

Abstract
LEM domain (LEM-D) proteins are conserved components of the nuclear lamina (NL) that contribute to stem cell maintenance through poorly understood mechanisms. The Drosophila emerin homolog Otefin (Ote) is required for maintenance of germline stem cells (GSCs) and gametogenesis. Here, we show that ote mutants carry germ cell-specific changes in nuclear architecture that are linked to GSC loss. Strikingly, we found that both GSC death and gametogenesis are rescued by inactivation of the DNA damage response (DDR) kinases, ATR and Chk2. Whereas the germline checkpoint draws from components of the DDR pathway, genetic and cytological features of the GSC checkpoint differ from the canonical pathway. Instead, structural deformation of the NL correlates with checkpoint activation. Despite remarkably normal oogenesis, rescued oocytes do not support embryogenesis. Taken together, these data suggest that NL dysfunction caused by Otefin loss triggers a GSC-specific checkpoint that contributes to maintenance of gamete quality.
Otefin is a nuclear lamina protein required for survival of Drosophila germ stem cells. Here the authors show that nuclear lamina dysfunction resulting from loss of Otefin activates a DNA damage-independent germ stem cell-specific checkpoint, mediated by the ATR and Chk2 kinases, which ensures that healthy gametes are passed on to the next generation.
Introduction
The nuclear lamina (NL) is an extensive protein network comprised of lamins and hundreds of associated proteins. Among these are the conserved family of LEM Domain (LEM-D) proteins, named for the founding human members LAP2, emerin, and MAN11,2. These proteins share a LEM-D that interacts with Barrier-to-Autointegration Factor (BAF), a conserved double-stranded DNA and histone-binding protein that promotes chromosome condensation important for nuclear assembly3,4. Interactions between LEM-D proteins and BAF tether chromatin to the nuclear periphery, organizing the genome for transcription, DNA replication, and repair5–7. In addition, LEM-D proteins interact with transcriptional regulators, including repressors and nuclear effectors of signaling cascades1,2. Although LEM-D proteins are globally expressed, mutations in LEM-D genes cause tissue-restricted diseases, termed laminopathies, that affect tissues such as skeletal muscle, skin, fat, and bone2,8. A unifying feature of laminopathies is the age-dependent worsening of disease phenotypes, leading to the suggestion that laminopathies result from defects in tissue homeostasis and a failure to maintain adult stem cell populations9,10. Despite the growing evidence that LEM-D proteins are required in multiple types of adult stem cells, it remains unclear how LEM-D proteins maintain healthy stem cell populations and promote tissue homeostasis.
Drosophila melanogaster has emerged as a powerful model to define the function of LEM-D proteins in tissue homeostasis. Drosophila encodes four LEM-D proteins11, of which three are NL-associated (Fig. 1a). These include Drosophila MAN1 (dMAN1) and two emerin homologs Otefin (Ote) and Bocksbeutel (Bocks). As in mammals, loss of individual Drosophila LEM-D proteins causes distinct tissue-restricted developmental defects that worsen with age, with little to no effect on viability11–15. Even so, LEM-D proteins have overlapping developmental functions, as loss of any two NL LEM-D proteins is lethal13. Studies of Ote revealed a key role for this LEM-D protein in stem cell homeostasis. Loss of Ote causes female and male sterility due to a failure in adult germline stem cell (GSC) maintenance12,15,16. As Drosophila GSC niches and germ cell properties are among the best characterized17,18, studies of Ote are instrumental for elucidating how LEM-D proteins support adult stem cells.
Fig. 1.

Loss of Ote causes germline stem cell-specific NL defects. a Schematic of the Drosophila nuclear lamina (NL). Underneath the inner nuclear envelope (yellow) resides the NL (blue) that contains three Drosophila LEM-D proteins, Otefin (Ote), Bocksbeutel (Bocks α and β), and dMAN1. Each protein localizes to the NL using a domain (orange, Peripheral localization, PL) that interacts with lamin or using transmembrane domains (orange, TM). Proteins in the LEM-D family carry a LEM-D (blue) that binds Barrier-to-Autointegration Factor (BAF, green), to promote chromatin assembly at the nuclear periphery. b Confocal images of ote+/+ and ote−/− (oteB279G/PK) nuclei stained with antibodies against Lamin Dm0 (red) and Heterochromatin protein 1a (HP1a; green). Scale bars, 2.5 μm
Loss of Ote blocks germ cell differentiation and causes progressive GSC death12,16. Here we investigate the mechanism behind these defects. We find that loss of Ote causes a thickening of the NL and reorganization of heterochromatin in GSC nuclei, but not in mutant somatic cell nuclei. Such architectural defects are reminiscent of phenotypes observed in human LMNA mutant progeroid (premature aging) models19, in which cellular senescence has been linked to compromised replication and DNA damage20–23. Prompted by these links, we investigated whether GSC death resulted from activation of a DNA damage response (DDR). We found that loss of Ataxia Telangiectasia and Rad3-related (ATR) or Checkpoint kinase 2 (Chk2) in ote mutants rescues oogenesis, with females laying eggs. Surprisingly, although rescue of oogenesis was achieved by loss of components in the DDR pathway, we demonstrate that canonical triggers are not responsible for pathway activation. Instead, we establish that Chk2 activation is linked to defects in NL structure. Rescued chk2, ote double-mutant oocytes do not support embryogenesis, suggesting that NL dysfunction in GSCs triggers a checkpoint pathway that maintains female gamete quality to safeguard offspring fitness. Our studies suggest that laminopathies might be linked to activation of similar quality control pathways that selectively impact specific adult stem cell populations.
Results
NL organization in GSCs is altered in the absence of Ote
Loss of Ote compromises GSC homeostasis in female and male gonads12,16. To define mechanisms responsible for these defects, we focused our studies on the ovary because ote−/− mutant GSC phenotypes are immediately apparent in newborn females but have an age-dependent component in males16. Drosophila ovaries are divided into 16 to 20 ovarioles, which are arranged in advancing stages of oocyte maturation. At the anterior end of the ovariole is the germarium, a specialized structure that contains a stem cell niche that supports two to three GSCs (Supplementary Fig. 1A). Upon division, GSCs produce one daughter cell that remains attached to the somatic niche and receives niche factors critical for GSC self-renewal. The second daughter cell is displaced from the niche, does not receive self-renewal signals and differentiates. Germ cell differentiation begins with four incomplete mitotic divisions to generate a 16-cell cyst, which develops into an egg chamber containing 1 oocyte and 15 supporting nurse cells. In wild-type Drosophila, GSCs are maintained and gametes are produced throughout the lifespan of the animal, making this defined and tractable system instrumental to understanding how LEM-D proteins support adult stem cell function.
Ote localizes to the NL in somatic and germ cells, yet loss of Ote specifically affects germ cells. To understand whether this tissue-restricted requirement is linked to nuclear structure, we dissected ovaries from young (<1-day-old) females and stained them with antibodies against the B-type lamin (Lamin Dm0) and Heterochromatin Protein 1a (HP1a). Notably, germline progenitors and adult GSCs have a distinct nuclear organization from somatic cells. In GSCs, homologous chromosomes and centromeres are unpaired24,25, whereas in other cell types, homologous chromosomes are paired and centric regions cluster to form an HP1a-containing chromocenter26. We found that ote−/− GSCs have a thick and irregular NL, as well as increased clustering of HP1a compared to wild-type GSCs (Fig. 1b). Strikingly, this altered nuclear organization is specific to ote−/− GSCs because ote−/− nuclei of imaginal disc tissues (2n) and salivary gland tissues (~1000n) are phenotypically indistinguishable from wild-type controls (Fig. 1b). Together, these findings imply that Ote loss is associated with germ cell-specific nuclear architectural defects.
The DDR transducer kinase Chk2 causes ote−/− GSC loss
Nuclear architecture defects are common in premature aging syndromes such as progeria27. Accumulating evidence suggests that compromised nuclear architecture coincides with altered DNA replication and damage repair, two processes regulated by lamins28,29. Indeed, studies of progeroid cells with defective maturation of prelamin A found evidence of DNA damage and activation of DDR-mediated cellular senescence20,22. Based on shared progeroid-like nuclear phenotypes, we tested whether ote−/− GSC death might result from DDR activation, by genetically compromising the DDR pathway. The DNA damage response in GSCs depends upon activation of the transducer kinase Chk230,31. For this reason, we tested the role of Chk2 on ote mutant phenotypes by generating double mutant animals carrying null mutations in both ote and chk2 (also known as mnk, loki in Drosophila; Supplementary Table 1). Ovaries from wild-type and mutant animals were stained with antibodies against Vasa to identify germ cells and DAPI to visualize DNA (Fig. 2a). As previously observed12,15, ote mutant ovaries were rudimentary and lacked the strings of developing egg chambers found in wild-type ovaries. Strikingly, loss of one or both copies of chk2 in an ote mutant background restored oogenesis, producing large ovaries filled with developing egg chambers (Fig. 2a). As Chk2 transduces the checkpoint through phosphorylation of downstream targets32, we tested whether the Chk2 kinase activity was required for execution of ote mutant phenotypes. To this end, we crossed a chk2 kinase dead [chk2KD] allele into an ote mutant background. We found that both heterozygous and homozygous chk2KD mutations rescued oogenesis (Fig. 2b). These data demonstrate that the kinase activity of Chk2 is crucial for ote mutant GSC loss.
Fig. 2.

Chk2 activation is responsible for oogenesis defects in ote mutants. a, b Confocal images of ovaries dissected from less than 2-day-old females stained for Vasa (red) and DAPI (white). Genotypes are noted on the top of each image, wherein ote−/− corresponds to oteB279G/PK. The chk2- alleles were either deletion alleles (a) or a kinase dead allele (Table S1, b). Scale bars represent 250 µm (a) or 100 µm (b). c Fecundity (eggs per female per day) of females mated to ote+/+ males as a function of female age in days. Bars indicate the standard deviation from a minimum of three independent experiments, assaying 7–15 females each time. Genotypes are noted to the right of each line. d Quantification of germarial phenotypic classes, as described on the right12. Genotypes are listed below the corresponding bar, with the number of germaria scored listed at the top. e Percentage of eggs that hatched within 48 h following deposition by females that were mated to ote+/+ males. Bars represent the standard deviation from at least two independent experiments. The number of eggs analyzed is noted above each bar. f Volcano plot of gene expression microarray results. The x-axis shows fold change between NULL (chk2P6, otePK/ oteB279G and otePK/ chk2P30, oteB279G), and HET (chk2P6, otePK/ CyO and chk2P30, oteB279G/ CyO) ovaries and the y-axis shows one-way ANOVA p value; areas corresponding to twofold change cutoff and p < 0.05 are shaded, points corresponding to significantly decreasing or increasing RNAs are colored blue and red, respectively
We next completed several experiments to define the scope of the dramatic rescue in chk2, ote mutants. First, egg production was quantified. These analyses revealed that fecundity remained strong throughout the entire 8-day assay period (Fig. 2c). These studies also revealed that the Chk2 rescue was dose dependent, wherein loss of one wild-type chk2 allele partially rescued fecundity and loss of both wild-type alleles provided full rescue. Second, the composition of germaria was determined. In these experiments, ovaries were stained with antibodies against Vasa and the cytoskeletal protein Spectrin (Supplementary Fig. 1A). Spectrin distinguishes the differentiation status of germ cells. In GSCs and their immediate daughters, Spectrin is found within a spherical structure called the spectrosome. Upon differentiation, Spectrin adopts a branched shape within a structure called the fusome33. Germaria from newly eclosed ote+/+ females carry approximately four germ cells with spectrosomes, corresponding to GSCs and daughter cystoblasts, as well as multiple developing cysts with fusomes (Supplementary Fig.1A, B). In contrast, newly eclosed ote−/− ovaries show a complex germarial phenotype12, including germaria without germ cells due to GSC death (~24%) and germaria containing only spectrosome-positive germ cells (~68%) due to blocked differentiation (Fig. 2d, Supplementary Fig. 1A). Loss of Chk2 alone has no effect on GSC homeostasis (Fig. 2d, Supplementary Fig. 1B), consistent with previous findings31. In chk2, ote double mutant ovaries, all germaria had GSCs, cystoblasts, and differentiating germ cells (Fig. 2d). Remarkably, even loss of one wild-type chk2 allele fully rescued GSC death and restored germline differentiation within the majority (~80%) of germaria (Fig. 2d). In chk2−/−, ote−/− germaria, there is a slight increase in the number of spectrosome-containing germ cells per germarium (Supplementary Fig. 1B), suggesting that germ cell differentiation might be delayed. Together, these data reveal loss of Chk2 largely rescues germarial phenotypes in the ote mutants.
Rescued oogenesis shows minimal changes in transcription
The chk2-dependent rescue provided the first opportunity to assess the Ote requirement in later stages of oogenesis. This multi-faceted developmental program includes rapid syncytial germ cell divisions, oocyte specification, meiosis, endocyclic DNA replication in nurse cells, axis specification, and active transcription to produce maternal products required for early embryogenesis. We stained chk2−/−, ote−/− ovaries with Vasa and DAPI to examine oocyte development. Overall, oogenesis appeared normal in chk2, ote mutant females, with only a small number (10%) of developing chk2−/−, ote−/− egg chambers containing a reduced number of nurse cells compared to wild-type controls. Next, we examined eggs laid by chk2, ote mutant females. Although these eggs were phenotypically normal, all failed to hatch even when the oocyte was fertilized with an ote+/+ sperm (Fig. 2e). These findings imply that Ote is either required for early events in embryogenesis or that underlying defects remain in ote, chk2 germ cells that prevent embryogenesis. To determine whether blocked embryonic development was caused by altered production of maternal RNAs, we defined effects of Ote loss on transcription. To control for inter-strain variability and ovary size, total RNA was isolated from ovaries of two chk2+/–, ote−/− (null) backgrounds and their chk2+/–, ote+/– (het) siblings. Surprisingly, Ote loss has minimal effects on transcription of protein-coding genes. We identified only 28 differentially expressed genes (5 downregulated and 23 upregulated) in ote null ovaries (Fig. 2f, Supplementary Fig. 1C). Gene ontology analyses revealed a modest enrichment for serine endopeptidases among the upregulated genes and unsupervised hierarchical clustering revealed no distinct ote mutant signature (Supplementary Fig. 1C, D). These data indicate that the maternal RNA pool is largely unaffected in ote mutants, with the exception of ote mRNA. Notably, early embryos possess a large pool of maternally deposited Otefin that was linked to nuclear envelope assembly34, suggesting that embryogenesis might fail due to the absence of Ote itself. Even so, oogenesis in chk2, ote mutant females proceeds with remarkably few detectable defects.
Loss of ATR rescues oogenesis in ote mutants
To understand the extent of the DDR pathway involvement in ote mutants, we generated double mutants lacking Ote and DDR components, and examined effects on female fecundity and germarial phenotypes. We tested mutations in genes encoding both responder kinases, ATM (tefu), and ATR (mei-41), the second transducer kinase Checkpoint kinase 1 (Chk1; grapes), and the downstream effector, p53 (Fig. 3a, Supplementary Table 1). We reasoned that if a DDR component contributed to signaling in the GSC checkpoint, then germ cell phenotypes would improve in the ote, ddr double mutants relative to ote single mutants. First, we examined effects of loss of ATM. As complete loss of ATM is lethal in Drosophila, temperature sensitive ATM alleles were used. We found no rescue of egg laying or germarial phenotypes in ATM-defective ote mutants (Fig. 3b–d), suggesting that ATM is not a component of the GSC checkpoint. Second, we tested effects of loss of the second responder kinase, ATR. Interestingly, oogenesis was rescued in the atr−/−; ote−/− females, with egg laying sustained throughout the 12-day measurement period (Fig. 3b, c). Germarial phenotypes in atr−/−; ote−/− ovaries were completely rescued, with all germaria carrying GSCs, cystoblasts and differentiating germ cells (Fig. 3d). These data indicate that ATR is the responder kinase of the checkpoint resulting from Ote loss. Third, we evaluated effects of loss of the second transducer kinase, Chk1. Whereas no rescue of fecundity was found in chk1−/−, ote−/− females (Fig. 3b), a modest improvement was observed in germline differentiation (Fig. 3d). These data suggest that Chk1 has a minor role. Finally, we tested effects of loss of the DDR effector p53. We found no rescue of fecundity or germarial phenotypes, including no improvement of GSC survival (Fig. 3b–d). These data are consistent with the absence of staining with standard apoptosis markers in ote−/− germaria12,15, and support previous studies showing that GSC death is distinct from classic apoptotic cell death35. Together, our data suggest that a checkpoint is activated in GSCs in the absence of Ote, wherein ATR is the critical responder kinase and Chk2 is the critical transducer kinase.
Fig. 3.

Select DDR components contribute to the checkpoint pathway in ote mutants. a Schematic representation of signaling through the DNA damage response (DDR) pathway. Common triggers that activate the ATM and ATR responder kinases are shown. The responder kinases canonically activate the Chk2 and Chk1 transducer kinases, which signal to p53 and cause cell death. b Quantification of peak fecundity (eggs per female per day) of ddr, ote double-mutant females crossed to ote+/+ males. Genotypes are noted below each bar, with ote−/− corresponding to oteB279G/PK. Error bars represent the standard deviation from a minimum of two independent experiments, each using 7–15 females. c Fecundity (eggs per female per day) of ddr, ote double-mutant females of indicated genotypes, crossed to wild-type males, with ote−/− corresponding to oteB279G/PK. Genotypes are noted above the graph. Error bars indicate the standard deviation from a minimum of three independent experiments with 7–15 females each. Wild-type data from Fig. 2c. were included in the graph as a reference. d. Quantification of germarial phenotypic classes (noted to the right) in 2-day-old ddr, ote double-mutant females, with ote−/− corresponding to oteB279G/PK. Number of germaria assessed from at least ten females is noted above each bar
The GSC checkpoint is activated by a non-canonical trigger
DNA double-strand breaks activate the DDR pathway in GSCs30. To test the role of DNA damage in Chk2-dependent GSC loss, we stained ote+/+ and ote−/− ovaries with antibodies against γ-H2Av (the homolog of mammalian γ-H2AX), a commonly used marker of DNA damage30,36,37. In wild type germaria, γ-H2Av accumulates in the nuclei of all meiotic germ cells but is infrequently detected in GSCs30,31. However, upon DNA damage, GSCs rapidly respond, such that nearly all (~95%) stain with γ-H2Av antibodies within 1 hour of induction of double-strand breaks (DSBs), persisting for several days30. Consistent with these studies, we found that γ-H2Av accumulated in all meiotic germ cells and confirmed a low frequency in wild-type GSCs (7.8%; Fig. 4a). In ote−/− GSCs, over half (54.3%) had γ-H2Av signal, indicating that these GSCs have increased levels of DNA damage.
Fig. 4.

DNA damage in ote−/− GSCs is downstream of Chk2. a Confocal images of germaria stained with antibodies against Vasa (red), DNA damage marker γ-H2Av (grayscale) and with DAPI (blue). Genotypes are noted above each image, wherein ote−/− corresponds to oteB279G/PK and chk2−/− corresponding to chk2P6/P30. Bottom: Image of the signal in the γ-H2Av channel. Boxes and brackets indicate GSCs and meiotic germ cells, respectively. Dashed line indicates the position of GSC niche. The percentage of γ-H2Av-positive GSCs is noted at the bottom of the γ-H2Av image, with the number of GSCs analyzed in parenthesis. All scale bars represent 20 µm. b Confocal images of whole mount ovaries dissected from ote mutants with heterozygous or homozygous loss of Claspin (Cla). Ovaries were stained for Vasa (red), Lamin Dm0 (green), and DNA (DAPI, blue), revealing the absence of suppression of ote mutant phenotypes. Genotypes are indicated at the top of each image, with ote−/− corresponding to oteB279G/PK; scale bars represent 100 µm
We conducted genetic epistasis studies to test the role of DNA damage in checkpoint activation. Phosphorylation of H2AX occurs upstream of Chk2 activity and depends upon the ATM/ATR kinases38. Thus, we reasoned that if DNA damage triggered the DDR pathway, then chk2−/−, ote−/− GSCs would maintain elevated frequencies of γ-H2Av accumulation similar to those found in ote−/− GSCs. Contrary to this prediction, we found that the frequency of γ-H2Av staining in chk2−/−, ote−/− GSCs reverted to wild-type (7.8%), even though all chk2−/−, ote−/− germaria were γ-H2Av-positive in the meiotic zone (Fig. 4a). We note that this response differs from one elicited by DNA damage, wherein chk2−/− GSCs retain elevated frequencies of γ-H2Av signal31. These surprising results were reproduced using a second, independently generated γ-H2Av antibody (Supplementary Fig. 2A). Our data indicate that increased levels of γ-H2Av in ote−/− GSCs are downstream of Chk2, suggesting that DNA damage is not the checkpoint trigger.
In complementary analyses, we investigated whether loss of Ote compromised DNA repair. Unrepaired meiotic DSBs cause retention of γ-H2Av staining within the oocyte nucleus of developing egg chambers [spnB mutants in Supplementary Fig. 2B)39. We examined γ-H2Av staining in chk2−/−, ote−/− oocyte nuclei of similarly aged egg chambers, finding that the γ-H2Av signal was lost (Supplementary Fig. 2B). These studies show that loss of Ote does not affect meiotic DSB repair.
Replication stress activates ATR and Chk2 in the Drosophila germline40. Notably, mutations in lamin disrupt the distribution of replication factories and induce replication fork arrest, causing replication stress28,41. Motivated from the deformed NL in ote−/− GSCs (Fig. 1b), we tested whether loss of Ote induces replication stress. To this end, we assessed the role of Claspin, a conserved component of DNA replication stress pathways42,43. In Drosophila, Claspin-mediated signaling occurs in response to incomplete DNA replication, but not DNA DSBs43. Furthermore, replication stress in the ovary produces atrophied ovaries, defects that are suppressed by loss of Claspin40. Using previously tested alleles40 (Supplementary Table 1), we generated ote−/−; claspin–/+ and ote−/−; claspin−/− females and assessed ovarian phenotypes. Unlike the dramatic rescue observed with ATR and Chk2 loss, the ote, claspin double-mutant ovaries remained rudimentary, with nearly 40% of germaria devoid of germ cells (Fig. 4b). Whereas GSC loss resulting from replication stress is partially dependent on p5340, GSC loss in ote−/− mutants is p53-independent (Fig. 3b, d). Based on these data, we conclude that replication stress is not responsible for induction of ATR/Chk2-dependent GSC loss in ote mutants.
We next explored other possible mechanisms known to activate ATR/Chk2. Germline activation of these kinases has been linked to transposon de-repression and mobilization44–46. The presence of transposon-mapped probes within our microarrays provided an opportunity to investigate effects of Ote loss on transcription of transposons. Of the 73 transposons annotated on the array, only two demonstrated modest differences in chk2, ote double-mutant ovaries, corresponding to gypsy (3.4-fold downregulation) and springer (2.1-fold upregulation). As with the protein-coding genes, unsupervised hierarchical clustering did not reveal an ote mutant signature in transposon expression (Fig. 5a), implying that transposons remain stable and do not activate the checkpoint in ote−/− ovaries. To complement these analyses, we directly examined the expression and function of several piRNA-regulated transposons (47,48; Fig. 5b). These included the specialized retrotransposons that maintain Drosophila telomeres (HeT-A and TART) because telomere damage activates Chk249 and increased transcription of telomeric transposons is a hallmark of telomere dysfunction50. We completed quantitative reverse transcriptase PCR (RT-PCR), using two wild-type and two ote mutant backgrounds, with animals of three of these genetic backgrounds generated from a cross with the same oteB279G stock. In this way, differences in transposon position and numbers were minimized. We reasoned that if loss of Ote caused transposon mis-regulation, then both ote mutant backgrounds should show consistent changes in gene expression. Our studies revealed largely unchanged levels of retrotransposon and telomeric RNAs in ote mutants relative to controls, with no transposon showing significant changes in both mutant backgrounds (Fig. 5b). Further, we examined telomere organization directly. Drosophila telomeres are capped with the Terminin complex, composed of HP1a, HipHop, and HOAP51. GSCs have 7–14 Terminin foci, depending on the stage of the cell cycle24,52. Using HipHop and HOAP antibodies, we observed no significant difference between ote–/+ and ote−/− profiles (Fig. 5c). Taken together, these data argue that ote mutants retain transposon stability and telomere function, implying these factors do not induce Chk2 activity in ote mutant GSCs.
Fig. 5.

Transposon transcription and telomere capping are unchanged in ote mutants. a Heat map of the log2 signal intensities of RNAs corresponding to D. melanogaster transposons (indicated to the left); unsupervised clustering reveals no segregation by genotype. The genotypes are shown below. b Quantitative RT-PCR analysis of transposon RNAs in the ovary. For Ote-containing genotypes (light and dark gray), ovaries were dissected from less than two-hour old females, and for ote null genotypes (blue and red), ovaries were dissected from 3-day-old females. Transposons analyzed are noted below (red indicates germline transposons and green indicates somatic transposons). RNAs were normalized to vasa and fold change is shown relative to ote+/+. Error bars indicate standard deviation from three biological replicates (*p < 0.05, Student’s t-test comparing RNAs from ote mutant ovaries to RNAs from oteB279G/+ ovaries). c Violin plots of the number of HipHop foci (left) or HOAP foci (right) per GSC nucleus in 1-day-old females. Numbers of GSCs analyzed are noted above each plot. Significance was assessed by Student’s t-test; n.s., not significant
NL defects persist in chk2, ote mutant GSCs
Altered chromatin and chromosome structures can promote DNA damage-independent DDR signaling53–55. These observations prompted our investigation of whether heterochromatin coalescence represents the trigger of Chk2-dependent GSC loss in ote mutants. Although tools for overexpression of HP1a in the male germline exist56, these did not establish overexpression of HP1a in the female germline. Instead, we used a complementary approach to promote heterochromatin aggregation. Overexpression of the multi-AT hook satellite DNA binding protein D1 induces ectopic chromosome associations57, suggesting that elevated levels of D1 in GSCs might promote centromeric heterochromatin interactions and coalescence. To this end, we overexpressed D1 using a nos-gal4:VP16 driver to direct expression of the D1EP473 responder allele (Supplementary Fig. 3A), an allele that carries a GAL4-dependent P{EP} element in the 5′ UTR of the D1 gene57. Strikingly, nos-gal4:VP16, D1EP473 GSCs had elevated D1 levels and HP1a aggregation, similar to levels found in ote-/- GSCs (Fig. 6a, b, Supplementary Fig. 3A). Even so, the NL structure was unaffected, as shown by Ote staining (Fig. 6a). Despite increased HP1a coalescence, GSCs survive, γH2Av levels are low (6.3%) and oogenesis is normal (Fig. 6a, Supplementary Fig. 3B). These observations indicate that HP1a coalescence alone does not cause Chk2-dependent germ cell death. Although D1 and Ote loss might affect HP1a chromatin structure in different ways, mechanistically distinct perturbations of chromatin structure elicit DDR responses53–55 suggesting that, in GSCs, chromatin-based mechanisms of DDR signaling are not responsible for checkpoint activation.
Fig. 6.

HP1a coalescence does not lead to increased γH2Av staining or GSC loss. a Confocal images of germaria and GSC nuclei in females carrying either the nosgal4:vp16 driver alone (top) or the nosgal4:vp16 and the D1EP473 allele (bottom). Ovaries were stained with antibodies against HP1a (green), γ-H2Av (white), and Ote (red). The percentage of γ-H2Av-positive GSCs is noted at the bottom of the GSC image, with the number of GSCs analyzed in parenthesis. Scale bars represent 5 μm. b Box plots of the quantification of HP1a foci per GSC nucleus found in GSCs of the indicated genotype. Genotypes are noted below each box plot, and the number of nuclei analyzed is noted above each top whisker. An A indicates absence of the nosgal4:vp16 driver or the D1EP allele, and P indicates their presence. For each box plot, the box represents the 25th to 75th percentile interval, the line represents the median and the whiskers represent the 5th to 95th percentile interval and non-outlier range. Asterisks indicate significance (one-way ANOVA, ns = not significant, **<0.01, ***<0.001, ****<0.0001). c Confocal images of a GSC nucleus from 1-day-old chk2−/−, ote−/− females stained for Lamin Dm0 (red) and HP1a (green), revealing that nuclear structural defects are upstream of Chk2. Genotypes are listed above each image. The chk2−/− corresponds to chk2P6/chk2P30 and ote−/− corresponds to oteB279G/PK. The box outlines GSCs. Scale bars represent 5 μm
We investigated whether nuclear defects remain in ote−/−, chk2−/− ovaries (Fig. 6c). Quantification of HP1a foci revealed an intermediate phenotype in ote−/−, chk2−/− GSCs (Fig. 6b). These data suggest that Chk2 is upstream of HP1a coalescence and might contribute to heterochromatin clustering. In contrast, NL deformities remain in ote−/−, chk2−/− GSCs (Fig. 6c), implying that these defects are upstream of Chk2. Taken together, our collective epistasis studies suggest that alteration of NL structure is the primary event that leads to checkpoint activation in ote mutants.
Chk2 rescue reveals cell-type specificity of NL checkpoint
Requirements for Ote extend beyond the female germline. Males lacking Ote also display age-dependent GSC loss and sterility16. In the testis, spermatogenesis initiates within a single stem cell niche, called the hub, that supports two stem cell populations, germline stem cells (GSCs) and cyst stem cells (CySCs). Both stem cell populations undergo asymmetric division to produce differentiating daughters that move away from the hub, forming a unit of two post-mitotic somatic cyst cells and one germ cell. Subsequent mitotic and meiotic germ cell divisions produce a unit comprised of 64 spermatids that proceed to differentiate into sperm. To determine whether male germline defects are caused by checkpoint activation, two assays were completed. First, testes phenotypes were defined. As reported previously16, 6-day-old wild-type testes had Vasa-positive germ cells at all stages of spermatogenesis, whereas 6-day-old ote−/− testes showed GSC loss and blocked differentiation (Fig. 7a). In the aged chk2−/−, ote−/− testes, we found that GSCs were retained and germ cells differentiated (Fig. 7a), indicating rescued gametogenesis. Second, male fertility was assessed using a sperm depletion assay16. Whereas only ~30% of 2-week-old ote−/− males remained fertile, 2-week-old chk2, ote double-mutant males showed dose-dependent improvement of fertility, with chk2−/−, ote−/− males reaching wild-type levels (Fig. 7b). These data reveal a Chk2-dependent GSC loss and sterility in ote−/− males. Strikingly, ote−/− male GSCs have an irregular and thickened NL (Supplementary Fig. 4), consistent with NL dysfunction as a primary event in checkpoint activation.
Fig. 7.

Checkpoint inhibition rescues male GSC, but not somatic defects in ote mutants. a Confocal images of testes from 6-day-old males stained for Vasa (red) and Spectrin (green). Genotypes are noted above the images. The GSC niche is denoted by an asterisk. b Percentage of fertile 12–15-day-old males mated to ote+/+ virgin females. Males were determined to be fertile if greater than 5 offspring were produced over a 3-day mating period as described in ref. 16. Genotypes are noted below, and the number of males analyzed is noted above each data set. Error bars indicate the standard deviation from a minimum of three independent experiments. c Confocal images of the central nervous system and surrounding imaginal discs dissected from wandering third instar larvae stained for DAPI (gray) and antibodies against phosphorylated serine 10 of Histone H3 (H3 S10ph; red). Genotypes are noted above each image. All scale bars are 100 µm. d Percent expected class from the following cross: y1,w67c23; chk2P6,otePK/CyO; bocksΔ66/Tm6b,Tb1 crossed to y1,w67c23; chk2P30,oteB279G/CyO; bocksΔ10/Tm6b,Tb1. The genotype of the progeny is noted below and the number of progeny eclosed over the expected number is noted above
Ote is required for the development of somatic tissues. This requirement was uncovered in genetic studies testing overlapping functions of NL LEM-D proteins13. Indeed, ote−/−; bocks−/− double mutants die during larval development, carrying rudimentary organs and imaginal discs, with few mitotic cells (Fig. 7c). To determine whether lethality in the ote, bocks double mutants is Chk2-dependent, we assessed effects of its loss. To this end, we generated chk2−/−, ote−/−; bocks−/− triple mutant animals. Based on the number of heterozygous sibling offspring (331) recovered from a cross of trans-heterozygous parents, 83 triple mutant adult offspring were expected if lethality were fully rescued. In contrast to this prediction, no chk2−/−, ote−/−; bocks−/− adults were found (Fig. 7d). In addition, chk2−/−, ote−/−; bocks−/− triple mutant larvae carried rudimentary brains and imaginal discs with reduced levels of mitotic nuclei, revealed with antibodies against phosphorylated serine 10 of Histone H3 (H3 S10ph; Fig. 7c). Indeed, chk2−/−, ote−/−; bocks−/− triple mutant phenotypes were indistinguishable from ote−/−; bocks−/− double mutants. These data show that Chk2 loss is not sufficient to rescue the somatic requirements of Ote and Bocks, demonstrating GSC-specific rescue.
Discussion
The Drosophila emerin homolog Ote has an essential requirement for GSC survival and germ cell differentiation12,16. Here, we show that Ote loss causes GSC-specific nuclear defects that include a thickened and irregular NL and aggregation of heterochromatin (Fig. 1). Strikingly, inactivation of two DDR kinases, ATR, and Chk2, rescues oogenesis in ote−/− females (Figs. 2, 3), a rescue that is cell-type specific (Fig. 7). Genetic and cytological features of the checkpoint pathway present in ote mutant GSCs differ from those found in canonical DDR pathways (Figs. 3, 4, 5;58). In addition, although heterochromatin coalesce is present, such defects by themselves do not trigger the checkpoint (Fig. 6, S3). Instead, our data correlate Chk2 activity with defects in NL structure (Fig.1, S4), indicating that NL dysfunction is responsible for the activation of a checkpoint pathway in GSCs (Fig. 8). Despite remarkably normal oogenesis, rescued oocytes do not support embryogenesis (Fig. 2e). We suggest this NL checkpoint pathway functions in GSCs to ensure that only healthy gametes are passed on to the next generation.
Fig. 8.
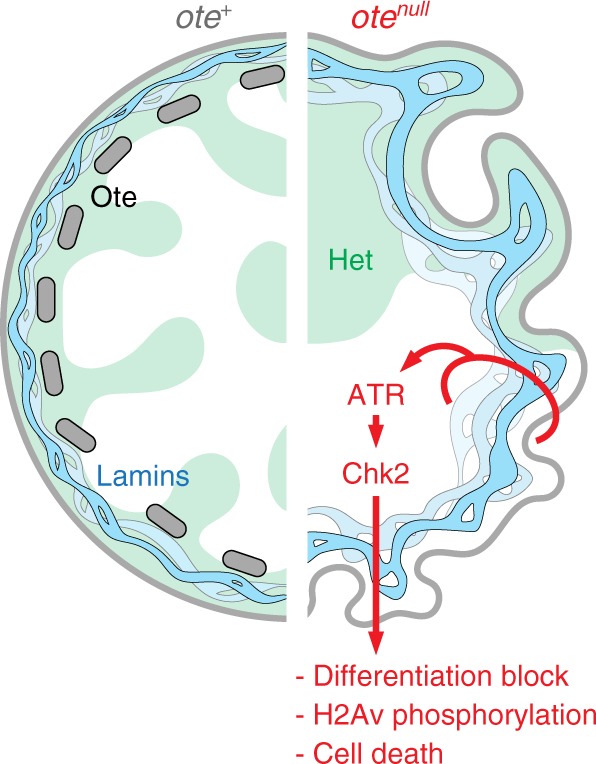
Loss of Otefin activates a NL checkpoint in GSCs. In ote+/+ GSCs (left side), Ote (gray bars) resides within the NL, stabilizing NL structure (blue ribbon) and heterochromatin (green shading) contacts. In ote−/− GSCs (right side), the NL becomes thick and deformed, causing NL dysfunction that activates a checkpoint pathway. This pathway uses the ATR and Chk2 kinases, leading to blocked differentiation, H2Av phosphorylation, and, ultimately, death of GSCs. As loss of ATR/Chk2 in ote mutants rescues oogenesis, but not embryogenesis, we suggest this NL checkpoint pathway functions to ensure that healthy gametes are passed on to the next generation
Our studies identify ATR as the critical responder kinase and Chk2 as the critical transducer kinase in the NL checkpoint (Figs. 2, 3, and 6). This signaling axis differs from the canonical ATM-to-Chk2 or ATR-to-Chk1 axes58. Several factors might contribute to the choice of responder and transducer kinase. First, species-specific constraints might exist. ATR, but not ATM is essential in mammals, whereas ATM, but not ATR, is essential in Drosophila59,60. Second, cell-type specific distinctions are apparent. In both the fly and mouse germline, persistent meiotic double-strand breaks activate ATR and Chk245,61, implying that the ATR–Chk2 axis might be dominant in germ cells. Third, the nature of the trigger might influence which proteins are involved in signaling. For example in Drosophila, ATR and Chk2 are both required for the patterning defects caused by a failure to repair meiotic double-strand breaks62. However, in DNA-damaged GSCs, ATR protects against GSC death, whereas Chk2 promotes it30. Our data suggest that in the case of the NL checkpoint, both ATR and Chk2 promote germ cell death. These studies add to growing evidence that the DDR pathway is modular, with selective use of pathway components in response to various cellular stresses58.
Activated Chk2 is commonly associated with phosphorylation and activation of p5363. Canonically, p53 activation leads to cell cycle arrest and apoptosis64. Our studies demonstrate that GSC loss persists in ote−/−; p53−/− females (Fig. 3b, d), suggesting that classical apoptosis is not responsible for GSC death. These findings are consistent with the absence of classic markers of apoptosis in ote mutants15 and observations that the p53 regulatory network differs in GSCs65. Recently, an alternative cell death pathway was identified in spermatogonia of Drosophila testes66. This pathway is responsible for spontaneous elimination of spermatogonia, using activated lysosomal and mitochondrial-associated factors. Additional studies are needed to determine whether ATR/Chk2-dependent GSC loss in ote mutants targets a similar pathway.
Our data suggest that NL dysfunction is the primary cause of the ATR/Chk2 checkpoint in GSCs. Notably, NL defects are found only in affected cells and persist in rescued chk2−/−, ote−/− double mutants (Figs. 1 and 6; Supplementary Fig. 4). Multiple mechanisms might connect nuclear architecture changes to ATR/Chk2 activation. First, altered NL structure might change genomic contacts needed for appropriate transcriptional regulation, with resulting gene expression changes prompting activation of the checkpoint. While we did not observe global transcriptional changes during oogenesis (Fig. 2f), identification of transcriptional changes specific to GSCs or early germ cells would have been masked in our studies. Second, disruptions in the NL might affect trafficking of products between the nucleus and cytoplasm. Notably, a recent study identified large ribonucleoparticles (megaRNPs) that exit the nucleus by egress or budding through the inner and outer nuclear membranes67, a process disrupted by defects in the NL68. As such, it remains possible that the thickened NL in ote−/− GSCs disrupts megaRNP egress, leading to cellular stress and ATR/Chk2 activation. Third, defects in the NL structure might alter scaffolding of components of the DDR pathway, leading to checkpoint activation. Indeed, proteomic studies from Drosophila cultured somatic cells found that Ote interacted with proteins involved in DNA replication and repair69, implying that Ote might assemble responder and transducer kinases complexes at the NL. However, observations that the ATR/Chk2-dependent checkpoint is GSC-specific (Fig. 7c, d), coupled with findings that meiotic double-strand breaks are repaired appropriately in chk2−/−, ote−/− germaria (Supplementary Fig. 2B), argue against this model. Fourth, structural alteration in the nuclear envelope itself might trigger ATR/Chk2 activation70. Indeed, emerging evidence implicates ATR as a general sensor of the structural integrity of cellular components71. Further studies are needed to identify how NL dysfunction triggers the GSC-specific checkpoint.
Mutations in NL LEM-D proteins cause dystrophic diseases. Much evidence suggests that these diseases result from compromised stem cell populations that underlie the defects in tissue homeostasis72–75. Indeed, a wealth of evidence links NL defects to increased DNA damage20,22,41,76,77. Our data are consistent with these reports, as we show that elevated accumulation of the commonly used DNA damage marker. However, we find that phosphorylation of the H2A variant occurs downstream of Chk2, suggesting that accumulation of DNA damage in cells with a dysfunctional NL might be a consequence of cells dying, not the primary cause. These unexpected results suggest that caution is needed in linking causation of γH2Av/H2X accumulation to DNA damage and a failure in DNA repair. Indeed, recent studies of progerin-expressing cells indicated that the cellular defect in Hutchinson–Gilford progeria cells does not lie in defective DNA repair and DNA damage, even though these cells accumulate phosphorylated H2AX78. Our findings establish a new context for consideration of mechanisms of laminopathic diseases, suggesting that detrimental effects of NL dysfunction are primary events that are linked to checkpoint activation and stem cell loss.
Methods
Animal culture conditions
Drosophila crosses were incubated at 25 °C at 70% humidity on standard cornmeal/agar medium with p-hydroxybenzoic acid methyl ester as a mold inhibitor. For fecundity assays, animals were supplied dry active yeast pellets. The wild-type reference strain was y1, w67c23. In all cases, ote mutant animals were y1, w67c23; oteB279G/PK, wherein both alleles fail to generate Ote protein12. The oteB279G allele carries an insertion of a Piggybac transposon at +764 and otePK carries a premature stop at codon 12712. Other alleles used are listed in Supplementary Table 1, and genotyping primers are listed in Supplementary Table 3. In all cases, mutant animals carried heteroallelic combinations of alleles to avoid complications associated with homozygous chromosomes that carry second site mutations.
Immunohistochemical analyses
Adult Drosophila ovaries were dissected and stained as described in ref. 79. Wandering third instar larval tissues were processed similarly to adult ovaries with one additional permeabilization step with 0.1% TritonX-100 in 1X PBS for 1 h at room temperature immediately following fixation. Primary antibodies were diluted in 5% BSA, 0.3% TritonX-100 in PBS and are listed in Supplementary Table 2. Alexa Fluor-conjugated secondary antibodies were used at a 1:500 dilution. All images were collected on a Zeiss LSM 710 Confocal Microscope, cropped and rotated using ImageJ software, size matched with Adobe Photoshop and assembled with Adobe Illustrator. The number of HP1a foci was quantified manually using ImageJ. Unless otherwise noted, all images are a single slice of a confocal stack.
Analyses of ovarian phenotypes
Germarial phenotypes were quantified in 2-day-old females as described in12. We analyzed four to five germaria from one ovary, at least five different pairs of ovaries for each biological replicate, and at least two to three biological replicates. For female fecundity assays, 7 to 15 1-day-old females and 4 to 8 males were placed in egg collection bottles with orange juice and agar egg collection plates (90% orange juice, 0.9% agar, 1% ethyl acetate) with wet yeast. Egg collection plates were changed every 24 h. Fecundity is reported as eggs laid per female per day. If a female died during the assay, it was assumed to die minutes before changing of the egg collection plate such that the number of females in the bottle changed in the subsequent 24-h collection period. Egg collection plates were incubated at 25 °C for an additional 30 to 40 h to quantify the number of hatched progeny.
RNA analyses
For qRT-PCR analyses, biological replicates of 25 ovary pairs were dissected in PBS on ice and stored at −80 °C. To normalize for the difference in ovary size, ovaries were dissected from <2-h-old Ote-containing and 3-day-old ote mutant females. RNA was isolated with TRIzol (Invitrogen) and treated with DNaseI using DNA-free kit (Ambion). cDNA was generated using the High Capacity cDNA kit with random hexamer primers (Applied Biosystems) and diluted fivefold prior to setting up qPCR reactions. Primers used in this study are listed in Supplementary Table 4. Cycle threshold levels were normalized to housekeeping gene RpL32, or the germline-restricted gene vasa, and fold enrichment was calculated using the ΔΔCt method80. Statistical significance was tested by comparing fold change of ote mutant samples to those of ote−/+ samples.
For microarray analyses, two biological replicates of 100 ovary pairs per genotype were dissected in PBS and stored at −80 °C. Four genotypes were studied, including two chk2+/−, ote−/− (null) backgrounds [chk2P30, oteB279G/ chk2+, otePK and chk2P6, otePK/ chk2+, oteB279G] and their chk2+/−, ote+/− (het) siblings [chk2P30, oteB279G/chk2+, ote+ and chk2P6, otePK/ chk2+, ote+]. Ovaries were dissected from 1-day-old females. RNA was isolated with TRIzol (Invitrogen). DNaseI treatment (Qiagen DNaseI) was done on RNeasy columns (Qiagen) during RNA purification. RNA sample quality was assessed with a Agilent 2100 Bioanalyzer prior to hybridization. Affymetrix Drosophila 2.0 arrays (900532) were used for microarray hybridization (University of Iowa Genomics Division). Array files were processed and analyzed using Affymetrix Transcriptome Analyses Console version 4.0, using default settings and RMA normalization, and contrasting four samples per group (otenull vs otehet), using two-fold change and p < 0.05 (one-way ANOVA) cutoffs.
Sperm exhaustion assay
A sperm exhaustion assay assessed male fertility, completed as described in ref. 16. Briefly, less than 1-day-old males were collected and individually crossed to three new newly eclosed wild-type virgin females every 3 days until the males reached an age of 15 days. A male that generated five or more progeny in 3 days was considered fertile at that age window.
Viability analysis
To determine whether Chk2 loss rescued somatic defects associated with the absence of Ote, we assessed the viability of chk2, ote; bocks triple mutant animals. In these studies, we crossed y1w67c23; chk2P6, otePk/CyO; bocksΔ66/Tm6b,Tb1 males to y1w67c23; chk2P30, oteB279-G/CyO; bocksΔ10/Tm6b,Tb1 females. Progeny were genotyped and counted from each vial, until no progeny eclosed for 48 h.
Statistical analysis
For microarray analyses and HP1 foci quantification, statistical significance was assessed using one-way ANOVA. For all other data, statistical significance was tested using a two-tailed Student’s t-test.
Electronic supplementary material
Acknowledgements
We thank Eric Schultz for technical assistance. We thank members of the Geyer and Lehmann labs for helpful discussions, especially Toby Leiber for critically reading the manuscript. We thank Y. Rong, A. Molla-Herman, J-R Huynh, Y, Yamashita for generously supplying fly stocks, antibodies, and advice. Imaging was supported by the University of Iowa Central Microscopy Research Facility and use of the Zeiss LSM 710 confocal microscope acquired via National Institutes of Health (NIH) funding (S10 RR025439-01). This work was supported by the NIH R01 funding (GM087341) to P.K.G. L.J.B. is a William Raveis Charitable Fund Fellow of the Damon Runyon Cancer Research Foundation (DRG-2235-15). A.A.S. is HHMI Fellow of the Damon Runyon Cancer Research Foundation (DRG-2185-14).
Author contributions
K.E.L., W.K., A.L., T.D., L.J.B., and P.K.G. designed the experiments and contributed to data analysis. K.E.L., W.K., T.D., L.J.B., A.L. carried out the experiments. A.A.S. assisted in microarray analyses and figure preparation. L.J.B. and P.K.G. wrote the manuscript.
Data availability
The microarray data set generated in the current study are available at Gene Expression Omnibus under the accession number GSE95309.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41467-018-06277-z.
References
- 1.Wilson KL, Foisner R. Lamin-binding proteins. Cold Spring Harb. Perspect. Biol. 2010;2:a000554. doi: 10.1101/cshperspect.a000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barton LJ, Soshnev AA, Geyer PK. Networking in the nucleus: a spotlight on LEM-domain proteins. Curr. Opin. Cell Biol. 2015;34:1–8. doi: 10.1016/j.ceb.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brachner A, Foisner R. Evolvement of LEM proteins as chromatin tethers at the nuclear periphery. Biochem. Soc. Trans. 2011;39:1735–1741. doi: 10.1042/BST20110724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samwer M, et al. DNA cross-bridging shapes a single nucleus from a set of mitotic chromosomes. Cell. 2017;170:956–972 e923. doi: 10.1016/j.cell.2017.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Aguilera C, et al. Genome-wide analysis links emerin to neuromuscular junction activity in Caenorhabditis elegans. Genome Biol. 2014;15:R21. doi: 10.1186/gb-2014-15-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrales RR, Forn M, Georgescu PR, Sarkadi Z, Braun S. Control of heterochromatin localization and silencing by the nuclear membrane protein Lem2. Genes Dev. 2016;30:133–148. doi: 10.1101/gad.271288.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jamin A, Wiebe MS. Barrier to autointegration factor (BANF1): interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr. Opin. Cell Biol. 2015;34:61–68. doi: 10.1016/j.ceb.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobrzynska A, Gonzalo S, Shanahan C, Askjaer P. The nuclear lamina in health and disease. Nucleus. 2016;7:233–248. doi: 10.1080/19491034.2016.1183848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vlcek S, Foisner R. A-type lamin networks in light of laminopathic diseases. Biochim. Biophys. Acta. 2007;1773:661–674. doi: 10.1016/j.bbamcr.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Gotzmann J, Foisner R. A-type lamin complexes and regenerative potential: a step towards understanding laminopathic diseases? Histochem. Cell Biol. 2006;125:33–41. doi: 10.1007/s00418-005-0050-8. [DOI] [PubMed] [Google Scholar]
- 11.Pinto BS, Wilmington SR, Hornick EE, Wallrath LL, Geyer PK. Tissue-specific defects are caused by loss of the drosophila MAN1 LEM domain protein. Genetics. 2008;180:133–145. doi: 10.1534/genetics.108.091371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barton LJ, Pinto BS, Wallrath LL, Geyer PK. The Drosophila nuclear lamina protein otefin is required for germline stem cell survival. Dev. Cell. 2013;25:645–654. doi: 10.1016/j.devcel.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barton LJ, et al. Unique and shared functions of nuclear lamina LEM domain proteins in Drosophila. Genetics. 2014;197:653–665. doi: 10.1534/genetics.114.162941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner N, Kagermeier B, Loserth S, Krohne G. The Drosophila melanogaster LEM-domain protein MAN1. Eur. J. Cell Biol. 2006;85:91–105. doi: 10.1016/j.ejcb.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Jiang X, et al. Otefin, a nuclear membrane protein, determines the fate of germline stem cells in Drosophila via interaction with Smad complexes. Dev. Cell. 2008;14:494–506. doi: 10.1016/j.devcel.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 16.Barton LJ, Lovander KE, Pinto BS, Geyer PK. Drosophila male and female germline stem cell niches require the nuclear lamina protein Otefin. Dev. Biol. 2016;415:75–86. doi: 10.1016/j.ydbio.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Losick VP, Morris LX, Fox DT, Spradling A. Drosophila stem cell niches: a decade of discovery suggests a unified view of stem cell regulation. Dev. Cell. 2011;21:159–171. doi: 10.1016/j.devcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taimen P, et al. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc. Natl Acad. Sci. USA. 2009;106:20788–20793. doi: 10.1073/pnas.0911895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J. Cell Sci. 2006;119:4644–4649. doi: 10.1242/jcs.03263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varela I, et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–568. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
- 22.Liu B, et al. Genomic instability in laminopathy-based premature aging. Nat. Med. 2005;11:780–785. doi: 10.1038/nm1266. [DOI] [PubMed] [Google Scholar]
- 23.Shimi T, Goldman RD. Nuclear lamins and oxidative stress in cell proliferation and longevity. Adv. Exp. Med. Biol. 2014;773:415–430. doi: 10.1007/978-1-4899-8032-8_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joyce EF, Apostolopoulos N, Beliveau BJ, Wu CT. Germline progenitors escape the widespread phenomenon of homolog pairing during Drosophila development. PLoS Genet. 2013;9:e1004013. doi: 10.1371/journal.pgen.1004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christophorou N, Rubin T, Huynh JR. Synaptonemal complex components promote centromere pairing in pre-meiotic germ cells. PLoS Genet. 2013;9:e1004012. doi: 10.1371/journal.pgen.1004012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fung JC, Marshall WF, Dernburg A, Agard DA, Sedat JW. Homologous chromosome pairing in Drosophila melanogaster proceeds through multiple independent initiations. J. Cell Biol. 1998;141:5–20. doi: 10.1083/jcb.141.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lans H, Hoeijmakers JH. Cell biology: ageing nucleus gets out of shape. Nature. 2006;440:32–34. doi: 10.1038/440032a. [DOI] [PubMed] [Google Scholar]
- 28.Spann TP, Moir RD, Goldman AE, Stick R, Goldman RD. Disruption of nuclear lamin organization alters the distribution of replication factors and inhibits DNA synthesis. J. Cell Biol. 1997;136:1201–1212. doi: 10.1083/jcb.136.6.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shumaker DK, et al. The highly conserved nuclear lamin Ig-fold binds to PCNA: its role in DNA replication. J. Cell. Biol. 2008;181:269–280. doi: 10.1083/jcb.200708155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma X, et al. DNA damage-induced CHK2 activation compromises germline stem cell self-renewal and lineage differentiation. Development. 2016;143:4312–4323. doi: 10.1242/dev.141069. [DOI] [PubMed] [Google Scholar]
- 31.Ma X, et al. Aubergine controls germline stem cell self-renewal and progeny differentiation via distinct mechanisms. Dev. Cell. 2017;41:157–169. doi: 10.1016/j.devcel.2017.03.023. [DOI] [PubMed] [Google Scholar]
- 32.Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014;6:442–457. doi: 10.1093/jmcb/mju045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin H, Yue L, Spradling AC. The Drosophila fusome, a germline-specific organelle, contains membrane skeletal proteins and functions in cyst formation. Development. 1994;120:947–956. doi: 10.1242/dev.120.4.947. [DOI] [PubMed] [Google Scholar]
- 34.Ashery-Padan R, et al. Localization and posttranslational modifications of otefin, a protein required for vesicle attachment to chromatin, during Drosophila melanogaster development. Mol. Cell. Biol. 1997;17:4114–4123. doi: 10.1128/MCB.17.7.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xing Y, Su TT, Ruohola-Baker H. Tie-mediated signal from apoptotic cells protects stem cells in Drosophila melanogaster. Nat. Commun. 2015;6:7058. doi: 10.1038/ncomms8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joyce EF, et al. Drosophila ATM and ATR have distinct activities in the regulation of meiotic DNA damage and repair. J. Cell Biol. 2011;195:359–367. doi: 10.1083/jcb.201104121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lukas J, Lukas C, Bartek J. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 38.Podhorecka, M., Skladanowski, A. & Bozko, P. H2AX phosphorylation: its role in DNA damage response and cancer therapy. J Nucleic Acids2010, 920161 (2010). [DOI] [PMC free article] [PubMed]
- 39.Klattenhoff C, et al. The Drosophila HP1 homolog Rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell. 2009;138:1137–1149. doi: 10.1016/j.cell.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molla-Herman A, Valles AM, Ganem-Elbaz C, Antoniewski C, Huynh JR. tRNA processing defects induce replication stress and Chk2-dependent disruption of piRNA transcription. EMBO J. 2015;34:3009–3027. doi: 10.15252/embj.201591006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh M, et al. Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Mol. Cell Biol. 2013;33:1210–1222. doi: 10.1128/MCB.01676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell. 2000;6:839–849. doi: 10.1016/S1097-2765(05)00092-4. [DOI] [PubMed] [Google Scholar]
- 43.Lee EM, et al. Drosophila Claspin is required for the G2 arrest that is induced by DNA replication stress but not by DNA double-strand breaks. DNA Repair. 2012;11:741–752. doi: 10.1016/j.dnarep.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 44.Theurkauf WE, et al. rasiRNAs, DNA damage, and embryonic axis specification. Cold Spring Harb. Symp. Quant. Biol. 2006;71:171–180. doi: 10.1101/sqb.2006.71.066. [DOI] [PubMed] [Google Scholar]
- 45.Klattenhoff C, et al. Drosophila rasiRNA pathway mutations disrupt embryonic axis specification through activation of an ATR/Chk2 DNA damage response. Dev. Cell. 2007;12:45–55. doi: 10.1016/j.devcel.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y, Pane A, Schupbach T. Cutoff and aubergine mutations result in retrotransposon upregulation and checkpoint activation in Drosophila. Curr. Biol. 2007;17:637–642. doi: 10.1016/j.cub.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sytnikova YA, Rahman R, Chirn GW, Clark JP, Lau NC. Transposable element dynamics and PIWI regulation impacts lncRNA and gene expression diversity in Drosophila ovarian cell cultures. Genome Res. 2014;24:1977–1990. doi: 10.1101/gr.178129.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang SH, Elgin SC. Drosophila Piwi functions downstream of piRNA production mediating a chromatin-based transposon silencing mechanism in female germ line. Proc. Natl Acad. Sci. USA. 2011;108:21164–21169. doi: 10.1073/pnas.1107892109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ciapponi L, Cenci G. Telomere capping and cellular checkpoints: clues from fruit flies. Cytogenet. Genome Res. 2008;122:365–373. doi: 10.1159/000167824. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, Beaucher M, Cheng Y, Rong YS. Coordination of transposon expression with DNA replication in the targeting of telomeric retrotransposons in Drosophila. EMBO J. 2014;33:1148–1158. doi: 10.1002/embj.201386940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raffa GD, Ciapponi L, Cenci G, Gatti M. Terminin: a protein complex that mediates epigenetic maintenance of Drosophila telomeres. Nucleus. 2011;2:383–391. doi: 10.4161/nucl.2.5.17873. [DOI] [PubMed] [Google Scholar]
- 52.Ables ET, Drummond-Barbosa D. The steroid hormone ecdysone functions with intrinsic chromatin remodeling factors to control female germline stem cells in Drosophila. Cell. Stem. Cell. 2010;7:581–592. doi: 10.1016/j.stem.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burgess RC, Burman B, Kruhlak MJ, Misteli T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014;9:1703–1717. doi: 10.1016/j.celrep.2014.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 55.Huyen Y, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 56.Xing Y, Li WX. Heterochromatin components in germline stem cell maintenance. Sci. Rep. 2015;5:17463. doi: 10.1038/srep17463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith MB, Weiler KS. Drosophila D1 overexpression induces ectopic pairing of polytene chromosomes and is deleterious to development. Chromosoma. 2010;119:287–309. doi: 10.1007/s00412-010-0257-x. [DOI] [PubMed] [Google Scholar]
- 58.Burgess RC, Misteli T. Not all DDRs are created equal: non-canonical DNA damage responses. Cell. 2015;162:944–947. doi: 10.1016/j.cell.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 60.Song YH, Mirey G, Betson M, Haber DA, Settleman J. The Drosophila ATM ortholog, dATM, mediates the response to ionizing radiation and to spontaneous DNA damage during development. Curr. Biol. 2004;14:1354–1359. doi: 10.1016/j.cub.2004.06.064. [DOI] [PubMed] [Google Scholar]
- 61.Bolcun-Filas E, Rinaldi VD, White ME, Schimenti JC. Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science. 2014;343:533–536. doi: 10.1126/science.1247671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abdu U, Brodsky M, Schupbach T. Activation of a meiotic checkpoint during Drosophila oogenesis regulates the translation of Gurken through Chk2/Mnk. Curr. Biol. 2002;12:1645–1651. doi: 10.1016/S0960-9822(02)01165-X. [DOI] [PubMed] [Google Scholar]
- 63.Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 2012;13:579–590. doi: 10.1038/nrm3420. [DOI] [PubMed] [Google Scholar]
- 64.Vousden KH, Lane DP. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 65.Wylie A, Lu WJ, D’Brot A, Buszczak M, Abrams JM. p53 activity is selectively licensed in the Drosophila stem cell compartment. eLife. 2014;3:e01530. doi: 10.7554/eLife.01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yacobi-Sharon K, Namdar Y, Arama E. Alternative germ cell death pathway in Drosophila involves HtrA2/Omi, lysosomes, and a caspase-9 counterpart. Dev. Cell. 2013;25:29–42. doi: 10.1016/j.devcel.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 67.Speese SD, et al. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell. 2012;149:832–846. doi: 10.1016/j.cell.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y, et al. Lamin mutations accelerate aging via defective export of mitochondrial mRNAs through nuclear envelope budding. Curr. Biol. 2016;26:2052–2059. doi: 10.1016/j.cub.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guruharsha KG, et al. A protein complex network of Drosophila melanogaster. Cell. 2011;147:690–703. doi: 10.1016/j.cell.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kumar A, et al. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell. 2014;158:633–646. doi: 10.1016/j.cell.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kidiyoor GR, Kumar A, Foiani M. ATR-mediated regulation of nuclear and cellular plasticity. DNA Repair. 2016;44:143–150. doi: 10.1016/j.dnarep.2016.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shimi T, et al. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011;25:2579–2593. doi: 10.1101/gad.179515.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chandra T, et al. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015;10:471–483. doi: 10.1016/j.celrep.2014.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barascu A, et al. Oxydative stress alters nuclear shape through lamins dysregulation: a route to senescence. Nucleus. 2012;3:411–417. doi: 10.4161/nucl.21674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dreesen O, et al. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013;200:605–617. doi: 10.1083/jcb.201206121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hutchison CJ. The role of DNA damage in laminopathy progeroid syndromes. Biochem. Soc. Trans. 2011;39:1715–1718. doi: 10.1042/BST20110700. [DOI] [PubMed] [Google Scholar]
- 77.Butin-Israeli V, et al. Role of lamin b1 in chromatin instability. Mol. Cell Biol. 2015;35:884–898. doi: 10.1128/MCB.01145-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deniaud E, Lemaitre C, Boyle S, Bickmore WA. Expression of progerin does not result in an increased mutation rate. Chromosome Res. 2017;25:227–239. doi: 10.1007/s10577-017-9556-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baxley RM, Soshnev AA, Koryakov DE, Zhimulev IF, Geyer PK. The role of the Suppressor of Hairy-wing insulator protein in Drosophila oogenesis. Dev. Biol. 2011;356:398–410. doi: 10.1016/j.ydbio.2011.05.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray data set generated in the current study are available at Gene Expression Omnibus under the accession number GSE95309.