifestations, muscle performance imaging, biopsy, and target next-generation sequencing (NGS) technology was utilized to identify pathogenic genetic variants in a 4-generation pedigree that includes 6 affected individuals.
The results revealed mild-to-moderate hypertrophy of bilateral gastrocnemii and slight weakness and atrophy in the proximal muscles of the lower limbs, with obviously increased serum creatine kinase levels. The symptoms were more serious in the male proband but were also observed in females. Obvious and symmetric atrophy and fat infiltration of posterior segments of the thigh was evident in muscle magnetic resonance imaging (MRI). The pathological changes included a small amount of atrophic and hypertrophic fibers, scattered necrotizing fibers, a small number of increased nuclei, inward migration, mild proliferation of interstitial connective tissue, and no inflammatory cell infiltration. The pathogenic allele was a c.220C > T mutation in the anoctamin 5 (ANO5) gene.
The LGMD2L family was characterized by mild chronic myopathy and bilateral gastrocnemius hypertrophy with obviously increased CK levels. Pathological changes included atrophy of fibers with interstitial connective tissues hyperplasia. The pathogenic allele was a c.220C> T mutation in the ANO5 gene.
Keywords: ANO5, anoctamin 5, LGMD2L, limb-girdle muscular dystrophy 2L, muscular dystrophy, next-generation sequencing
1. Introduction
Anoctaminopathies are a group of autosomal recessive skeletal muscle disorders with various clinical phenotypes, caused by the abnormal expression of anoctamin 5 (ANO5) protein due to ANO5 gene mutations, mainly divided into 3 clinical phenotypes: limb-girdle muscular dystrophy 2L (LGMD2L), Miyoshi phenotype of distal muscular dystrophy 3 (MMD3) and asymptomatic hyper-CK-emia.[1] LGMD2L is the most common phenotype of anoctaminopathy, mainly characterized by late adult onset, slowly progressing asymmetric weakness and atrophy of proximal muscles, accompanied by increased creatine kinase (CK) levels, dystrophic pathological changes and electromyography (EMG) showing myogenic damage.[2] To date, the diagnosis mainly depends on genetic testing.
In 2007, Jarry et al initially described a new type of autosomal recessive limb-girdle muscular dystrophy (LGMD), named LGMD2L, characterized by asymmetric atrophy, and weakness of quadriceps and biceps.[3] In 2010, Bolduc et al confirmed that ANO5 was the pathogenic gene of LGMD2L.[4] To date, most LGMD2L and ANO5 gene mutations have been reported in European countries.[5] In Asia, it wasn’t until 2016 that Kadoya et al confirmed the first case of LGMD2L in Japan,[6] and then Yun Yuan et al reported the first LGMD2L patient in China in 2017.[7] However, the case report was without detailed clinical data such as clinical symptoms, muscle imaging, and gene mutation analysis. In this study, we used carefully collected clinical data of the proband and his family members in conjunction with next-generation sequencing (NGS) technology to detect the pathogenic mutation. We finally diagnosed an LGMD2L family in 2016. By searching the PubMed database and the Chinese knowledge network (CNKI) database, it could be confirmed that this is the first known LGMD2L family in China. Based on preliminary work that summarized the research progress on anoctaminopathy,[8] and by analyzing the clinical, imaging, pathological and genetic data, we finally uncovered the distinct characteristics of this first Chinese LGMD2L family.
2. Materials and methods
2.1. Family pedigree and patient evaluation
The probands constitute a consanguineous LGMD2L family of Han nationality from Nanchang, Jiangxi Province. As shown in Figure 1, there were a total of 4 generations with 33 persons in this family at the time of writing. This 4-generational Han family included 5 patients. Physical examination of the proband showed no obvious abnormalities in routine examination. He was conscious with fluent speech and normal orientation. The strength of eye muscles, facial muscles, masseter muscle, and diaphragm muscle were 5 (5/5). The mandible was not skewed, and the tongue was centered. There was no atrophy and fibrillation of the lingual muscles, and thus no gastric reflex. Among the cervical muscles, the bilateral sternocleidomastoid muscles had a strength of 5 (5/5), with powerful cough. The bilateral deltoid muscle-, biceps brachii-, triceps-, wrist extension flexor muscle-, and grip strength were 5 (5/5). The iliopsoas and quadriceps muscles on both sides were slightly weakened, while the bilateral biceps brachii and the anterior tibialis muscle and gastrocnemius muscle strength were all 5 (5/5). There was slight atrophy of bilateral quadriceps, biceps femoris, and light to moderate hypertrophy of bilateral gastrocnemii (Fig. 2). Muscle tension was normal. Bilateral biceps-, triceps-, knee-, and Achilles tendon reflexes were positive (++). Pathological sign was negative. There were no abnormalities in the sensory examination accompanied with coordinating movement. Examination of the III2, III11, III13, and III15 members presented only bilateral hypertrophy of the gastrocnemius muscles, without other abnormities such as muscle weakness or atrophy.
Figure 1.
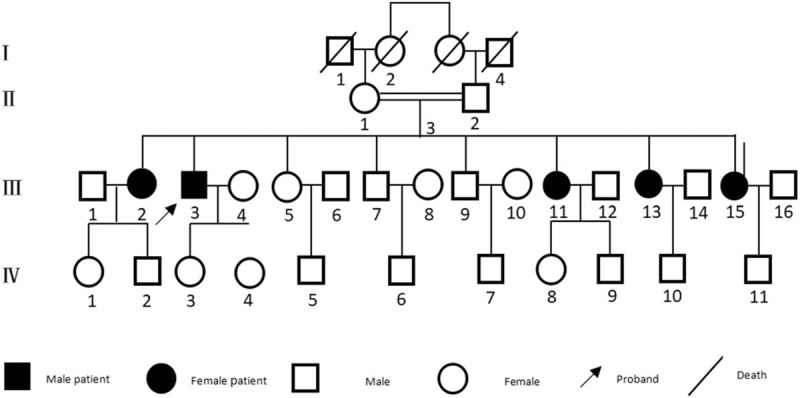
The genogram of the LGMD2L family. LGMD2L = limb girdle muscular dystrophy 2L.
Figure 2.
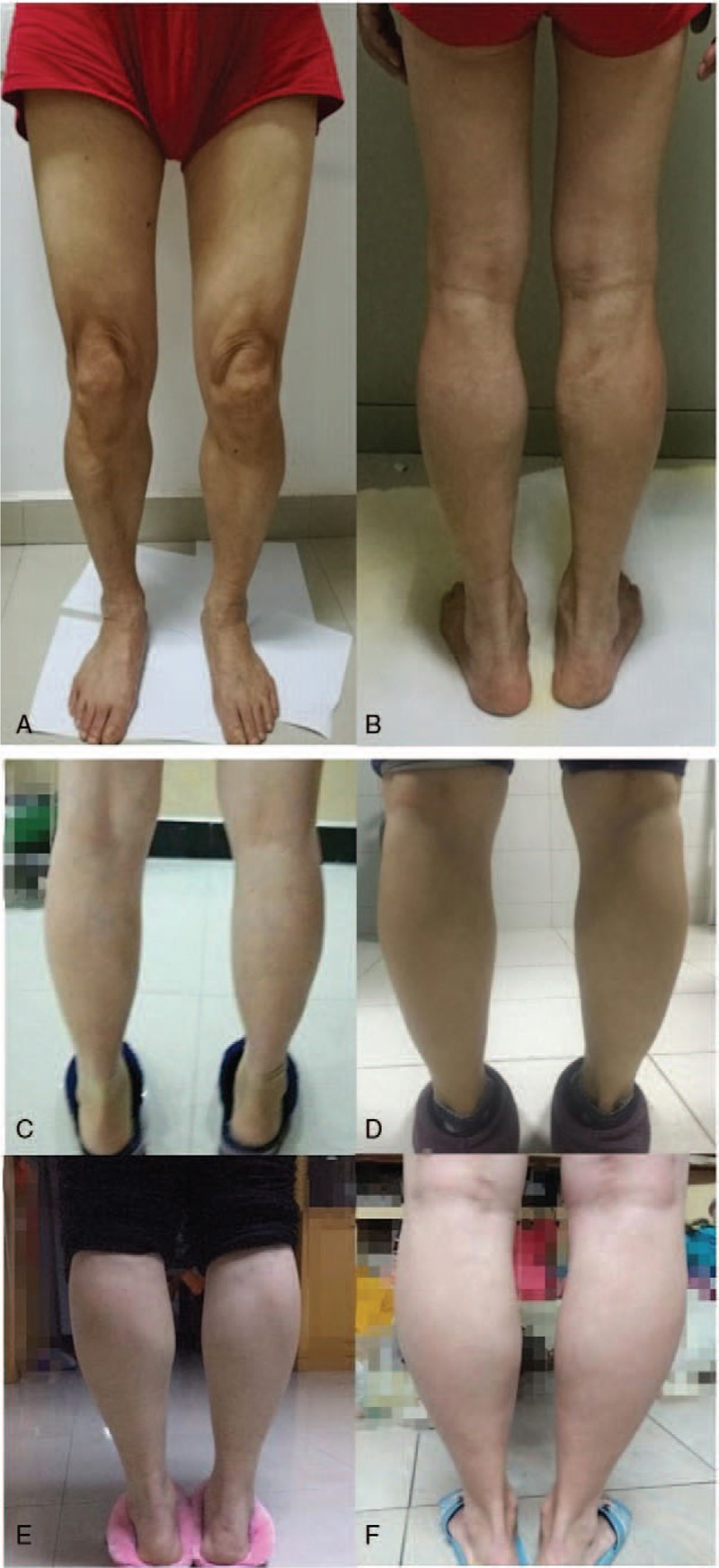
Pictures A and B showed slight atrophy in bilateral quadriceps and biceps, mild-to-moderate hypertrophy of bilateral gastrocnemius muscles of the proband. Pictures C, D, E, and F showed different degrees of bilateral calf muscles hypertrophy of 4 family members(C: III2; D: III11; E: III13; and F: III15).
2.2. Clinical data collection
The probands underwent serum CK testing, EMG (Denmark Keypoint EMG /evoked potential instrument), skeletal muscle magnetic resonance imaging (MRI) (SingaHDxt 3.0T magnetic resonance scanner; GE) after signing the informed consent document. Four family members (III2, III11, III13, and IV4) completed serum CK testing after signing the informed consent document. Four family members (III5, III7, III9, and III15) refused serum CK testing.
2.3. Muscle biopsy and pathohistological staining
After signing the informed consent document, the biopsy was performed in the right biceps of the proband. Subsequently, a series of stainings was conducted, covering routine histological staining including Hematoxylin and Eosin (HE), modified Gomori trichrome (MGT), Oil Red O (ORO), and periodic acid-Schiff (PAS) staining; enzyme histological staining for nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR); and further immunohistochemical stains were applied using antibodies against dystrophin at a dilution of 1:50 (dystrophin C, N, and R, Novocastra).
2.4. NGS
After signing the informed consent, Genomic DNA was extracted from peripheral blood by standard methods from whole blood samples of the proband and 5 family members (III2, III11, III13, III15, and IV4). Genetic investigation by NGS Illumina MiSeq panel designed to screen for 195 relevant genes associated with muscular dystrophy, metabolic myopathy, myofibrosis, ion channel disease, congenital myopathy, congenital muscular dystrophy, myotonia syndrome, congenital myasthenic syndrome, distal myopathy and other common skeletal muscle diseases, such as DMD, EMD, SGCA, SGCB, SGCD, SGCG, ANO5, DYSF, FKTN, CAV3, CAPN3, ETFA, ETFB, GAA, COQ2, COQ6, DES, CHAT, DOK7, AGRN, MUSK, COLQ, and PLEC was performed on the proband. We confirmed ANO5 homozygous mutation in exon 11. And then Coding exons of ANO5 were amplified using polymerase chain reaction (PCR) with intronic primers. Primers were designed according to the published ANO5 sequence. After purification, the PCR products were processed with Nextera XT DNA sample preparation kit (Illumina). Sequencing was performed on an Illumina MiSeq instrument. The results obtained were compared with the ANO5 gene standard sequences to verify the results of gene sequencing.
3. Results
3.1. Clinical characteristics
The proband was a 60-year-old man whose mother and father were cousins. He presented at our hospital in April 2016 due to weakness in both lower extremities for 3 years. Three years ago, the patient started to develop apparent chronic progressive symptoms of slight muscle strength reduction when walking upstairs, with slight chest tightness. The symptoms improved after taking a few minutes of rest. There was no muscle pain, muscle stiffness, drooping eyelids, diplopia, difficulty in swallowing, or other discomforts. The physical examination showed slight weakness of the bilateral iliopsoas, quadriceps femoris, and biceps, but normal muscle strength of the remaining skeletal muscles, with a normal response (++) of the tendon reflexes and sensory function, negative response of the pathology sign, normal coordination of movement, with slight atrophy in bilateral quadriceps and biceps, and mild-to-moderate hypertrophy of bilateral gastrocnemius muscles (Fig. 2). There was no obvious muscle weakness or muscular atrophy in III2, III11, III13, and III15 pedigree members, with only different degrees of bilateral calf muscle hypertrophy (Fig. 2). The remaining family members, III5, III7, III9, and IV4, were completely normal.
3.2. Muscle imaging
MRI performed on the lower limb muscles of the proband revealed apparent atrophy of bilateral posterior muscles of the thigh, of which the right quadriceps femoris, adductor muscle, semitendinosus, and semimembranosus were involved more seriously than their left-side counterparts, revealing evident asymmetry. Simultaneously, obvious fat infiltration was observed in the right semimembranosus and semitendinosus (Fig. 3).
Figure 3.
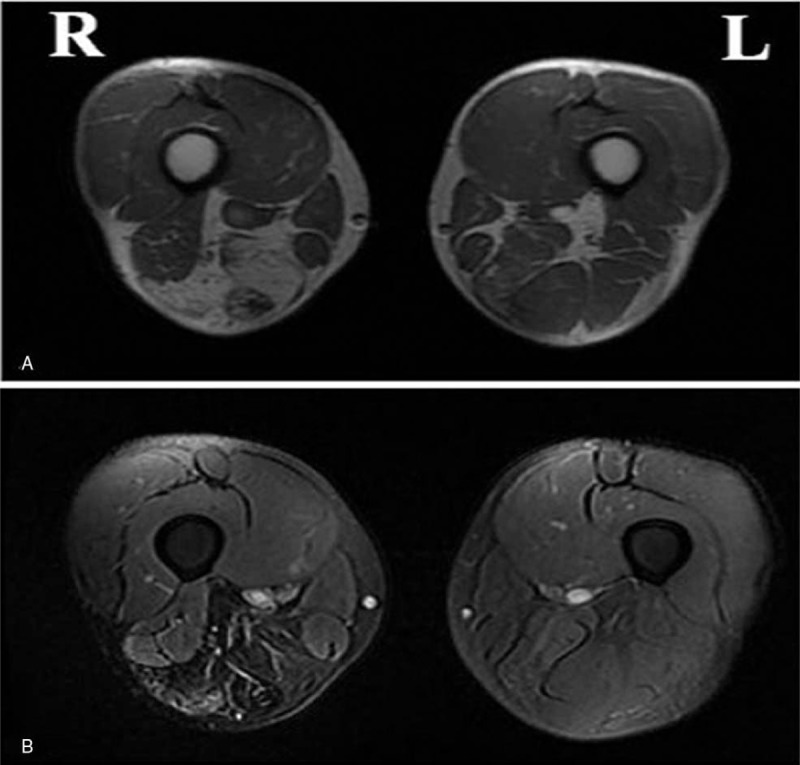
Pictures A and B showed apparent atrophy of bilateral posterior muscles of the thigh, of which quadriceps femoris, adductor muscle, semitendinosus and semimembranosus were involved more serious than the left side, obvious fatty infiltration in right semimembranosus and semtendinosus (A: T1WI; B: T2 Fat suppression).
3.3. Laboratory examinations
Laboratory examination of the proband revealed serum CK of 3468 IU/L (normal range 38–174 IU/L), Creatine Kinase-MB Isoenzyme of 52.70 IU/L (normal range 0–25IU/L), lactate dehydrogenase (LDH) of 298.1 IU/L (normal range 80–190IU/L), alanine aminotransferase (ALT) of 72.70 U/L (normal range 5–40U/L), aspartate aminotransferase (AST) of 89.70 U/L (normal range 5–40 U/L), and myoglobin of 797.51 ng/mL (normal range 0–100 ng/ml), with normal thyroid function. Serum CK activity was found to be elevated to various degrees (582.72–2630.48 IU/L) (normal range 38–174 IU/L) in theIII2, III11, and III13 family members, while a normal CK of 111.75 IU/L was found in member IV4. Detailed clinical data of the family members are summarized in Table 1.
Table 1.
Main clinical data of members of the investigated family.

3.4. Electrophysiological examination
A test of nerve conduction in the proband showed that motor and sensory conduction velocity and wave amplitude of the left median, ulnar, tibial, and peroneus nerves were normal. Needle EMG showed no significant abnormalities in the left median nerve F wave. A small amount of spontaneous potential of left quadriceps muscle, the amplitude of the motor unit potential narrowed and the duration shorted, increased multiphase potential, with atypical pathological interference phase when strongly contracting in left quadriceps, deltoid, and extensor muscles. According to the aforementioned findings, the presentation was considered abnormal myogenic damage. The electrocardiogram (ECG) hinted at sinus bradycardia with a heart rate of 51 beats/min. Echocardiography showed a dilated right atrium with a right and left diameter of the right atrium(RALR) of 43 mm (normal range 30–40 mm), mild regurgitation of the mitral and tricuspid valves, with a disorder of left ventricular diastolic function.
3.5. Pathological examination
HE staining (Fig. 4) detected a small amount of atrophy and hypertrophic muscle fibers, scattered with necrotic muscle fibers, accompanied with increased myonuclei and migration, mild hyperplasia of interstitial connective tissues, and no inflammatory cell infiltration. MGT staining showed no rimmed vacuoles and ragged-red fibers. ORO staining showed no significantly increase of intramuscular lipid droplets. PAS staining revealed no significant increase of intramuscular glycogen. NADH-TR staining showed no obvious structural changes in the muscle fibers. Immunohistochemical staining with the monoclonal anti-dystrophin antibody was positive.
Figure 4.

HE staining showed a small amount of atrophy and hypertrophic muscle fibers, scattered with necrotic muscle fibers, accompanied with myoneucleolus increased and migration, interstitial connective tissues mild hyperplasia, without inflammatory cell infiltration. (A: HE × 100; B: HE × 200; and C: HE × 400). HE = Hematoxylin and Eosin.
3.6. Genetic analysis
Targeted NGS detected the same mutation in the proband (III3) and his family membersIII2, III11, III13, and III15. We thus identified a characteristic homozygous nucleotide mutation variation of nucleotide 220 in the coding region of the ANO5 gene from C to T (c.220C>T), leading to premature termination, with a stop codon in place of codon 74 that normally codes for arginine (p.R74X). A compound heterozygous mutation in the same region resulted in equivalent consequences in member IV4. No other pathogenic mutations were found. The results of gene sequencing of the proband and family members are shown in Figure 5.
Figure 5.
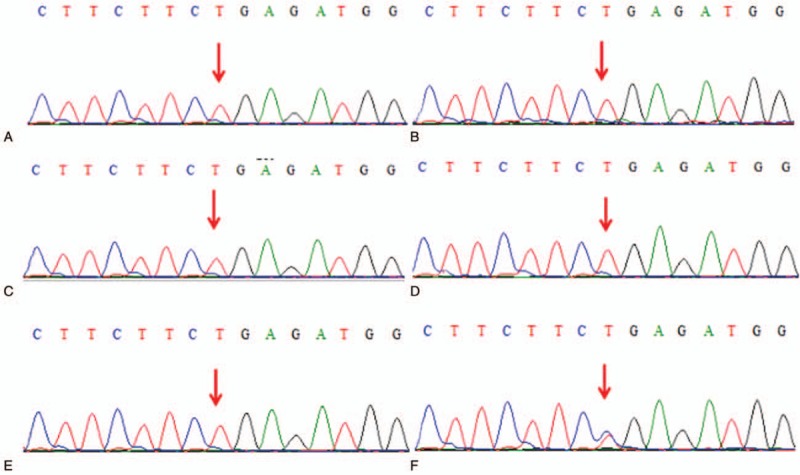
Mutated ANO5. Sequencing analysis detects homozygous mutation variation of c.220C > T in exon 11 of ANO5 in the proband and 4 family members III2, III11, III13, and III15 (A–E); compound heterozygous mutation of c.220C > T in exon 11 of ANO5 in the family member IV4 (F). ANO5 = anoctamin 5.
4. Discussion
Anoctaminopathies are autosomal recessive skeletal muscle disorders caused by abnormal expression of ANO5 protein due to mutations in the ANO5 gene. They present in various forms, such as asymptomatic hyper-CK-emia, exercise intolerance, myalgia, or weakness.[9] The clinical phenotypes are divided into 3 types, LGMD2L, MMD3, and asymptomatic hyper-CK-emia,[1] which differ in families, even in 1 family.[9] Patients with anoctaminopathy usually have only mild clinical symptoms, and even if serious muscle involvement is evident in muscle MRI, the ambulation of the patients can be preserved for decades after onset.[2] Most anoctaminopathy patients are male, and males are frequently more seriously affected than females.[10] The diagnosis can thus be quite difficult, especially for females who are frequently asymptomatic or present with minor symptoms and slow disease progression. The reason for this gender difference in anoctaminopathy remains unknown.[2] A significantly increased serum CK concentration is a pervasive manifestation of muscle diseases, and if lymphocytic infiltration or marginal vacuoles are found in the muscles simultaneously, the syndrome may be misdiagnosed as myositis or optionally inclusion body myositis.[11] Timely and accurate diagnosis can thus help patients avoid immunosuppressive therapy. Similarly, it is necessary to differentiate anoctaminopathy from the other subtypes of LGM, such as dystrophinopathies, FSHD or metabolic muscle diseases.
The most typical presentation of anoctaminopathy is limb-girdle muscular dystrophy type 2L (LGMD2L), which is characterized by a late onset with benign progression of weakness and atrophy in the proximal limbs. LGMD2L, caused by mutations in the ANO5 gene, is the third most common LGMD2 etiology in northern and central Europe,[5] where the c.191dupA mutation causes the majority of cases. By contrast, only a few cases were reported in non-European countries.[6,12,13] Typically, physical examination and neuroimaging suggest asymmetric muscle fatigue, and EMG reveals myogenic changes accompanied by increased CK blood values. Patients may be asymptomatic or show nonspecific symptoms such as muscle stiffness, cramps, exercise intolerance, or exercise myalgia rather than notable muscle weakness, especially in females. There are also reports of patients who presented with recurrent myoglobinuria.[14] Invariably, LGMD2L patients show asymmetrical atrophy or hypertrophy of quadriceps femoris,[5] accompanied by gastrocnemius hypertrophy. The involvement of upper limbs is rare, but involvement of proximal muscles in both upper and lower extremities has been reported in some patients.[2]
LGMD2L patients characterized by proximal limb weakness and atrophy, with or without gastrocnemius hypertrophy, accompanied by significantly elevated serum CK, are mostly aged from 20 to 70. Because LGMD2L is an indolent disease, that is, it progresses slowly, patients can retain their walking ability for decades, with normal heart and respiratory function, and have the same overall life expectancy as the general population.[15] In 2016, Kadoya et al[6] confirmed the first LGMD2L patient in Japan, who was a 60-year-old male, with initial symptoms of muscle stiffness and muscle cramps, accompanied by significantly elevated CK. However, with further progression of the disease, proximal limb weakness and bilateral gastrocnemius hypertrophy gradually appeared. The age of onset of the proband in this study was 57 years, which belongs to the high paroxysmal age, with main classical clinical manifestations of mild proximal muscular weakness, atrophy of lower extremities and light to moderate hypertrophy of bilateral calf muscles. Although the clinical manifestations of the proband were consistent with those reported for LGMD2L patients outside Asia, they were more similar but milder compared to the patients reported in Japan. Unfortunately, we cannot compare the proband with the first LGMD2L case in China due to the lack of a detailed history. Based on current information, we hazarded a guess that the similarity of symptoms of our LGMD2L patient with those of Japanese patients is associated with similarities of the environment. Because Japan and China are close to each other geographically and historically, certain climatic and dietary conditions may be similar. Simultaneously, 4 female family members (III2, III11, III13, and III15) revealed varying degrees of bilateral gastrocnemius hypertrophy which was less severe than that of the male proband. This result may indicate that LGMD2L in China is less likely to happen in males, even if the symptoms in the male patient were more serious than in the females. However, this conjecture needs further study.
The NGS results of the proband and 4 relatives (III 2, III 11, III 13, III 15) revealed the same homozygous c.220C>T mutation in the ANO5 gene as reported in the LGMD2L patients in Europe,[16] without other or novel pathogenic mutations. ANO5 was also reported to be expressed in cardiomyocytes, but related case reports are rare. Wahbi et al found 2 patients with concurrent dilated cardiomyopathy among 19 anoctaminopathy patients,[17] suggesting that dilated cardiomyopathy may be a potential complication of ANO5 gene mutations. Witting et al performed cardiac examinations on 40 patients with anoctaminopathy, which showed an increased risk of ventricular arrhythmias in these patients.[5] A Dutch study found hypertrophic cardiomyopathy and intraventricular septum thickening in patients with ANO5 mutations,[18] and a shortened PQ interval was also reported in individuals with anoctaminopathy.[9] However, the complete cardiac investigations performed by Bohlega et al showed that the hearts of the patients were normal.[12] In our study, the proband's ECG only hinted at sinus bradycardia, with a dilated right atrium in cardiac ultrasound. However, whether this is associated with the ANO5 mutation remains unknown. Regrettably, no ECG and cardiac investigations of the proband's family members were completed.
The typical MRI manifestations of LGMD2L are the overt asymmetric involvement of posterior segments of the thigh- and paravertebral muscles, presenting as high signal in the short T1 inversion recovery (STIR) sequence, while the anterior thigh-, gracilis and sartorius muscles remain intact.[19] In general, the semitendinosus muscle is not or rarely involved in skeletal muscle diseases, but in some cases, LGMD2L patients have been reported to present with bilateral solitary semitendinosus in the initial stage.[20] In this study, the proband's muscle MRI showed apparent atrophy and fat infiltration of bilateral posterior muscles of the thigh, of which the right side was involved more seriously than the left side, revealing evident asymmetry. The anterior thigh group was relatively well retained, which was in accordance with the typical LGMD2L muscle MRI features but was different from common muscular diseases. Research has shown completely normal muscle MRI presentation in asymptomatic young patients, varying degrees of muscle involvement in most older patients, and, as the condition progresses, an expanding scope and extent of involvement of muscles.[19] However, in our study, we observed that the degree of the calf muscle hypertrophy was not correlated with age.
From the perspective of muscle biopsy pathohistology, the proband's result was consistent with the typical pathological changes of LGMD2L, which presented as a small amount of atrophy and hypertrophic muscle fibers, scattered with necrotic muscle fibers, accompanied with increased myonuclei and migration, with mild hyperplasia of interstitial connective tissues,[2] which is different from other muscular dystrophies. The first case report of LGMD2L in China by Yu et al has been verified to present with lobulated fibers,[7] which was not found in this family. To date, a number of studies have reported the presence of rimmed vacuoles[2,15] and necrotic changes[11,12,21] in LGMD2L patients. There were also studies indicating a small amount of undefined amyloid deposits around perivascular and interstitial locations within skeletal muscles in anoctaminopathy patients,[12,13,22,23] which merits further study.
Multiple mutations have been found in LGMD2L patients, including missense-, nonsense-, and point-mutations, as well as small deletions, insertions and splice site mutations.[4,9] So far, the homozygous mutation c. 191dupA in the 5th exon (ANO5) has been regarded as the founder mutation in most of the British and German LGMD2L patients,[5] while the most common mutation in Finland is c.2272C> T, also in the 5th exon.[15] Recently, there has been a report of a homozygous c.2394dupmutation in the ANO5 gene in patients with LGMD2L.[6] The ANO5 gene shows a wide array of equivalent mutations without a hotspot region, resulting in difficult diagnosis by screening only 1 or 2 common mutations.[2] Screening genes associated with myopathy by NGS demonstratedthat the homozygous c.220C> T mutation in the proband's ANO5 gene resulted in a change of the 74th codon, which encodes an arginine, into a stop codon, leading to premature termination of protein translation. Verifying the proband's pedigree by targeted NGS revealed the presence ofthe same homozygous mutation in family membersIII2, III11, III13, and III15, and a compound heterozygous c.220C> T mutation in the ANO5 gene in family member IV4, suggesting that the mutatedallele of the proband was inherited from his parents. After a careful investigation of his asymptomatic parents, we speculated that the heterozygous c.220C> T mutation of the proband's parents complied with autosomal recessive genetic rules. In the first published LGMD2L case in China, regrettably, it is only known that the patient carried a compound heterozygous mutation, without a definitely identified gene. Nevertheless, absent or decreased dystrophin expression was found by immunohistochemical analysis.[7] Unfortunately, we were not able to detect the ANO5 protein at that time. To date, we still have not found the correlations between genotype and phenotype, because the phenotype profile is broader than expected, from asymptomatic/mild symptoms to pronounced muscle weakness.[9]
No definitive treatment interventions have been established for this condition. Vissing et al reported that supervised aerobic exercise may be beneficial to patients with ANO5 deficiency,[10] but this requires further study. In this case, we did not perform tentative treatment, but we informed the patient and his family to avoid heavy muscle force training and the use of statins, which can induce muscle pain and worsen muscle weakness. After a follow-up for more than 1.5 years, the symptoms of the proband and the 4 members did not deteriorate.During the same time,no additional patients appeared in this family.
5. Conclusions
In part due to its pioneering character, there were some short comings in our study. We did characterize the first LGMD2L pedigree in China and analyzed the characteristics of this pedigree. The Chinese LGMD2L family members mainly presented mild weakness and atrophy in proximal muscles of lower limbs and mild to moderate hypertrophy of bilateral gastrocnemii, with obviously increased serum CK levels. The symptoms may be associated with the environment and were much more serious in males. However, the epidemiological features may not be similar to previous reports which indicated a higher incidence in males. The case was characterized by overt and asymmetric atrophy and fat infiltration of posterior segments of the thigh in the muscle MRI. Pathological changes included a small amount of atrophy and hypertrophic muscle fibers, scattered with necrotic muscle fibers, accompanied with increased myonuclei and migration, and mild hyperplasia of interstitial connective tissues. The pathogenic allele was a c.220C> T mutation in the ANO5 gene. This study lends support to the hypothesis that LGMD2L is likely to affect a broader population than has been reported previously, and clinicians should, therefore, take the diagnosis of LGMD2L into consideration when patients only display elevated CK values without apparent muscle weakness but with gastrocnemius hypertrophy, especially for female patients. However, reports of LGMD2L are rare even in Europe, and especially in China, considering that it is home to about 1/5 of the world's population. We, therefore, believe that the actual incidence of the disease is higher than currently reported, and this question warrants more research.
Acknowledgments
The authors wish to thank the patient and his family members for cooperation.
Author contributions
BH and LX performed the analytical studies and wrote the manuscript; YZ, XL, QX, and QL conducted and performed the periodic clinical monitoring; XQ and WD planned the study, checked the final form of the manuscript and approved the final manuscript. All authors read and approved the final manuscript.
Data curation: Bolin Hu, Li Xiong.
Formal analysis: Bolin Hu, Li Xiong, Yibiao Zhou.
Funding acquisition: Bolin Hu, Yibiao Zhou.
Investigation: Bolin Hu, Li Xiong, Yibiao Zhou, Xiaoqing Lu.
Methodology: Xiaoqing Lu, Qianqian Xiong, Qing Liu.
Resources: Bolin Hu, Li Xiong, Weijiang Ding.
Supervision: Xueliang Qi, Weijiang Ding.
Visualization: Xueliang Qi, Weijiang Ding.
Writing – original draft: Bolin Hu, Li Xiong.
Writing – review & editing: Xueliang Qi, Weijiang Ding.
Footnotes
Abbreviations: ANO5 = anoctamin 5, CK = creatine kinase, EMG = electromyography, LGMD = Limb girdle muscular dystrophy, LGMD = limb girdle muscular dystrophy, LGMD2L = limb girdle muscular dystrophy 2L, MGT = modified Gomori trichrome, MMD3 = Miyoshi phenotype of distal muscular dystrophy 3, MRI = magnetic resonance imaging, NADH-TR = nicotinamide adenine dinucleotide-tetrazolium reductase, NGS = next-generation sequencing, PAS = periodic acid-Schiff, PCR = polymerase chain reaction.
BH and LX contributed equally to this work and should be considered to be co-first authors.
The datasets generated and analyzed during the present study are available from the corresponding author on reasonable request.
This study was approved by the ethics committee of The Second Affiliated Hospital of Nanchang University. A written informed consent form was obtained from each study participant.
The authors declare that they have no competing interests.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors have no conflicts of interest to disclose
References
- [1].Schessl J, Kress W, Schoser B. Novel ANO5 mutations causing hyper-CK-emia, limb girdle muscular weakness and Miyoshi type of muscular dystrophy. Muscle Nerve 2012;45:740–2. [DOI] [PubMed] [Google Scholar]
- [2].Penttilä S, Palmio J, Suominen T, et al. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology 2012;78:897–903. [DOI] [PubMed] [Google Scholar]
- [3].Jarry J, Rioux MF, Bolduc V, et al. A novel autosomal recessive limb-girdle muscular dystrophy with quadriceps atrophy maps to 11p13-p12. Brain 2007;130:368–80. [DOI] [PubMed] [Google Scholar]
- [4].Bolduc V, Marlow G, Boycott KM, et al. Recessive mutations in the putative calcium- activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am J Hum Genet 2010;86:213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Witting N, Duno M, Petri H, et al. Anoctamin 5 muscular dystrophy in Denmark: prevalence, genotypes, phenotypes, cardiac findings, and muscle protein expression. J Neurol 2013;260:2084–93. [DOI] [PubMed] [Google Scholar]
- [6].Kadoya M, Ogata K, Suzuki M, et al. A Japanese male with a novel ANO5 mutation with minimal muscle weakness and muscle pain till his late fifties. Neuromuscul Disord 2017;27:477–80. [DOI] [PubMed] [Google Scholar]
- [7].Yu M, Zheng Y, Jin S, et al. Mutational spectrum of Chinese LGMD patients by targeted next-generation sequencing. PLoS One 2017;12:e0175343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xiong L, Liu Q, Xiong QQ, et al. The research progress of anoctaminopathy. Chin J Neurol 2017;50:794–6. [Google Scholar]
- [9].Savarese M, Di Fruscio G, Tasca G, et al. Next generation sequencing on patients with LGMD and nonspecific myopathies: findings associated with ANO5 mutations. Neuromuscul Disord 2015;25:533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vissing CR, Preisler N, Husu E, et al. Aerobic training in patients with anoctamin 5 myopathy and hyperckemia. Muscle Nerve 2014;50:119–23. [DOI] [PubMed] [Google Scholar]
- [11].Leung DG, Taylor HA, Lindy AS, et al. A case of progressive quadriceps weakness and elevated creatine kinase level mimicking inclusion body myositis. Arthritis Care Res (Hoboken) 2014;66:328–33. [DOI] [PubMed] [Google Scholar]
- [12].Bohlega S, Monies DM, Abulaban AA, et al. Clinical and genetic features of anoctaminopathy in Saudi Arabia. Neurosciences (Riyadh) 2015;20:173–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lahoria R, Winder TL, Lui J, et al. Novel ANO5 homozygous microdeletion causing myalgia and unprovoked rhabdomyolysis in an Arabic man. Muscle Nerve 2014;50:610–3. [DOI] [PubMed] [Google Scholar]
- [14].Blackburn PR, Selcen D, Jackson JL, et al. Early-onset limb-girdle muscular dystrophy-2L in a female athlete. Muscle Nerve 2017;55:E19–21. [DOI] [PubMed] [Google Scholar]
- [15].Hicks D, Sarkozy A, Muelas N, et al. A founder mutation in anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain 2011;134:171–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Magri F, Del Bo R, D’Angelo MG, et al. Frequency and characterisation of anoctamin 5 mutations in a cohort of Italian limb-girdle muscular dystrophy patients. Neuromuscul Disord 2012;22:934–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wahbi K, Béhin A, Bécane HM, et al. Dilated cardiomyopathy in patients with mutations in anoctamin 5. Int J Cardiol 2013;168:76–9. [DOI] [PubMed] [Google Scholar]
- [18].van der Kooi AJ, Ten Dam L, Frankhuizen WS, et al. ANO5 mutations in the Dutch limb girdle muscular dystrophy population. Neuromuscul Disord 2013;23:456–60. [DOI] [PubMed] [Google Scholar]
- [19].Sarkozy A, Deschauer M, Carlier RY, et al. Muscle MRI findings in limb girdle muscular dystrophy type 2L. Neuromuscul Disord 2012;22(suppl 2):S122–9. [DOI] [PubMed] [Google Scholar]
- [20].Tasca G, Evilä A, Pane M, et al. Isolated semitendinosus involvement in the initial stages of limb-girdle muscular dystrophy 2L. Neuromuscul Disord 2014;24:1118–9. [DOI] [PubMed] [Google Scholar]
- [21].Schneider I, Stoltenburg G, Deschauer M, et al. Limb girdle muscular dystrophy type 2L presenting as necrotizing myopathy. Acta Myol 2014;33:19–21. [PMC free article] [PubMed] [Google Scholar]
- [22].Milone M, Liewluck T, Winder TL, et al. Amyloidosis and exercise intolerance in ANO5 muscular dystrophy. Neuromuscul Disord 2012;22:13–5. [DOI] [PubMed] [Google Scholar]
- [23].Liewluck T, Winder TL, Dimberg EL, et al. ANO5-muscular dystrophy: clinical, pathological and molecular findings. Eur J Neurol 2013;20:1383–9. [DOI] [PubMed] [Google Scholar]