

Abstract
Calcineurin (CN) is a Ca2+/calmodulin-dependent protein phosphatase with high abundance in nervous tissue. Though enriched in neurons, CN can become strongly induced in subsets of activated astrocytes under different pathological conditions where it interacts extensively with the nuclear factor of activated T cells (NFATs). Recent work has shown that regions of small vessel damage are associated with the upregulation of a proteolized, highly active form of CN in nearby astrocytes, suggesting a link between the CN/NFAT pathway and chronic cerebrovascular disease. In this Mini Review article, we discuss CN/NFAT signaling properties in the context of vascular disease and use previous cell type-specific intervention studies in Alzheimer’s disease and traumatic brain injury models as a framework to understand how astrocytic CN/NFATs may couple vascular pathology to neurodegeneration and cognitive loss.
Keywords: vascular contributions to cognitive impairment and dementia, Ca2+, glia, excitotoxicity, Alzheimer’s disease
Introduction
Cerebrovascular pathology is one of the leading causes of cognitive loss and mortality. While stroke is usually the most devastating form of cerebrovascular disease, other forms of vascular damage and dysfunction including microinfarcts, microhemorrhages, cerebral amyloid angiopathy and cerebral hypoperfusion are more insidious and can lead to chronic and progressive cognitive loss, especially in aged individuals. These vascular contributions to cognitive impairment and dementia (VCID) are the second leading cause of dementia, behind Alzheimer’s disease, and frequently co-exist with other neurodegenerative conditions (O’Brien et al., 2003). Importantly, VCID comorbidities appear to interfere with the treatment of Alzheimer’s disease-related functional deficits in animal models (Weekman et al., 2016), highlighting the need to understand the cellular mechanisms that link vascular dysfunction to neurodegeneration and impaired cognition (Snyder et al., 2015; Horsburgh et al., 2018).
Brain ischemia results when stroke or other forms of VCID block the blood supply to parts of the brain, resulting in depletion of oxygen and glucose. This depletion rapidly exhausts the energy production of neural cells and their ability to maintain the normal balance of ions across cellular membranes, thus causing excitotoxicity and Ca2+ overload, among other adverse effects (Choi, 1988; Horst and Postigo, 1996; Szydlowska and Tymianskia, 2010). Ca2+ overload originates from a variety of sources and directly affects numerous intracellular signaling cascades, many of which have been explored as potential treatment targets for stroke and other forms of cerebrovascular disease (Harris et al., 1982; Infeld et al., 1999; Ray, 2006; Mattson, 2007; Rostas et al., 2017; Wu and Tymianski, 2018). In most cases, Ca2+-signaling pathways have been investigated in neurons, which are the primary target of excitotoxic damage. In the following Mini Review article, we will discuss the importance of the Ca2+/calmodulin dependent protein phosphatase, calcineurin (CN) and its dysregulation in astrocytes as a pathological mechanism and potential target for neurodegeneration and cognitive loss due to cerebrovascular damage.
CN Dysregulation in Stroke Models
CN, or protein phosphatase 3, is the only phosphatase in mammals that is directly activated by Ca2+/calmodulin. CN consists of a catalytic subunit (PPP3CA) and a Ca2+ binding regulatory subunit (PPP3R1). When cellular Ca2+ levels are low, the phosphatase activity of CN is held in check by an autoinhibitory domain located near the C terminus of the catalytic subunit. The interaction of Ca2+ with the CN regulatory subunit and calmodulin leads to a physical interaction between the CN catalytic subunit and Ca2+/calmodulin, which, in turn, displaces the AID and frees the catalytic core from inhibition. When cellular Ca2+ levels fall, calmodulin is released from the catalytic subunit and AID-mediated inhibition of phosphatase activity is reinstated (Klee et al., 1998; Aramburu et al., 2000). In healthy nervous tissue, CN provides an essential mechanism for bidirectional synaptic plasticity through the induction and maintenance of activity-dependent synaptic depression (Mansuy, 2003). In this capacity, CN is widely thought to link Ca2+ signaling to several forms of learning and memory, including extinction learning (Baumgärtel et al., 2008; de la Fuente et al., 2011; Rivera-Olvera et al., 2018). However, due to its exquisite sensitivity to Ca2+, CN is also frequently identified as a central player in numerous deleterious or maladaptive processes arising from Ca2+ overload and/or dysregulation (Uchino et al., 2008; Mukherjee and Soto, 2011; Reese and Taglialatela, 2011; Furman and Norris, 2014; Sompol and Norris, 2018).
Large and/or sustained surges in Ca2+ can lead to calpain or caspase-mediated proteolytic disruption of the CN AID (Wang et al., 1989; Wu et al., 2004), which partially and irreversibly uncouples CN from Ca2+, resulting in constitutive phosphatase activity. Several acute and chronic neurodegenerative conditions are associated with the generation of high activity CN proteolytic fragments (ΔCN), thus perpetuating de-phosphorylation of the myriad of CN targets (Norris, 2014). Hypoxic/ischemic insults appear to be particularly effective at triggering the proteolysis of CN from its full length highly-regulated form (60 kDa), to high activity fragments (ΔCN) ranging in size from 45 to 57 kDa (Shioda et al., 2006, 2007; Rosenkranz et al., 2012). Conversely, blockade of CN typically provides considerable neuroprotection during ischemia and other adverse consequences of cerebrovascular damage. For instance, the CN inhibiting immunosuppressant drug, tacrolimus (or FK506), has been shown to reduce infarct size (Sharkey and Butcher, 1994; Butcher et al., 1997), suppress neuroinflammation (Zawadzka and Kaminska, 2005) and promote recovery of function (Sharkey et al., 1996) in middle cerebral artery occlusion models of ischemic stroke. More recently, a CN modulatory protein, known as regulator of CN (RCAN), was found to favorably affect the pathogenesis of stroke in vivo and hypoxia in vitro using both gene overexpression and knockout approaches (Brait et al., 2012; Sobrado et al., 2012). Together, these results suggest that CN proteolysis (hyperactivation) is not only a biomarker, but also an important mediator, of neurodegeneration resulting from vascular damage.
NFATs
The exact mechanisms through which CN acts are complex and multifaceted. CN has a broad and diverse range of substrates, many of which have been implicated as downstream targets in CN-mediated cellular dysfunction and neurotoxicity (Uchino et al., 2008; Mukherjee and Soto, 2011; Reese and Taglialatela, 2011; Furman and Norris, 2014). Perhaps the best characterized substrate of CN is the nuclear factor of activated T cells (NFATs), a transcription factor related to NFκB/Rel-family proteins (Rao et al., 1997). There are four CN-dependent NFAT family members (NFATs 1–4), all of which are expressed in nervous tissue (Nguyen and Di Giovanni, 2008; Vihma et al., 2008). NFATs reside in the cytosol in their resting state, but upon de-phosphorylation by CN, they translocate to the nucleus where they can activate or suppress numerous gene expression programs linked to immune/inflammatory signaling, Ca2+ regulation, and cell survival, among other things (Im and Rao, 2004). NFAT isoforms have different cellular distributions inside and outside of the nervous system (Horsley and Pavlath, 2002; Abdul et al., 2010) and appear to engage in both overlapping and distinct transcriptional programs through interactions with multiple other transcription factor families (Rao et al., 1997; Im and Rao, 2004; Wu et al., 2006). Of the four isoforms, NFATs 1 and 4 seem to show a greater bias for glial cells where they respond to many different kinds of inflammatory factors and other noxious stimuli, including blood derived factors (Canellada et al., 2008; Sama et al., 2008; Abdul et al., 2009; Nagamoto-Combs and Combs, 2010; Serrano-Pérez et al., 2011; Neria et al., 2013; Furman et al., 2016; Manocha et al., 2017; Sompol et al., 2017).
Hyperactive Astrocytic CN/NFAT Signaling: Biomarker for Vascular Damage?
Astrocytic CN/NFAT signaling may provide, and give rise to, useful biomarkers for cerebrovascular damage. One of the most striking changes in CN/NFAT expression following CNS injury and disease is strong and selective expression in subsets of activated astrocytes (Hashimoto et al., 1998; Norris et al., 2005; Celsi et al., 2007; Serrano-Pérez et al., 2011; Lim et al., 2013; Neria et al., 2013; Furman et al., 2016; Pleiss et al., 2016; Sompol et al., 2017). For instance, the NFAT4 isoform, which is weakly expressed in healthy nervous tissue, appears at elevated levels in many activated astrocytes following kainic acid lesions, cortical stab wounds and controlled cortical contusion injuries (Serrano-Pérez et al., 2011; Neria et al., 2013; Furman et al., 2016). NFAT4 expression in a mouse model of Alzheimer’s disease also exhibited extensive co-localization with activated astrocytes, increasing directly in proportion to the expression of GFAP (Sompol et al., 2017). Using a custom antibody to CN, based on calpain-dependent cleavage sites, our lab recently observed intense labeling of a 45–48 kDa ΔCN fragment in activated astrocytes surrounding microinfarcts in human neocortex (Pleiss et al., 2016). Labeling for ΔCN was very faint throughout most brain areas examined, but increased dramatically in GFAP-positive astrocytes around the periphery of the lesion (Figure 1). These observations suggest considerable molecular heterogeneity in astrocytes depending on distance from vascular injury, consistent with studies in other injury/disease models (Zamanian et al., 2012; Itoh et al., 2018).
Figure 1.
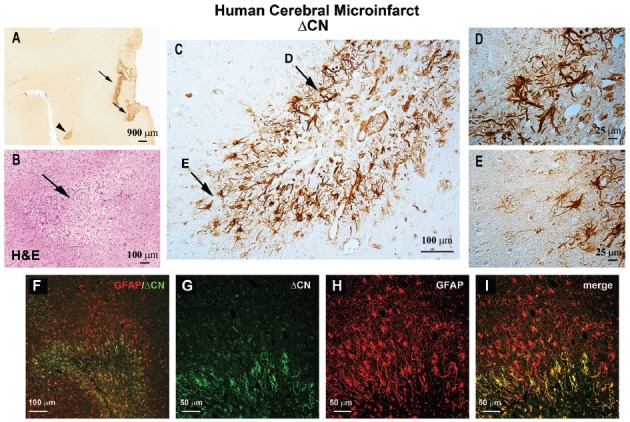
ΔCN is intensely expressed in activated astrocytes surrounding microinfarcts in human neocortex. (A) Representative low magnification photomicrograph from superior and middle temporal gyrus (SMTG) of a 90 year old human subject with multiple microinfarcts, but little-to-no Alzheimer’s pathology (Braak stage II) ΔCN labeling is present around several microinfarcts (arrows and arrowhead). (B) Serial section through STMG stained by H&E to confirm the presence of microinfarcts. The image shown is a high magnification of the region denoted by the arrowhead in Panel (A). (C) High power photomicrograph of the region in (A; arrowhead) showing intense ΔCN antibody labeling of astrocytes. Higher magnification of the areas denoted by arrows are shown in panels (D,E). (F) Merged confocal micrograph showing the colocalization of ΔCN (green) with GFAP around a microinfarct in human SMTG (red). (G–I) High magnification images of the infarct in Panel (F) shown in individual channels (G,H) and merged (I). Co-localization of ΔCN with GFAP was most extensive in the region immediately adjacent to the infarct. From Pleiss et al. (2016) used with permission.
Several outstanding issues regarding the relationship between astrocytic CN/NFAT and microinfarcts require further clarification. Presently, it is unknown whether CN/NFAT alterations occur immediately following microinfarct induction, or are more characteristic of chronic changes that arise with the formation of glial scars. The molecular phenotype of ΔCN-positive astrocytes has also yet to be elucidated. In primary neural cultures, forced overexpression of ΔCN in astrocytes induces the expression of numerous transcripts associated with morphogenesis and immune response (Norris et al., 2005). Studies are presently underway in our lab to determine the time course of ΔCN expression in photothrombosis models of microinfarct pathology (Risher et al., 2010; Masuda et al., 2011; Summers et al., 2017; Underly and Shih, 2017) and to determine if endogenous expression of ΔCN is associated with transcriptional changes, reminiscent of forced overexpression studies.
It deserves noting that many of the transcripts induced by CN/NFAT activity in glial cells, and in other cell types, encode releasable factors, such as cytokines and chemokines (Norris et al., 2005; Canellada et al., 2008; Sama et al., 2008; Nagamoto-Combs and Combs, 2010; Neria et al., 2013). Given the intimate structural and functional interactions between astrocytes and cerebral blood vessels, it seems likely that many CN/NFAT-dependent factors released from activated astrocytes could find their way into the bloodstream near regions of vascular damage. Presence of these factors (or ΔCN itself) in blood could then be used as potential biomarkers for the presence of microinfarcts or other forms of vascular pathology. Indeed, given the insidious nature of microinfarcts, the identification of peripheral biomarkers would be most helpful for diagnostic and/or prognostic screening purposes. Of course, additional research will be necessary to assess these possibilities.
Functional Impact of CN Signaling in Activated Astrocytes
Astrocyte activation is a complex process associated with both neuroprotective and deleterious consequences for surrounding nervous tissue (Khakh and Sofroniew, 2015; Pekny et al., 2016; Verkhratsky et al., 2016). The increased expression of CN/NFAT components in astrocytes associated with vascular pathology may offer important targets that could be exploited for determining the functional impact of these cells. Overexpression of ΔCN in hippocampal astrocytes of intact healthy adult rats causes reduced synaptic strength and hyperexcitability in nearby neurons, which is consistent with other studies linking activated astrocytes with impaired neuronal connectivity in acute injury models (Wilhelmsson et al., 2004). In contrast, astrocytic expression of ΔCN has also been found to reduce amyloid pathology and improve cognitive function in mouse models of Alzhieimer’s disease, consistent with other reports that have found protective roles of activated astrocytes in neurodegenerative conditions (Okada et al., 2006; Kraft et al., 2013; Wanner et al., 2013; Tyzack et al., 2014). Whether CN gives rise to beneficial or detrimental processes may depend critically on the presence of different activating factors and/or the recruitment of different transcription factor families (Furman and Norris, 2014). For instance, the pro-inflammatory cytokine TNF was shown to trigger the association of CN with the transcription factors NFκB and FOXO3, which, in turn, induced pro-inflammatory responses for promoting neurodegeneration (Fernandez et al., 2012, 2016). In contrast, CN stimulation by the insulin-like growth factor (IGF-I), has been proposed to mediate neuroprotective responses of activated astrocytes via interactions between NFκB and PPARγ (Fernandez et al., 2012).
Blockade of CN interactions with NFAT transcription factors, using the peptide VIVIT, has been associated with many beneficial effects in cell culture and intact animal models of neurodegeneration. VIVIT mimics the CN-binding PxIxIT motif found in the regulatory region of NFATs 1–4 (Aramburu et al., 1999). When delivered to numerous cell types, VIVIT prevents CN from binding to NFATs and therefore inhibits NFAT nuclear localization, without inhibiting CN catalytic activity per se. Expression of VIVIT in hippocampal astrocytes, using adeno-associated virus (AAV) vectors equipped with the human GFAP promoter Gfa2 (Lee et al., 2008), improved synaptic strength and/or normalized synaptic plasticity in animal models of Alzheimer’s disease and traumatic brain injury (Furman et al., 2012, 2016; Sompol et al., 2017). Where tested, AAV-Gfa2-VIVIT delivery to the hippocampus also improved hippocampal-dependent cognitive function (Furman et al., 2012; Sompol et al., 2017). In primary neural cultures, VIVIT prevented the loss of astrocyte-enriched glutamate transporters, primarily GLT1, in response to pro-inflammatory cytokines and oligomeric Aβ, leading to reduced extracellular glutamate levels, reduced neuronal excitability and greater neuronal survival (Sama et al., 2008; Abdul et al., 2009). VIVIT similarly restored GLT1 levels in intact 5xFAD mice—an aggressive mouse model for Alzheimer’s disease (Sompol et al., 2017). Mice treated with AAV-Gfa2-VIVIT showed greater GLT1 expression, measured via immunofluorescent microscopy and Western blot. VIVIT-treated 5xFAD mice also exhibited fewer and shorter-duration spontaneous glutamate transients (measured in vivo), healthier neurite morphology, reduced synaptic hyperexcitability, and normalized NMDA-to-AMPA receptor activity ratios (Sompol et al., 2017). Together, these observations suggest that hyperactive CN/NFAT signaling underlies a neurotoxic activated astrocyte phenotype characterized by glutamate dysregulation and excitotoxicity.
Interestingly, many of the same telltale signs of glutamate toxicity, including a loss of GLT1 and neuronal hyperactivity, have been noted in experimental models of ischemia and stroke (Maragakis and Rothstein, 2004; Soni et al., 2014). Moreover, glutamate dysregulation would not only influence the behavior and viability of surrounding neurons, but may also be expected to negatively affect the cerebrovascular unit as well. For instance, functional knockdown of GLT1 in otherwise healthy animals can lead to reduced cerebral blood flow and/or impaired neurovascular coupling (Petzold et al., 2008). Other work has shown that hyperexcitable neural networks and/or excitotoxic insults compromise the structural integrity of vascular endothelial cells and perivascular astrocyte endfeet, and precipitate blood brain barrier (BBB) leakage (Bolton and Perry, 1998; Parathath et al., 2006; Alvestad et al., 2013; Gondo et al., 2014; Ryu and McLarnon, 2016) leading to perivascular and parenchymal neuroinflammation.
Summary and Future Directions
Cerebrovascular pathology is one of the leading causes of dementia and a frequently identified comorbid factor in many neurologic diseases, such as Alzheimer’s disease. Numerous studies have reported a role for CN hyperactivity in the pathophysiologic sequelae coupling vascular disruption and damage to neuronal death and cognitive loss. Mounting evidence suggests that CN/NFAT signaling may play a particularly important role in neural changes that arise with astrocyte activation in many different neurodegenerative diseases, including cerebrovascular disease. However, no studies to date have tested the specific involvement of astrocytic CN/NFAT signaling in either global ischemia models, models characterized by localized damage to microvessels, or in models that develop chronic vascular inflammation and microhemhorrages. Based on the observations discussed above, we hypothesize that acutely and chronically developing vascular damage will lead to the activation of astrocytes and hyperactivation of CN/NFAT signaling (Figure 2). In this scenario, increased CN/NFAT activity would lead to the induction and release of numerous immune/inflammatory factors and/or to the dysregulation of astrocytic glutamate uptake, resulting in impaired synaptic function, excitotoxicity, impaired neuronal viability and neuroinflammation. These deleterious actions, could, in turn, promote further vascular damage and inflammation and hasten neurodegeneration and cognitive loss as part of vicious positive feedback cycle. Of course, this hypothesis will require extensive testing using astrocyte-specific targeting strategies in experimental models of stroke and/or VCID.
Figure 2.
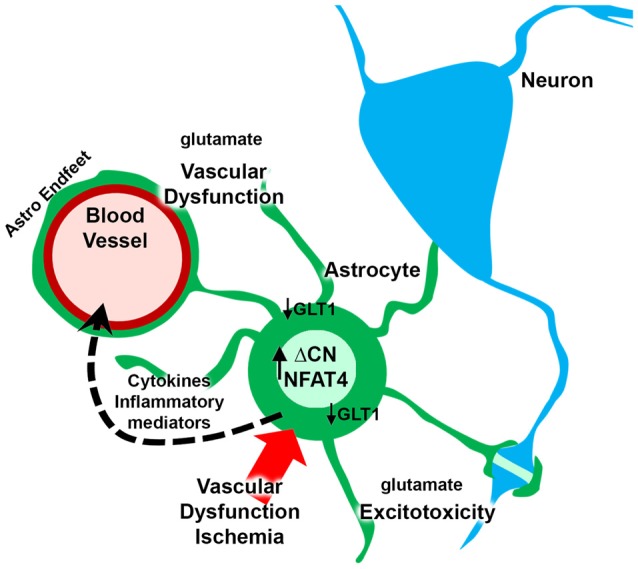
Putative role for astrocytic CN/nuclear factor of activated T cell (NFAT) in vascular dysfunction and neurodegeneration. Ischemia arising from vascular degeneration or disruption leads to increased expression of ΔCN and hyperactivation of NFAT4 in astrocytes. The CN/NFAT pathway induces numerous cytokines and other inflammatory mediators linked to neuroinflammation. Some of these factors may target blood vessels, leading to perivascular inflammation. CN/NFAT signaling also leads to the downregulation of GLT1 glutamate transporters resulting in elevated extracellular glutamate levels. Glutamate causes excitotoxicity at synaptic connections and disrupts astrocyte endfeet and/or blood brain barrier (BBB) integrity, leading to further vascular dysfunction and/or degeneration.
Author Contributions
SK and CN researched and wrote this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by National Institutes of Health Grants AG027297, AG056998, AG051945 and a gift from the Hazel Embry Research Trust.
References
- Abdul H. M., Furman J. L., Sama M. A., Mathis D. M., Norris C. M. (2010). NFATs and Alzheimer’s disease. Mol. Cell. Pharmacol. 2, 7–14. [PMC free article] [PubMed] [Google Scholar]
- Abdul H. M., Sama M. A., Furman J. L., Mathis D. M., Beckett T. L., Weidner A. M., et al. (2009). Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 29, 12957–12969. 10.1523/jneurosci.1064-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvestad S., Hammer J., Hoddevik E. H., Skare Ø., Sonnewald U., Amiry-Moghaddam M., et al. (2013). Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res. 105, 30–41. 10.1016/j.eplepsyres.2013.01.006 [DOI] [PubMed] [Google Scholar]
- Aramburu J., Rao A., Klee C. B. (2000). Calcineurin: from structure to function. Curr. Top. Cell. Regul. 36, 237–295. 10.1016/s0070-2137(01)80011-x [DOI] [PubMed] [Google Scholar]
- Aramburu J., Yaffe M. B., López-Rodriguez C., Cantley L. C., Hogan P. G., Rao A. (1999). Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 285, 2129–2133. 10.1126/science.285.5436.2129 [DOI] [PubMed] [Google Scholar]
- Baumgärtel K., Genoux D., Welzl H., Tweedie-Cullen R. Y., Koshibu K., Livingstone-Zatchej M., et al. (2008). Control of the establishment of aversive memory by calcineurin and Zif268. Nat. Neurosci. 11, 572–578. 10.1038/nn.2113 [DOI] [PubMed] [Google Scholar]
- Bolton S. J., Perry V. H. (1998). Differential blood-brain barrier breakdown and leucocyte recruitment following excitotoxic lesions in juvenile and adult rats. Exp. Neurol. 154, 231–240. 10.1006/exnr.1998.6927 [DOI] [PubMed] [Google Scholar]
- Brait V. H., Martin K. R., Corlett A., Broughton B. R., Kim H. A., Thundyil J., et al. (2012). Over-expression of DSCR1 protects against post-ischemic neuronal injury. PLoS One 7:e47841. 10.1371/journal.pone.0047841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher S. P., Henshall D. C., Teramura Y., Iwasaki K., Sharkey J. (1997). Neuroprotective actions of FK506 in experimental stroke: in vivo evidence against an antiexcitotoxic mechanism. J. Neurosci. 17, 6939–6946. 10.1523/jneurosci.17-18-06939.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canellada A., Ramirez B. G., Minami T., Redondo J. M., Cano E. (2008). Calcium/calcineurin signaling in primary cortical astrocyte cultures: Rcan1–4 and cyclooxygenase-2 as NFAT target genes. Glia 56, 709–722. 10.1002/glia.20647 [DOI] [PubMed] [Google Scholar]
- Celsi F., Svedberg M., Unger C., Cotman C. W., Carri M. T., Ottersen O. P., et al. (2007). β-amyloid causes downregulation of calcineurin in neurons through induction of oxidative stress. Neurobiol. Dis. 26, 342–352. 10.1016/j.nbd.2006.12.022 [DOI] [PubMed] [Google Scholar]
- Choi D. W. (1988). Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 11, 465–469. 10.1016/0166-2236(88)90200-7 [DOI] [PubMed] [Google Scholar]
- de la Fuente V., Freudenthal R., Romano A. (2011). Reconsolidation or extinction: transcription factor switch in the determination of memory course after retrieval. J. Neurosci. 31, 5562–5573. 10.1523/JNEUROSCI.6066-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A. M., Hervas R., Dominguez-Fraile M., Garrido V. N., Gomez-Gutierrez P., Vega M., et al. (2016). Blockade of the interaction of calcineurin with FOXO in astrocytes protects against amyloid-β-induced neuronal death. J. Alzheimers Dis. 52, 1471–1478. 10.3233/jad-160149 [DOI] [PubMed] [Google Scholar]
- Fernandez A. M., Jimenez S., Mecha M., Davila D., Guaza C., Vitorica J., et al. (2012). Regulation of the phosphatase calcineurin by insulin-like growth factor I unveils a key role of astrocytes in Alzheimer’s pathology. Mol. Psychiatry 17, 705–718. 10.1038/mp.2011.128 [DOI] [PubMed] [Google Scholar]
- Furman J. L., Norris C. M. (2014). Calcineurin and glial signaling: neuroinflammation and beyond. J. Neuroinflammation 11:158. 10.1186/s12974-014-0158-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman J. L., Sama D. M., Gant J. C., Beckett T. L., Murphy M. P., Bachstetter A. D., et al. (2012). Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 16129–16140. 10.1523/jneurosci.2323-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman J. L., Sompol P., Kraner S. D., Pleiss M. M., Putman E. J., Dunkerson J., et al. (2016). Blockade of astrocytic calcineurin/NFAT signaling helps to normalize hippocampal synaptic function and plasticity in a rat model of traumatic brain injury. J. Neurosci. 36, 1502–1515. 10.1523/jneurosci.1930-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondo A., Shinotsuka T., Morita A., Abe Y., Yasui M., Nuriya M. (2014). Sustained down-regulation of β-dystroglycan and associated dysfunctions of astrocytic endfeet in epileptic cerebral cortex. J. Biol. Chem. 289, 30279–30288. 10.1074/jbc.m114.588384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R. J., Branston N. M., Symon L., Bayhan M., Watson A. (1982). The effects of a calcium antagonist, nimodipine, upon physiological responses of the cerebral vasculature and its possible influence upon focal cerebral ischaemia. Stroke 13, 759–766. 10.1161/01.str.13.6.759 [DOI] [PubMed] [Google Scholar]
- Hashimoto T., Kawamata T., Saito N., Sasaki M., Nakai M., Niu S., et al. (1998). Isoform-specific redistribution of calcineurin Aα and Aβ in the hippocampal CA1 region of gerbils after transient ischemia. J. Neurochem. 70, 1289–1298. 10.1046/j.1471-4159.1998.70031289.x [DOI] [PubMed] [Google Scholar]
- Horsburgh K., Wardlaw J. M., Van Agtmael T., Allan S. M., Ashford M. L. J., Bath P. M., et al. (2018). Small vessels, dementia and chronic diseases—molecular mechanisms and pathophysiology. Clin. Sci. 132, 851–868. 10.1042/CS20171620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsley V., Pavlath G. K. (2002). NFAT: ubiquitous regulator of cell differentiation and adaptation. J. Cell Biol. 156, 771–774. 10.1083/jcb.200111073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst G. J. T., Postigo A. (1996). “Stroke: prevalence and mechanism of cell death,” in Clinical Pharmacology of Cerebral Ischemia, eds Ter Horst G. J., Korf J. (New York, NY: Springer Science and Business Media; ), 1–30. [Google Scholar]
- Im S. H., Rao A. (2004). Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling. Mol. Cells 18, 1–9. [PubMed] [Google Scholar]
- Infeld B., Davis S. M., Donnan G. A., Yasaka M., Lichtenstein M., Mitchell P. J., et al. (1999). Nimodipine and perfusion changes after stroke. Stroke 30, 1417–1423. 10.1161/01.str.30.7.1417 [DOI] [PubMed] [Google Scholar]
- Itoh N., Itoh Y., Tassoni A., Ren E., Kaito M., Ohno A., et al. (2018). Cell-specific and region-specific transcriptomics in the multiple sclerosis model: focus on astrocytes. Proc. Natl. Acad. Sci. U S A 115, E302–E309. 10.1073/pnas.1716032115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh B. S., Sofroniew M. V. (2015). Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 18, 942–952. 10.1038/nn.4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee C. B., Ren H., Wang X. (1998). Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J. Biol. Chem. 273, 13367–13370. 10.1074/jbc.273.22.13367 [DOI] [PubMed] [Google Scholar]
- Kraft A. W., Hu X., Yoon H., Yan P., Xiao Q., Wang Y., et al. (2013). Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 27, 187–198. 10.1096/fj.12-208660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Messing A., Su M., Brenner M. (2008). GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia 56, 481–493. 10.1002/glia.20622 [DOI] [PubMed] [Google Scholar]
- Lim D., Iyer A., Ronco V., Grolla A. A., Canonico P. L., Aronica E., et al. (2013). Amyloid β deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 61, 1134–1145. 10.1002/glia.22502 [DOI] [PubMed] [Google Scholar]
- Manocha G. D., Ghatak A., Puig K. L., Kraner S. D., Norris C. M., Combs C. K. (2017). NFATc2 modulates microglial activation in the AβPP/PS1 mouse model of Alzheimer’s disease. J. Alzheimers Dis. 58, 775–787. 10.3233/jad-151203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansuy I. M. (2003). Calcineurin in memory and bidirectional plasticity. Biochem. Biophys. Res. Commun. 311, 1195–1208. 10.1016/j.bbrc.2003.10.046 [DOI] [PubMed] [Google Scholar]
- Maragakis N. J., Rothstein J. D. (2004). Glutamate transporters: animal models to neurologic disease. Neurobiol. Dis. 15, 461–473. 10.1016/j.nbd.2003.12.007 [DOI] [PubMed] [Google Scholar]
- Masuda T., Croom D., Hida H., Kirov S. A. (2011). Capillary blood flow around microglial somata determines dynamics of microglial processes in ischemic conditions. Glia 59, 1744–1753. 10.1002/glia.21220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. (2007). Calcium and neurodegeneration. Aging Cell 6, 337–350. 10.1111/j.1474-9726.2007.00275.x [DOI] [PubMed] [Google Scholar]
- Mukherjee A., Soto C. (2011). Role of calcineurin in neurodegeneration produced by misfolded proteins and endoplasmic reticulum stress. Curr. Opin. Cell Biol. 23, 223–230. 10.1016/j.ceb.2010.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamoto-Combs K., Combs C. K. (2010). Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells). J. Neurosci. 30, 9641–9646. 10.1523/jneurosci.0828-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neria F., Del Carmen Serrano-Perez M., Velasco P., Urso K., Tranque P., Cano E. (2013). NFATc3 promotes Ca2+-dependent MMP3 expression in astroglial cells. Glia 61, 1052–1066. 10.1002/glia.22494 [DOI] [PubMed] [Google Scholar]
- Nguyen T., Di Giovanni S. (2008). NFAT signaling in neural development and axon growth. Int. J. Dev. Neurosci. 26, 141–145. 10.1016/j.ijdevneu.2007.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris C. M. (2014). Calpain interactions with the protein phosphatase calcineurin in neurodegeneration. Adv. Biochem. Health Dis. 8, 17–45. 10.1007/978-1-4614-9099-9_2 [DOI] [Google Scholar]
- Norris C. M., Kadish I., Blalock E. M., Chen K. C., Thibault V., Porter N. M., et al. (2005). Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J. Neurosci. 25, 4649–4658. 10.1523/JNEUROSCI.0365-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J. T., Erkinjuntti T., Reisberg B., Roman G., Sawada T., Pantoni L., et al. (2003). Vascular cognitive impairment. Lancet Neurol. 2, 89–98. 10.1016/S1474-4422(03)00305-3 [DOI] [PubMed] [Google Scholar]
- Okada S., Nakamura M., Katoh H., Miyao T., Shimazaki T., Ishii K., et al. (2006). Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 12, 829–834. 10.1038/nm1425 [DOI] [PubMed] [Google Scholar]
- Parathath S. R., Parathath S., Tsirka S. E. (2006). Nitric oxide mediates neurodegeneration and breakdown of the blood-brain barrier in tPA-dependent excitotoxic injury in mice. J. Cell Sci. 119, 339–349. 10.1242/jcs.02734 [DOI] [PubMed] [Google Scholar]
- Pekny M., Pekna M., Messing A., Steinhäuser C., Lee J. M., Parpura V., et al. (2016). Astrocytes: a central element in neurological diseases. Acta Neuropathol. 131, 323–345. 10.1007/s00401-015-1513-1 [DOI] [PubMed] [Google Scholar]
- Petzold G. C., Albeanu D. F., Sato T. F., Murthy V. N. (2008). Coupling of neural activity to blood flow in olfactory glomeruli is mediated by astrocytic pathways. Neuron 58, 897–910. 10.1016/j.neuron.2008.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss M. M., Sompol P., Kraner S. D., Abdul H. M., Furman J. L., Guttmann R. P., et al. (2016). Calcineurin proteolysis in astrocytes: implications for impaired synaptic function. Biochim. Biophys. Acta 1862, 1521–1532. 10.1016/j.bbadis.2016.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A., Luo C., Hogan P. G. (1997). Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 15, 707–747. 10.1146/annurev.immunol.15.1.707 [DOI] [PubMed] [Google Scholar]
- Ray S. K. (2006). Currently evaluated calpain and caspase inhibitors for neuroprotection in experimental brain ischemia. Curr. Med. Chem. 13, 3425–3440. 10.2174/092986706779010342 [DOI] [PubMed] [Google Scholar]
- Reese L. C., Taglialatela G. (2011). A role for calcineurin in Alzheimer’s disease. Curr. Neuropharmacol. 9, 685–692. 10.2174/157015911798376316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher W. C., Ard D., Yuan J., Kirov S. A. (2010). Recurrent spontaneous spreading depolarizations facilitate acute dendritic injury in the ischemic penumbra. J. Neurosci. 30, 9859–9868. 10.1523/JNEUROSCI.1917-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Olvera A., Nelson-Mora J., Gonsebatt M. E., Escobar M. L. (2018). Extinction of aversive taste memory homeostatically prevents the maintenance of in vivo insular cortex LTP: calcineurin participation. Neurobiol. Learn. Mem. [Epub ahead of print]. 10.1016/j.nlm.2018.04.005 [DOI] [PubMed] [Google Scholar]
- Rosenkranz K., May C., Meier C., Marcus K. (2012). Proteomic analysis of alterations induced by perinatal hypoxic-ischemic brain injury. J. Proteome Res. 11, 5794–5803. 10.1021/pr3005869 [DOI] [PubMed] [Google Scholar]
- Rostas J. A. P., Spratt N. J., Dickson P. W., Skelding K. A. (2017). The role of Ca2+-calmodulin stimulated protein kinase II in ischaemic stroke—A potential target for neuroprotective therapies. Neurochem. Int. 107, 33–42. 10.1016/j.neuint.2017.01.012 [DOI] [PubMed] [Google Scholar]
- Ryu J. K., McLarnon J. G. (2016). Pyruvate blocks blood-brain barrier disruption, lymphocyte infiltration and immune response in excitotoxic brain injury. Am. J. Neurodegener. Dis. 5, 69–73. [PMC free article] [PubMed] [Google Scholar]
- Sama M. A., Mathis D. M., Furman J. L., Abdul H. M., Artiushin I. A., Kraner S. D., et al. (2008). Interleukin-1β-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J. Biol. Chem. 283, 21953–21964. 10.1074/jbc.M800148200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pérez M. C., Martín E. D., Vaquero C. F., Azcoitia I., Calvo S., Cano E., et al. (2011). Response of transcription factor NFATc3 to excitotoxic and traumatic brain insults: identification of a subpopulation of reactive astrocytes. Glia 59, 94–107. 10.1002/glia.21079 [DOI] [PubMed] [Google Scholar]
- Sharkey J., Butcher S. P. (1994). Immunophilins mediate the neuroprotective effects of FK506 in focal cerebral ischaemia. Nature 371, 336–339. 10.1038/371336a0 [DOI] [PubMed] [Google Scholar]
- Sharkey J., Crawford J. H., Butcher S. P., Marston H. M. (1996). Tacrolimus (FK506) ameliorates skilled motor deficits produced by middle cerebral artery occlusion in rats. Stroke 27, 2282–2286. 10.1161/01.str.27.12.2282 [DOI] [PubMed] [Google Scholar]
- Shioda N., Han F., Moriguchi S., Fukunaga K. (2007). Constitutively active calcineurin mediates delayed neuronal death through Fas-ligand expression via activation of NFAT and FKHR transcriptional activities in mouse brain ischemia. J. Neurochem. 102, 1506–1517. 10.1111/j.1471-4159.2007.04600.x [DOI] [PubMed] [Google Scholar]
- Shioda N., Moriguchi S., Shirasaki Y., Fukunaga K. (2006). Generation of constitutively active calcineurin by calpain contributes to delayed neuronal death following mouse brain ischemia. J. Neurochem. 98, 310–320. 10.1111/j.1471-4159.2006.03874.x [DOI] [PubMed] [Google Scholar]
- Snyder H. M., Corriveau R. A., Craft S., Faber J. E., Greenberg S. M., Knopman D., et al. (2015). Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimers Dement. 11, 710–717. 10.1016/j.jalz.2014.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrado M., Ramirez B. G., Neria F., Lizasoain I., Arbones M. L., Minami T., et al. (2012). Regulator of calcineurin 1 (Rcan1) has a protective role in brain ischemia/reperfusion injury. J. Neuroinflammation 9:48. 10.1186/1742-2094-9-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sompol P., Furman J. L., Pleiss M. M., Kraner S. D., Artiushin I. A., Batten S. R., et al. (2017). Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in Aβ-bearing mice. J. Neurosci. 37, 6132–6148. 10.1523/JNEUROSCI.0877-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sompol P., Norris C. M. (2018). Ca2+, astrocyte activation and calcineurin/NFAT signaling in age-related neurodegenerative diseases. Front. Aging Neurosci. 10:199. 10.3389/fnagi.2018.00199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni N., Reddy B. V., Kumar P. (2014). GLT-1 transporter: an effective pharmacological target for various neurological disorders. Pharmacol. Biochem. Behav. 127, 70–81. 10.1016/j.pbb.2014.10.001 [DOI] [PubMed] [Google Scholar]
- Summers P. M., Hartmann D. A., Hui E. S., Nie X., Deardorff R. L., Mckinnon E. T., et al. (2017). Functional deficits induced by cortical microinfarcts. J. Cereb. Blood Flow Metab. 37, 3599–3614. 10.1177/0271678x16685573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szydlowska K., Tymianskia M. (2010). Calcium, ischemia and excitotoxicity. Cell Calcium 47, 122–129. 10.1016/j.ceca.2010.01.003 [DOI] [PubMed] [Google Scholar]
- Tyzack G. E., Sitnikov S., Barson D., Adams-Carr K. L., Lau N. K., Kwok J. C., et al. (2014). Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nat. Commun. 5:4294. 10.1038/ncomms5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchino H., Kuroda Y., Morota S., Hirabayashi G., Ishii N., Shibasaki F., et al. (2008). Probing the molecular mechanisms of neuronal degeneration: importance of mitochondrial dysfunction and calcineurin activation. J. Anesth. 22, 253–262. 10.1007/s00540-008-0617-3 [DOI] [PubMed] [Google Scholar]
- Underly R. G., Shih A. Y. (2017). Photothrombotic induction of capillary ischemia in the mouse cortex during in vivo two-photon imaging. Bio. Protoc. 7:e2378. 10.21769/bioprotoc.2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A., Steardo L., Parpura V., Montana V. (2016). Translational potential of astrocytes in brain disorders. Prog. Neurobiol. 144, 188–205. 10.1016/j.pneurobio.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihma H., Pruunsild P., Timmusk T. (2008). Alternative splicing and expression of human and mouse NFAT genes. Genomics 92, 279–291. 10.1016/j.ygeno.2008.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. K., Roufogalis B. D., Villalobo A. (1989). Characterization of the fragmented forms of calcineurin produced by calpain I. Biochem. Cell Biol. 67, 703–711. 10.1139/o89-105 [DOI] [PubMed] [Google Scholar]
- Wanner I. B., Anderson M. A., Song B., Levine J., Fernandez A., Gray-Thompson Z., et al. (2013). Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 33, 12870–12886. 10.1523/JNEUROSCI.2121-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekman E. M., Sudduth T. L., Caverly C. N., Kopper T. J., Phillips O. W., Powell D. K., et al. (2016). Reduced Efficacy of anti-aβ immunotherapy in a mouse model of amyloid deposition and vascular cognitive impairment comorbidity. J. Neurosci. 36, 9896–9907. 10.1523/JNEUROSCI.1762-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsson U., Li L., Pekna M., Berthold C. H., Blom S., Eliasson C., et al. (2004). Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J. Neurosci. 24, 5016–5021. 10.1523/JNEUROSCI.0820-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Borde M., Heissmeyer V., Feuerer M., Lapan A. D., Stroud J. C., et al. (2006). FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 126, 375–387. 10.1016/j.cell.2006.05.042 [DOI] [PubMed] [Google Scholar]
- Wu H. Y., Tomizawa K., Oda Y., Wei F. Y., Lu Y. F., Matsushita M., et al. (2004). Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J. Biol. Chem. 279, 4929–4940. 10.1074/jbc.M309767200 [DOI] [PubMed] [Google Scholar]
- Wu Q. J., Tymianski M. (2018). Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol. Brain 11:15. 10.1186/s13041-018-0357-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian J. L., Xu L., Foo L. C., Nouri N., Zhou L., Giffard R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. 10.1523/JNEUROSCI.6221-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawadzka M., Kaminska B. (2005). A novel mechanism of FK506-mediated neuroprotection: downregulation of cytokine expression in glial cells. Glia 49, 36–51. 10.1002/glia.20092 [DOI] [PubMed] [Google Scholar]