

Development of drug resistance1,2 and clonal evolution3–5 is a major problem in multiple myeloma (MM) but its comprehensive longitudinal genetic characterization has been impractical thus far. We asked if copy number variations (CNV) and somatic mutations can be called robustly across the entire genome or exome, from cell free DNA (cfDNA) in patients with active disease, and if clonal somatic mutations and CNVs found in the bone marrow (BM) are reliably reproduced by cfDNA. We hypothesized that i) the subclonal composition of somatic mutations and CNVs differs, indicating that cfDNA and BM MM cells reveal distinct genetic information6,7, and ii) cfDNA can be used to track disease load and clonal evolution of MM, to provide longitudinal genetic information about disease evolution that is not accessible by bone marrow biopsy8,9.
To first demonstrate whether low-pass whole genome sequencing of cfDNA from MM patients is feasible, cfDNA was extracted from a total of 147 samples obtained from blood of 93 randomly selected MM patients, as well as from 12 healthy donors (Supplementary Table 1). We performed library construction, low-pass whole genome sequencing (average depth of 0.22; range 0.01× to 1×) and predicted segments of CNVs and estimates of tumor fraction (fraction of MM-derived cfDNA) using the ichorCNA10 algorithm (Supplementary Figure 1). While CNVs were readily detected in cfDNA from MM patients, no CNVs were detected in cfDNA from healthy blood donors (predicted tumor fraction <0.05). Clinical data was available for 67 of 93 patients. Of those, we found that tumor fraction was ≥ 0.05 (detectable) in 16/26 (62%) cfDNA samples from patients with relapsed / refractory MM and 3/4 (75%) in cfDNA samples from patients with newly diagnosed MM, while tumor fraction in cfDNA from MM patients receiving treatment (35 patients with response and 2 patients with stable disease) was < 0.05 (Figure 1A).
Figure 1. Concordance of clonal genomic landscape in cfDNA and BM-derived Myeloma.
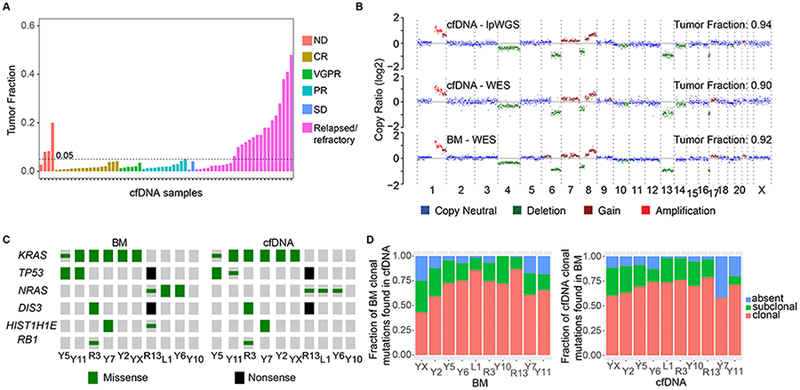
(A) Tumor fractions estimated by lpWGS of cfDNA from 67 patients (out of 93 patients) for whom clinical information was available. ND, newly diagnosed; CR, complete response; VGPR, very good partial response; PR, partial response; SD, stable disease. (B) Copy number profiles and tumor fraction estimated by lpWGS or WES of cfDNA and WES of BM MM from a patient. (C) Distribution of MM putative driver somatic mutations from WES of BM-derived myeloma and matched cfDNA samples. (Clonal = large symbols, subclonal = small symbols). (D) Left panel: Fraction of clonal mutations in BM-derived myeloma of ten MM patients that were also identified in matched cfDNA, either as clonal (red), as subclonal (green) or not detected (blue). Right panel: Fraction of clonal mutations detected in cfDNA that were also identified in the matched BM-derived myeloma.
To evaluate the concordance of CNVs and mutations between cfDNA and matched BM myeloma, we performed whole exome sequencing (WES) of cfDNA, BM-derived CD138+CD45− sorted MM cells and CD138−CD45+ white blood cells as matched normal control from 10 patients to a median depth of 142× (range 97 – 250×), 57× (11× - 137×) and 53× (34× - 80×), respectively (Supplementary Figure 1, Supplementary Table 2). In an index patient (R13) with high cfDNA tumor fraction, CNVs were highly concordant between low-pass whole genome (lpWGS) of cfDNA, WES of cfDNA, and WES of BM (Figure 1B, Supplementary Figure 2). High concordance of CNVs was also seen between cfDNA and BM samples of nine other MM patients, even when compared between low-pass whole genome (lpWGS) and WES. On average, 90.5% of all CNV segments in BM were concordant with cfDNA, whereas 9.5% were discordant (Supplementary Figure 3).
To determine if mutation calls were reproducible between cfDNA and BM-derived MM, we identified all coding somatic mutations (Supplementary Table 3) by WES of cfDNA and BM-derived MM from 10 patients followed by determining their clonality and the fraction of MM cells harboring these mutations (cancer cell fraction). All clonal and subclonal mutations in putative myeloma driver genes (KRAS, DIS3, TP53, NRAS, RBI) 3–5 were found in cfDNA and BM, except for one subclonal HIST1H1E mutation that was detected only in the bone marrow (Figure 1C). We also asked what fraction of these mutations could be detected in cfDNA from the same patients. We found a median of 93% (range 75%−100%) of clonal mutations in the bone marrow was detected in the cfDNA, either in a clonal or subclonal fashion (Figure 1D, left panel). Conversely, 91% (58%−98%) of clonal mutations in cfDNA were also detected in BM MM cells (Figure 1D, right panel). These data suggest that cfDNA represents a useful proxy for CNVs and the clonal mutational landscape of MM in the BM.
We next tested if the subclonal composition in cfDNA is different from bone marrow, perhaps revealing or de-prioritizing relevant subclones. Individual clones were distinguished by performing a two-dimensional kernel density estimation and displaying the results with contours (Materials and Methods). In the first patient, R13, there was a strong correlation of the cancer cell fractions (Pearson’s correlation r=0.901, P <2.2×10−16) of individual mutations between cfDNA and BM, as evidenced by two dominant subclones with a similar CCF in cfDNA and BM (Figure 2A). In another patient (Y2), there was a single dominant subclone comprising nearly half of the circulating myeloma DNA. Despite greater effective sequencing depth (Supplementary Table 2), this subclone was only partially detectable in the BM at much lower CCF (Figure 2B), suggesting a different subclonal composition of cfDNA compared to BM-derived MM. In another patient Y5, we detected 4 independent subclones, each harboring a nonoverlapping KRAS mutations (G12S, G12D, G12A, G12C) (Figure 2C). Lastly, in one patient we found that the clonal composition of BM and cfDNA differed by CNVs with BM-specific allelic amplification of chromosome 5q, 7p and 9 only in BM but not cfDNA (Supplementary Figure 4). These data demonstrate that differences in the subclonal composition between cfDNA and MM samples obtained from a single site bone marrow biopsy exist in some cases.
Figure 2. Subclonal composition and clonal evolution in cfDNA and matched BM-derived myeloma.
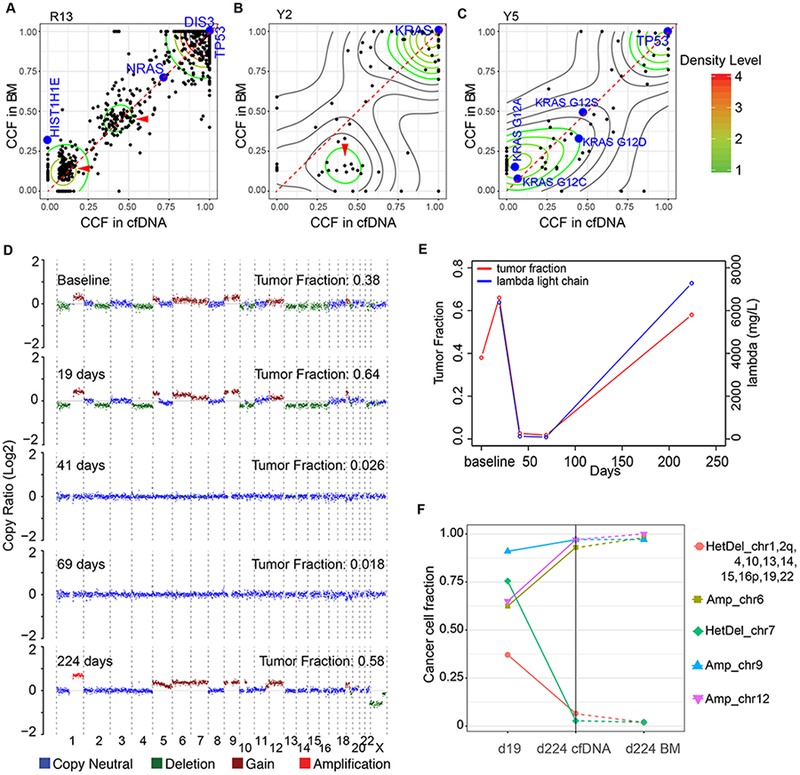
CCF of somatic mutations detected in cfDNA and matched BM-derived myeloma samples in patient R13 (A), Y2 (B) and Y5 (C). (D) Genome-wide CNVs and tumor fractions across five time-points, estimated from lpWGS data of cfDNA from patient R3. (E) Tumor fraction of myeloma-derived cfDNA (left vertical axis) and monoclonal lambda light chain concentration (right vertical axis) that were detected in patient R3 are plotted over time. (F) Percentage of myeloma cells harboring a particular arm-level chromosomal amplification or deletion in cfDNA on days 19 and 224) and in BM (day 224, BM), demonstrating concordance between bone marrow and cfDNA on day 224.
Although obtaining blood for genetic examination of cfDNA is safer, less costly and more convenient for the patient than BM biopsy, it is essential to estimate the success rate of comprehensive sequencing by low-pass WGS and WES of cfDNA beforehand in order to determine when to utilize this technique. Our data indicate that the cfDNA concentration can be used to estimate the expected tumor fraction in cfDNA and that the tumor fraction can be used to estimate the fraction of clonal bone marrow mutations that can be detected in cfDNA (Supplementary Figure 5).
We finally determined the potential of cfDNA as a longitudinal marker for disease progression and therapy response. We focused on a patient responding well to the indicated therapy initially, which was evident by decreasing levels of serum free light-chains (sFLC) and concordant trajectory of tumor fraction in cfDNA (Figure 2D,E). We then determined the CNVs in MM-derived cfDNA before treatment (day 19, prior to cytoreduction with cyclophosphamide, bortezomib, dexamethasone), and after relapse (day 224, after maintenance with ixazomib, dexamethasone). The cfDNA copy number profiles on day 0 and day 19 (with no change in management) were concordant. Tumor fraction became undetectable with response to treatment (days 41, 69). However, with relapse extensive clonal evolution occurred (day 224, after relapse) (Figure 2D,F) as drug resistance developed. Importantly, the copy number profile of cfDNA and BM on day 224 were concordant (Figure 2F). WES with allelic resolution revealed clonal or near clonal outgrowth of a subclone harboring allelic amplification of chromosome 6 and 12, as well as disappearance of subclones harboring allelic deletion of chromosome 7, 1, 2q, 4, 10, 13, 14, 15, 16p, 19 and 22 (Supplementary Figure 6). Two possible solutions for the observed clonal evolution are reflected as phylogenetic trees (Supplementary Figure 7), suggesting the selection and outgrowth of a drug-resistant subclone. These data demonstrate that cfDNA profiling may provide a powerful tool to longitudinally follow the clonal evolution of MM and the competitive emergence of drug-resistant subclones.
The studies we present demonstrate that comprehensive genomic interrogation by WES and WGS of MM-derived cfDNA is feasible and allows detailed genomic insight into MM evolution and progression over time. This may be particularly useful for elderly patients with significant comorbidities, as these patients are less likely to be subjected to frequent bone marrow biopsies or to be enrolled in clinical trials. cfDNA allows shipping from remote locations at room temperature in preservative-containing tubes and is therefore a particularly attractive alternative for this population. While the success rate of comprehensive genomic characterization by lpWGS and WES of cfDNA is high in patients with active disease, it is currently not cost-effective for patients in remission. Targeted sequencing approaches with limited genomic territory have been described 11–13 and allow greater sequencing depth at reduced cost in these cases. MRD negativity is of great interest as an endpoint for treatment success in MM. While the sensitivity of MRD detection in the BM is unlikely to be achieved even by targeted sequencing of cfDNA, liquid biopsy approaches, including circulating MM cells 14,15, may add further accuracy to progression free survival (PFS) prediction by defining the duration of MRD negativity. lpWGS and WES of cfDNA enable detailed genomic characterization of MM to identify putative resistance mechanisms and precision medicine targets as an alternative for repeated BM biopsies.
Supplementary Material
Footnotes
Conflicts of interest: For competing interests, M.M. is a founder and consultant to Foundation Medicine, Inc., a provider of cancer genome-based diagnostic testing.
References
- 1.Durie BGM. Role of new treatment approaches in defining treatment goals in multiple myeloma--the ultimate goal is extended survival. Cancer Treat Rev 2010; 36 Suppl 2: S18–23. [DOI] [PubMed] [Google Scholar]
- 2.Mateos M-V, Ocio EM, Paiva B, Rosiñol L, Martínez-López J, Bladé J et al. Treatment for patients with newly diagnosed multiple myeloma in 2015. Blood Rev 2015; 29: 387–403. [DOI] [PubMed] [Google Scholar]
- 3.Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun 2014; 5: 2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014; 25: 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J Clin Oncol Off J Am Soc Clin Oncol 2015; 33: 3911–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun 2017; 8: 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinhold N, Rasche L, Meissner T, Morgan G. Spatial-Temporal Genomic Analyses Reveal a Component of ‘Big Bang’ Kinetics in Multiple Myeloma Evolution. Blood 2016; 128: 239.27151888 [Google Scholar]
- 8.Egan JB, Shi C-X, Tembe W, Christoforides A, Kurdoglu A, Sinari S et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012; 120: 1060–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012; 120: 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 2017; 8: 1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kis O, Kaedbey R, Chow S, Danesh A, Dowar M, Li T et al. Circulating tumour DNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates. Nat Commun 2017; 8: 15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oberle A, Brandt A, Voigtlaender M, Thiele B, Radloff J, Schulenkorf A et al. Monitoring multiple myeloma by next-generation sequencing of V(D)J rearrangements from circulating myeloma cells and cell-free myeloma DNA. Haematologica 2017; 102: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mithraprabhu S, Khong T, Ramachandran M, Chow A, Klarica D, Mai L et al. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia 2017. doi: 10.1038/leu.2016.366. [DOI] [PubMed] [Google Scholar]
- 14.Lohr JG, Kim S, Gould J, Knoechel B, Drier Y, Cotton MJ et al. Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci Transl Med 2016; 8: 363ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mishima Y, Paiva B, Shi J, Park J, Manier S, Takagi S et al. The Mutational Landscape of Circulating Tumor Cells in Multiple Myeloma. Cell Rep 2017; 19: 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.