

Influenza virus is a long-lasting and severe threat to human health. Seasonal flu epidemics, which are caused by the co-circulating influenza A viruses (IAVs) and influenza B viruses (IBVs), occur annually and lead to tens of millions of respiratory illnesses and up to half a million human deaths worldwide each year (Ginsberg et al., 2009). Influenza pandemics are more devastating. The 2009 swine-originated H1N1 virus, which caused the latest influenza pandemic, spread from Mexico and U.S. to virtually all countries throughout the world within only several months and was associated much higher mortality among children, young adults, and pregnant women than typical seasonal influenza viruses (Fineberg, 2014). The zoonotic avian influenza viruses, including H5N1, H5N6, H7N9 and H10N8, cause alarmingly high fatality rate in human cases, raising a public concern of pandemic influenza outbreak of avian origin (Poovorawan et al., 2013; Barr, 2017; Bui et al., 2017).
Innate immune system is an important barrier of defending against influenza virus infection. According to the traditional paradigm, after IAV gets across the mucus that covers the respiratory epithelium, it first invades and infects respiratory epithelial cells, from where it spreads to other non-immune and immune cells (e.g., macrophages and dendritic cells). In these cells, the virus can be sensed by the pattern recognition receptors (PRRs), triggering the production of type I interferons (IFNs) which induce the expression of hundreds of IFN-stimulated genes (ISGs) that block viral replication and further virus spread. Simultaneously, activation of PRRs also leads to production of pro-inflammatory cytokines (IL-6, IL-1β, IL-18, TNF, etc.) and chemokines. Pro-inflammatory cytokines induce topical and systemic inflammation, cause fever and anorexia, and direct the adaptive immune response against the virus. Chemokines, on the other hand, recruit innate immune cells (neutrophils, monocytes, and NK cells) which engulf and inactivate the virus, kill virally infected cells, and guide subsequent innate and adaptive immune responses that mediate ultimate viral clearance (Iwasaki and Pillai, 2014).
However, effective protection from influenza virus infection is provided by finely tuned antiviral immunity, while excessive innate immunity causes detrimental inflammation. Infection with influenza viruses is usually self-limited, though the severe cases, especially caused by highly virulent strains (e.g., the 2009 pandemic H1N1 virus, H5N1 and H7N9) are characterized by severe pulmonary disease and lethal acute respiratory distress syndrome (ARDS) (Bauer et al., 2006; Ramsey and Kumar, 2011; Ma et al., 2017). Influenza-induced ARDS, which involves the damage to the epithelial-endothelial barrier of the pulmonary alveolus, flute leakage to the alveolar lumen, and respiratory insufficiency, is associated not only with direct viral damage to epithelial-endothelial barrier, but also with inflammation mediated by components of the innate immune response. The cytokines produced in influenza virus infected epithelial and endothelial cells, and cytokines and reactive oxygen species produced by neutrophils and macrophages recruited to pulmonary alveolus all contribute to damage to the epithelial-endothelial barrier (Short et al., 2014).
Both the protective and pathological roles of innate immunity have been evidenced in human and experimental animals. On one hand, genetic deficiency in production of interferon or certain ISGs (e.g., MX and IFITM proteins) increases the vulnerability to IAV infection (Ciancanelli et al., 2015, 2016). On the other hand, genetic ablation of the pro-inflammatory cytokine IL-6 (Dienz et al., 2012), or chemical depletion of alveolar macrophages (Abboud et al., 2015), NK (Abboud et al., 2015) or neutrophils (Tate et al., 2009) in mice can exacerbate lung injury during IAV infection. These data support that both IFN response and inflammatory innate immune response are essential in protecting the host against IAV infections. However, gene expression analysis displays that early innate immunity signatures including pro-inflammatory cytokines (TNF, IL-1β, IL-6), chemokines (CCL2, CCL3, CCL4, CXCL1) and neutrophil infiltration are strongly associated with acute death of the mice infected with IAV (Brandes et al., 2013). In addition, deletion of pro-inflammatory cytokines IL-1, TNF and various chemokines decrease mortality in IAV-infected mice (Teijaro, 2015). Furthermore, hyper expression of pro-inflammatory cytokines and chemokines are linked to the high mortality in human cases infected with highly pathogenic influenza virus strains such as 1918 pandemic H1N1 virus and H5N1 virus (Loo and Gale, 2007; Peiris et al., 2009). These data suggest that exaggerate inflammation during lethal infection with IAV can have fatal consequences. Therefore, benign outcomes of influenza virus infection results from inducing sufficient IFN-mediated antiviral immunity while avoiding harmful inflammatory response.
Most current knowledge about innate immunity in influenza virus infection has come from IAV. Although IBV contributes a significant part to the burden arising from the worldwide epidemics of seasonal influenza, much less attention has been paid to IBV than IAV because IBV does not have a potential to cause a pandemic (Chai et al., 2017). IBV is characterized by strict host range limits, initiates local epidemics with a lower evolutionary rate and causes milder clinical syndrome than IAV (Jiang et al., 2016). However, little is known about the innate response against the IBV infection.
Recently, we investigated the global transcriptome of the human lung A549 cell line infected with IBV with RNA-seq. We identified 340 differentially expressed genes (DEGs) in these cells at 8 h after infection. Among them, we found that type III IFNs (including IFN-λ1, IFN-λ2, and IFN-λ3) were among the top highly up-regulated genes. However, type I IFNs (IFN-α/β) was not significantly up-regulated and not listed among the DEGs. Besides, various antiviral ISGs including almost all known antiviral effectors in the innate immune system, including OAS proteins, MX proteins, IFITM proteins, and viperin (Schneider et al., 2014), are also highly up-regulated.
To verify this, we performed a qPCR analysis to determine the expression level of IFNs genes, as well as three important antiviral ISGs: Mx1, ISG20 and IFITM1 at 4, 8, and 12 h post infection. Consistent with the RNA-seq data analysis, IFN-λ expression was significantly increased at all the time points. The Mx1, ISG20 and IFITM1 also displayed markedly altered mRNA levels during IBV infection. In contrast, the expression of IFN-α was not altered at all points (Fig. 1A). Since the function of both IFN-λ and IFN-α is induction of ISG expression via the same intracellular signaling pathways, we speculate that it is IFN-λ rather than IFN-α that dominates the production of ISGs in IBV-infected A549 cells. Indeed, the IBV NP protein levels were obviously decreased in IFN-λ stimulated A549 cells, indicating the potent antiviral activity during IBV replication in A549 cell (Fig. 1B).
Figure 1.
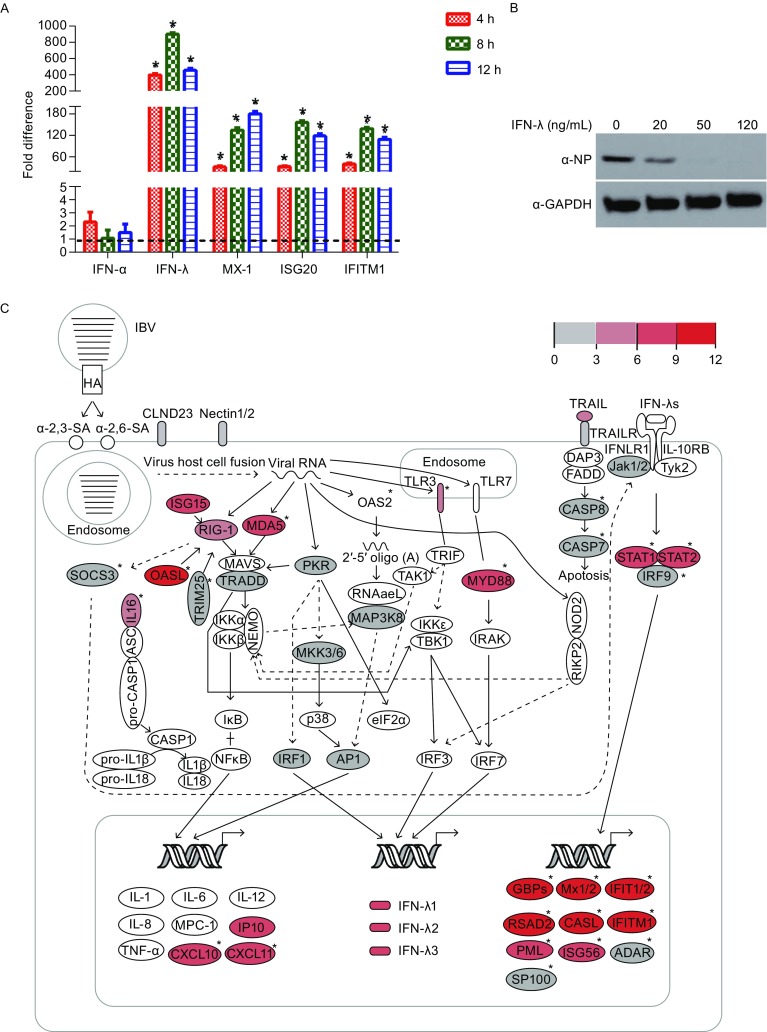
The integrated signaling pathway for IBV infection in A549 cells. (A) qRT-PCR analysis of the expression of selected genes in IBV infected A549 cells compared with uninfected controls. The fold difference was determined using the 2−∆∆Ct method, and RNA levels were normalized to GAPDH. Error bars represent the standard deviation. (B) A549 cells were treated with recombinant human IFN-λ (R&D systems) at final concentration of 0, 20, 50 and 100 ng/mL, followed by infection with IBV for 12 h. Cell lysates were analyzed by western blot using indicated antibodies. (C) The pathways for IBV infection in A549 cells. The relative expression values are indicated by the color gradient. The full lines represent direct interactions, and the dashed lines represent indirect interactions. The uncolored ellipses represent genes that are not DEGs. ISGs are marked with an asterisk
Based on the above results and KEGG pathways assay, we built the intracellular signaling pathway related to IBV infection in A549 cells (Fig. 1C). IBV infection begins with viral HA binding to α-2,3 or α-2,6 sialic acid on the host cell surface. After binding to the receptor, the virus enters the endosome, where the acidic environment triggers virus-host cell membrane fusion, after which the viral RNA is released into the cytoplasm. In the cytoplasm, the virus-derived RNA is recognized by multiple canonical pattern recognition receptors (PRRs), including the RIG-I-like receptors (RLRs, RIG-I, and MD5), the Toll-like receptors (TLRs, TLR-3, and 7), the NOD-like receptors (NLR and NOD1/2), and the PKR, and activates their respective signaling pathways. Activation of these pathways leads to the expression of IFN-λs, which then interact with their receptor, the heterogeneous dimer IFNLR1/IL-10R, and activate the expression of ISGs through the JAK/STAT pathway.
Interferons (IFNs) are classified into three categories (type I IFNs, type II IFNs, and type III IFNs). All IFNs signal through JAK/STAT pathway which leads to transcription of ISGs, but their production cells and receptor specificity are different (Chow and Gale, 2015). Type I IFNs include IFN-α, β, ε, κ, and ω. They can be produced by almost all cell types and their receptor is the widely-expressed heterodimer composed of IFNR1 and IFNR2. Type II IFN is INF-γ, which is produced by immune cells and forms dimmer to act on the receptor complex consisting of two IFNR1 and two IFNR2. Type III IFNs include IFN-λ1 (IL-29), IFN-λ2 (IL-28A), IFN-λ3 (IL-28B), and IFN-λ4. Their receptor is the heterodimeric complex composed of IFN-λR1 and IL-10Rβ. IL-10Rβ is broadly distributed, but the expression of IFN-λR1 is mainly restricted to epithelial cells (Broggi et al., 2017).
Although discovered later than type I and type II IFNs, type III IFNs in innate immunity are attracting more and more attention. The potential importance of type III IFNs has been suggested by life-threatening influenza developed in genetically deficiency that leads to impaired production of both type I and type III IFN production in a 9-year-old child (Ciancanelli et al., 2015). IFNLR is expressed by human hepatocytes, and IFNL polymorphisms are associated with ameliorated outcome from HCV and HBV infection (Bibert et al., 2013; Galmozzi et al., 2014). Clinical trials have displayed that the pegylated IFN-λ can reduce the level of HCV and HBV in patients infected with chronic HCV and HBV, respectively (Zeuzem et al., 2011; Phillips et al., 2017). IFN-λs are also highly expressed in gastrointestinal tracts, and can cure chronic murine norovirus infection without the presence of adaptive immunity (Nice et al., 2015).
Most recently, several important breakthroughs have increased the brightness of IFN-λs as a new spotlight in innate immunity. One of them is the determination of the crystal structure of an engineered high-affinity IFN-λ3 (H11) in complex with its two receptors, IFN-λR1 and IL-10R. In this structure, the IFN-λ3 helical bundle binds to the gorge formed by the two domains at the distal ends of its two receptors to the cellular membrane. In contrast, the two receptors bind to the opposite sides of the helical bundles of type I IFNs (Mendoza et al., 2017).
Another breakthrough is the definition of the differential roles of IFN-λs in contrast to type I IFNs in IAV infection using Ifnlr1−/−, Ifnαr1−/− and Ifnlr1−/− Ifnαr1−/− mice (Galani et al., 2017). This study shows that in mice infected with IAV, IFN-λs emerge earlier than type I IFNs and show potent protective function during the early phase of the infection, when type I IFNs are still scarce. Furthermore, IFN-λs treatment induces expression of antiviral genes but not pro-inflammatory cytokines in neutrophils, while type I IFNs induce the expression of both types of genes and are involved in causing inflammation and immunopathology.
The different functions between IFN-λs and type I IFNs can be explained by the different STAT complex they activated. When engaged with their receptor complexes, both IFN-λs and type I IFNs activate JAK-family kinases, which phosphorylate STAT1 and STAT2. The phosphorylated STAT1 and STAT2 dimerize and translocate to the nucleus where they associate with IRF9 to form IFN-stimulated gene factor 3 (ISGF3), bind to IFN-stimulated response elements (ISREs) and induces the expression of antiviral ISGs. However, engagement of type I IFNs with their receptor can also activate the STAT1 homodimer which binds to gamma-activated sequence (GAS) and induces the expression of pro-inflammatory cytokines and chemokines (Ivashkiv and Donlin, 2014). Thus, type I IFNs have the potential of inducing inflammation in addition to antiviral function, while IFN-λs promote the production of antiviral ISGs without the function of inducing inflammation (Fig. 2).
Figure 2.
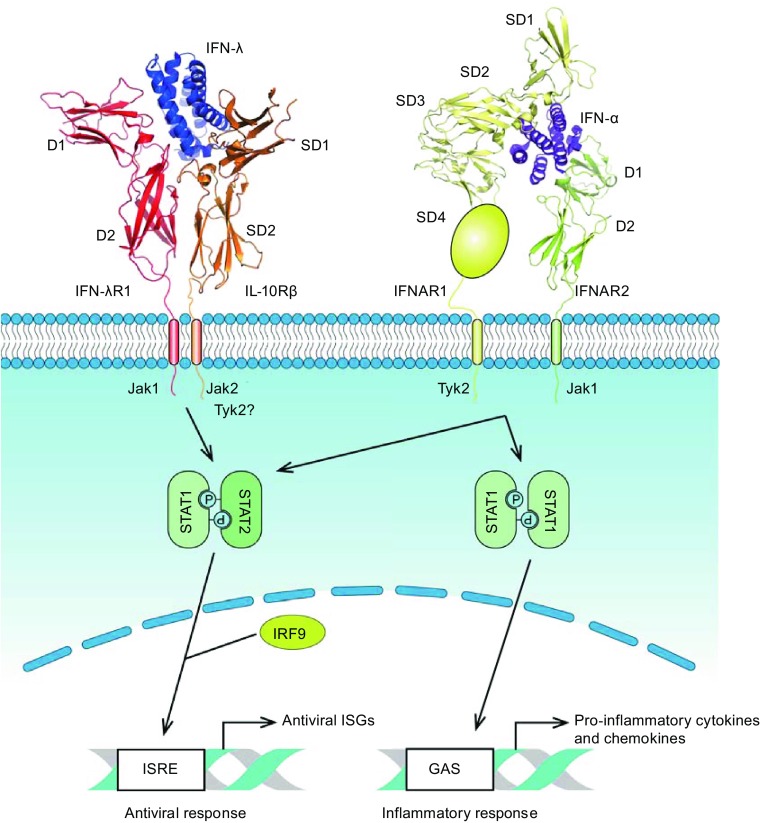
Different gene expression pattern triggered by type I and type III IFNs. Type III IFNs (IFN-λ3, PDB ID: 5T5W) binding to their heterodimeric receptor composed of IFNλR1 and IL-10Rβ triggers the formation of heterdimeric complex of STAT1 and STAT2 which translocates to the nucleus where it forms ternary complex with IRF9 and form IFN-stimulated gene factor 3 (IGF3). IGF3 binds to IFN-stimulated response elements (ISREs) and induces the expression of antiviral ISGs. Engagement of type I IFNs (IFN-α2, PDB ID: 3SE3) with their receptor, the heterodimer composed of IFNAR1 and IFNAR2, can also activate the STAT1 homodimer which binds to gamma-activated sequence (GAS) and induces the expression of pro-inflammatory cytokines and chemokines
In summary, IFN-λs are more advantageous than type I IFNs in defending against influenza virus for several reasons: (1) IFN-λs are produced earlier and in larger amount during influenza virus infection; (2) unlike type I IFNs, IFN-λs are produced and act specifically in respiratory tract, so they do not induce systematic side effects; (3) type III IFNs have potent antiviral activity without mediating inflammation. Therefore, IFN-λs share the therapeutic benefits but eschews many side effects associated with the clinical use of IFN-α/β (Lazear et al., 2015). In the future, scientists should explore the possibility that whether IFN-λs are more ideal therapeutic reagents for dealing with respiratory tract infection by viruses like IBV or not.
Acknowledgements
We gratefully thank Dr. Jun Wu of the Institute of Microbiology, Chinese Academy of Sciences, for helpful suggestions. This work was supported by grants from the National Key R&D Program of China (2016YFD0500206, 2017YFD051105, 2016YFC1200803, and 2016YFC1201303), National Natural Science Foundation of China (Grant Nos. 31572526, 31402216 and 31630079), National Defense Foundation of China (Grant No. 17-163-12-ZT-005-041-01), the Southeast Asia Biodiversity Research Institute, Chinese Academy of Sciences (Y4ZK111B01). Wenjun Liu and Po Tian are the principal investigators of the NSFC Innovative Research Group (Grant No. 81621091).
JL and YS designed the experiments, performed RNA-sequencing and other experimental data analysis, prepared the manuscript, and completed its revision. JL and JJ performed the viral replication ability tests. WL and PT provided helpful suggestions about the study. All authors read and approved the final manuscript. All of the authors declare that they have no conflict of interest.
Contributor Information
Wenjun Liu, Email: liuwj@im.ac.cn.
Jing Li, Email: lj418@163.com.
References
- Abboud G, Tahiliani V, Desai P, Varkoly K, Driver J, Hutchinson TE, Salek-Ardakani S. Natural killer cells and innate interferon gamma participate in the host defense against respiratory vaccinia virus infection. J Virol. 2015;90:129–141. doi: 10.1128/JVI.01894-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr IG. Assessing the potential pandemic risk of recent avian influenza viruses. Eur Respir J. 2017;49:1602517. doi: 10.1183/13993003.02517-2016. [DOI] [PubMed] [Google Scholar]
- Bauer TT, Ewig S, Rodloff AC, Muller EE. Acute respiratory distress syndrome and pneumonia: a comprehensive review of clinical data. Clin Infect Dis. 2006;43:748–756. doi: 10.1086/506430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibert S, Roger T, Calandra T, Bochud M, Cerny A, Semmo N, Duong FHT, Gerlach T, Malinverni R, Moradpour D, et al. IL28B expression depends on a novel TT/-G polymorphism which improves HCV clearance prediction. J Exp Med. 2013;210:1109–1116. doi: 10.1084/jem.20130012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes M, Klauschen F, Kuchen S, Germain RN. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. 2013;154:197–212. doi: 10.1016/j.cell.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broggi A, Tan Y, Granucci F, Zanoni I. IFN-lambda suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat Immunol. 2017;18:1084–1093. doi: 10.1038/ni.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui CM, Chughtai AA, Adam DC, MacIntyre CR. An overview of the epidemiology and emergence of influenza A infection in humans over time. Arch Public Health. 2017;75:15. doi: 10.1186/s13690-017-0182-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai N, Swem LR, Park S, Nakamura G, Chiang N, Estevez A, Fong R, Kamen L, Kho E, Reichelt M, et al. A broadly protective therapeutic antibody against influenza B virus with two mechanisms of action. Nat Commun. 2017;8:14234. doi: 10.1038/ncomms14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow KT, Gale M., Jr SnapShot: interferon signaling. Cell. 2015;163(1808–1808):e1801. doi: 10.1016/j.cell.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, Lafaille FG, Trouillet C, Schmolke M, Albrecht RA, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. 2015;348:448–453. doi: 10.1126/science.aaa1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciancanelli MJ, Abel L, Zhang SY, Casanova JL. Host genetics of severe influenza: from mouse Mx1 to human IRF7. Curr Opin Immunol. 2016;38:109–120. doi: 10.1016/j.coi.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienz O, Rud JG, Eaton SM, Lanthier PA, Burg E, Drew A, Bunn J, Suratt BT, Haynes L, Rincon M. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. 2012;5:258–266. doi: 10.1038/mi.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineberg HV. Pandemic preparedness and response—lessons from the H1N1 influenza of 2009. N Engl J Med. 2014;370:1335–1342. doi: 10.1056/NEJMra1208802. [DOI] [PubMed] [Google Scholar]
- Galani IE, Triantafyllia V, Eleminiadou EE, Koltsida O, Stavropoulos A, Manioudaki M, Thanos D, Doyle SE, Kotenko SV, Thanopoulou K, et al. Interferon-lambda mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity. 2017;46(875–890):e876. doi: 10.1016/j.immuni.2017.04.025. [DOI] [PubMed] [Google Scholar]
- Galmozzi E, Vigano M, Lampertico P. Systematic review with meta-analysis: do interferon lambda 3 polymorphisms predict the outcome of interferon-therapy in hepatitis B infection? Aliment Pharmacol Ther. 2014;39:569–578. doi: 10.1111/apt.12631. [DOI] [PubMed] [Google Scholar]
- Ginsberg J, Mohebbi MH, Patel RS, Brammer L, Smolinski MS, Brilliant L. Detecting influenza epidemics using search engine query data. Nature. 2009;457:U1012–U1014. doi: 10.1038/nature07634. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JW, Li J, Fan WH, Zheng WN, Yu M, Chen C, Sun L, Bi YH, Ding C, Gao GF, et al. Robust Lys63-linked ubiquitination of RIG-I promotes cytokine eruption in early influenza B virus infection. J Virol. 2016;90:6263–6275. doi: 10.1128/JVI.00549-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Nice TJ, Diamond MS. Interferon-lambda: immune functions at barrier surfaces and beyond. Immunity. 2015;43:15–28. doi: 10.1016/j.immuni.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Gale M., Jr Influenza: fatal immunity and the 1918 virus. Nature. 2007;445:267–268. doi: 10.1038/445267a. [DOI] [PubMed] [Google Scholar]
- Ma W, Huang H, Chen J, Xu K, Dai Q, Yu H, Deng F, Qi X, Wang S, Hong J, et al. Predictors for fatal human infections with avian H7N9 influenza, evidence from four epidemic waves in Jiangsu Province, Eastern China, 2013–2016. Influenza Other Respir Viruses. 2017;11:418–424. doi: 10.1111/irv.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza JL, Schneider WM, Hoffmann HH, Vercauteren K, Jude KM, Xiong A, Moraga I, Horton TM, Glenn JS, de Jong YP, et al. The IFN-lambda-IFN-lambdaR1-IL-10Rbeta complex reveals structural features underlying type III IFN functional plasticity. Immunity. 2017;46:379–392. doi: 10.1016/j.immuni.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, Diamond MS, Virgin HW. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science. 2015;347:269–273. doi: 10.1126/science.1258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris JS, Cheung CY, Leung CY, Nicholls JM. Innate immune responses to influenza A H5N1: friend or foe? Trends Immunol. 2009;30:574–584. doi: 10.1016/j.it.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips S, Mistry S, Riva A, Cooksley H, Hadzhiolova-Lebeau T, Plavova S, Katzarov K, Simonova M, Zeuzem S, Woffendin C, et al. Peg-interferon lambda treatment induces robust innate and adaptive immunity in chronic hepatitis B patients. Front Immunol. 2017;8:621. doi: 10.3389/fimmu.2017.00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poovorawan Y, Pyungporn S, Prachayangprecha S, Makkoch J. Global alert to avian influenza virus infection: from H5N1 to H7N9. Pathog Glob Health. 2013;107:217–223. doi: 10.1179/2047773213Y.0000000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey C, Kumar A. H1N1: viral pneumonia as a cause of acute respiratory distress syndrome. Curr Opin Crit Care. 2011;17:64–71. doi: 10.1097/MCC.0b013e3283427259. [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Kroeze E, Fouchier RAM, Kuiken T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis. 2014;14:57–69. doi: 10.1016/S1473-3099(13)70286-X. [DOI] [PubMed] [Google Scholar]
- Tate MD, Deng YM, Jones JE, Anderson GP, Brooks AG, Reading PC. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J Immunol. 2009;183:7441–7450. doi: 10.4049/jimmunol.0902497. [DOI] [PubMed] [Google Scholar]
- Teijaro JR. The role of cytokine responses during influenza virus pathogenesis and potential therapeutic options. Curr Top Microbiol Immunol. 2015;386:3–22. doi: 10.1007/82_2014_411. [DOI] [PubMed] [Google Scholar]
- Zeuzem S, Arora S, Bacon B, Box T, Charlton M, Diago M, Dieterich D, Mur RE, Everson G, Fallon M, et al. Pegylated interferon-Lambda (PEGIFN-lambda) shows superior viral response with improved safety and tolerability versus PEGIFN alpha-2A in HCV patients (G1/2/3/4): emerge phase IIB through week 12. J Hepatol. 2011;54:S538–S539. doi: 10.1016/S0168-8278(11)61362-7. [DOI] [Google Scholar]