

Abstract
Mucin-type O-glycosylation is initiated by a family of polypeptide GalNAc-transferases (GalNAc-Ts) which are type-II transmembrane proteins that contain Golgi luminal catalytic and lectin domains that are connected by a flexible linker. Several GalNAc-Ts, including GalNAc-T4, show both long-range and short-range prior glycosylation specificity, governed by their lectin and catalytic domains, respectively. While the mechanism of the lectin-domain-dependent glycosylation is well-known, the molecular basis for the catalytic-domain-dependent glycosylation of glycopeptides is unclear. Herein, we report the crystal structure of GalNAc-T4 bound to the diglycopeptide GAT*GAGAGAGT*TPGPG (containing two α-GalNAc glycosylated Thr (T*), the PXP motif and a “naked” Thr acceptor site) that describes its catalytic domain glycopeptide GalNAc binding site. Kinetic studies of wild-type and GalNAc binding site mutant enzymes show the lectin domain GalNAc binding activity dominates over the catalytic domain GalNAc binding activity and that these activities can be independently eliminated. Surprisingly, a flexible loop protruding from the lectin domain was found essential for the optimal activity of the catalytic domain. This work provides the first structural basis for the short-range glycosylation preferences of a GalNAc-T.
Short abstract
First characterization and visualization of the catalytic domain glycopeptide GalNAc binding site of a GalNAc-T revealing the molecular origins for GalNAc-T4’s neighboring O-glycosylation activity.
Mucin-type O-glycosylation (O-GalNAc-type) is one of the most diverse and complex types of protein O-glycosylation found in higher eukaryotes and is likely the most abundant, with over 80% of all proteins passing through the secretory pathway predicted to be O-glycosylated.1 This post-translational modification is important in a wide range of cellular/biological processes,1 including phosphate homeostasis and bone mineralization,2 regulation of HDL and LDL,3,4 tumorigenesis and formation of metastasis,5,6 organogenesis and development,7 and the fundamental process of ectodomain shedding and cell signaling.8,9 In addition, alteration of the cellular location and regulation of the activity of the GalNAc-T isoenzymes have clear implications in disease.1 Mucin-type O-glycosylation (henceforth called O-glycosylation) is initiated by a large family of GalNAc-T isoenzymes (20 members in humans and similar numbers in most mammals) that are suggested to act in a hierarchical manner. GalNAc-Ts are retaining glycosyltransferases (GTs) that transfer a GalNAc moiety from uridine diphosphate N-acetylgalactosamine (UDP-GalNAc) onto Ser/Thr residues of proteins.1 The GalNAc-Ts are unique among metazoan GTs because in addition to their N-terminal catalytic domain adopting a GT-A fold, they possess a C-terminal GalNAc binding lectin domain with a β-trefoil fold, which provides additional functions to these enzymes.10−12 The two domains are linked through a short flexible linker whose motion has been suggested to be responsible for the dynamic conformational landscape of these enzymes.13 Three distinctive modes of substrate glycosylation have been reported:10 (1) glycosylation of “naked” unglycosylated peptides; (2) glycosylation of GalNAc-containing glycopeptides where the sites of glycosylation are 1–3 residues away from the prior glycosite, hereafter termed short-range or neighboring glycosylation; and (3) glycosylation of GalNAc glycopeptides where the sites of glycosylation are ∼5–17 residues away from the prior glycosite, termed long-range or remote glycosylation.10 The latter, long-range glycosylation activity is due to glycopeptide GalNAc binding at the lectin domain, directing the acceptor peptide onto the catalytic domain in an N- and/or C-terminal direction.10,14 Most isoforms that contain a lectin domain have been found to possess this remote glycosylation activity.10 On the other hand, sites adjacent and neighboring to an existing GalNAc glycosite are glycosylated in a catalytic-domain-dependent manner, with GalNAc-T4 being part of a small number of GalNAc-Ts (including GalNAc-T7, -T10, and -T12) that have been shown to glycosylate contiguous or nearby sites.10,11,15 While GalNAc-T4 glycosylates acceptor sites that are directly C-terminal to a prior GalNAc glycosite, GalNAc-T7 and T10 glycosylate sites that are directly N-terminal from a prior site of GalNAc glycosylation. In addition, GalNAc-T12 glycosylates sites located three residues N-terminal from a prior GalNAc glycosite.10 Thus, it is likely that a GalNAc binding site should exist for those isoenzymes that possess short-range glycosylation preferences on glycopeptides. While the lectin domain GalNAc binding sites have been well-characterized by others and us for several GalNAc-Ts,16,17 the catalytic domain GalNAc binding site has never been revealed at the structural level. In addition, it is not known how GalNAc-T4, -T7, and -T10 may glycosylate contiguous acceptor sites in glycopeptides exclusively using a catalytic-domain-dependent manner.
Previous structural work from our lab with GalNAc-T2 in complex with “naked” peptides has provided insight into the catalytic-domain-mediated transfer of GalNAc from the UDP-GalNAc donor to the peptide acceptor. In addition, our studies on remotely glycosylated glycopeptides have demonstrated how the GalNAc-T2 lectin domain guides N-terminal acceptor sites into the catalytic domain for catalysis to take place, by binding to a distant prior GalNAc glycosite located at the C-terminus of the substrate.12,13,18 Recently, we have also demonstrated how other GalNAc-T isoforms such as GalNAc-T3, -T4, -T6, and -T12 achieve the opposite long-range glycosylation preferences.10,19 In that study we showed that the connecting interdomain flexible linker dictates the orientation of the lectin domain with respect to the catalytic domain. Thus, the different positions of the lectin domain GalNAc binding site of GalNAc-T4 relative to GalNAcT2 readily explained how these two isoenzymes achieved their opposite and distinct long-range glycosylation preferences.14
To date however no structural studies have revealed how the catalytic domains of any of the GalNAc-Ts that possess neighboring or adjacent glycopeptide activities actually accommodate an acceptor peptide with a GalNAc residue immediately adjacent to the acceptor site. Such knowledge would help us to understand how these transferases perform their so-called filling-in activities, i.e., completing the glycosylation of heavily O-glycosylated mucin domains. The GalNAc-T4 isoform is of particular interest as it is the only isoform shown capable of glycosylating two out of the five acceptor sites in the partially glycosylated MUC1 mucin tandem repeat.11 The density of glycosylation, together with the structure of O-glycans in the mucin tandem repeat regions, are important features for the exploitation of the cancer related glycoforms of MUC1 as diagnostic as well as therapeutic purposes.20 Generally, GalNAc-T4 glycosylates very few isolated sites in “naked” peptide acceptors.21
We report herein a multidisciplinary approach combining different structural and biophysical techniques, with enzyme kinetics analysis and computational studies on GalNAc-T4. Our findings combined with the kinetic characterization of a library of (glyco)peptides, against the wild-type and inactivating catalytic and lectin domain mutant transferases, have begun to reveal the molecular basis of the short-range glycosylation preferences of the GalNAc-T4 on glycopeptide substrates. The experiments further reveal the dominant role of long-range lectin domain glycopeptide binding over short-range catalytic domain glycopeptide binding in the overall glycosylation process of GalNAc-T4. In addition, we determine the role of a flexible loop protruding from the lectin domain as an important structural feature essential for the optimal activity of the catalytic domain.
Results and Discussion
Kinetics of GalNAc-T4 against Glycopeptide Substrates
We have previously used three model (glyco)peptides (Table 1) to compare the long-range glycosylation preferences and enzyme kinetics of GalNAc-T2 and -T4.14 Those were the “naked” peptide 1 (denoted -TT-, for simplicity), and the C- and N-terminal glycosylated monoglycopeptides, 2 and 3 (denoted -TT--T*- and -T*--TT-, respectively, where T* represents a GalNAc-glycosylated Thr, see Table 1 and Figure 1a).14 Here, we have expanded this (glyco)peptide library with monoglycopeptide 4 (denoted -T*T-) and diglycopeptides 5 and 6 (denoted -T*T--T*- and -T*--T*T-, respectively) (Table 1). The diglycopeptides were designed to evaluate the combined effects of having long-range and short-range prior glycosylation in the same substrate. Note that all substrates in Table 1 have one or two potential Thr acceptor sites (i.e., -T*T- or -TT-) that also contain an adjacent C-terminal PXP motif (as -PGP-), which is recognized by most GalNAc-Ts.10,13 These peptides also share the same amino acid sequence around the acceptor site, thus eliminating the effects of peptide sequence variation on catalysis (Table 1). The short sequences surrounding the remote prior T* glycosylation site are also identical, thus ensuring nearly identical lectin domain binding properties. Note that the remote prior T* glycosylation site is located 7–8 residues away from the potential Thr acceptor sites, which is in agreement with the optimal distance of 7–11 residues observed for the long-range glycosylation of GalNAc-T4.10 Likewise the neighboring N-terminal prior glycosylation preference of GalNAc-T4,10 i.e., -T*T-, was included in glycopeptides 5 and 6 in order to examine the combined effects of having both long- and short-range prior glycosylation in one glycopeptide. For simplicity we did not include in our present studies peptides bearing Ser acceptor sites or S*, since Ser is usually less efficiently glycosylated than Thr,18 and S* displays distinct conformational preferences in contrast to T*, both in the free state and bound to proteins.22 According to our previous crystal structure of GalNAc-T4 complexed with peptide 3 (-T*--TT-),14 the remote N-terminal glycosites (i.e., T*) of both peptides 3 and 6 (-T*--TT- and -T*--T*T-) would bind to the GalNAc binding site of the lectin domain, while directing the C-terminal of the peptide acceptor onto the catalytic domain for catalysis. In contrast, the remote C-terminal glycosite of peptides 2 (-TT—T*-) and 5 (-T*T—T*-), when bound to the lectin domain, would not be expected to correctly orient the N-terminal portion of the acceptor peptide onto the catalytic domain for efficient GalNAc transfer.
Table 1. Peptide Acceptor Substrates Used in This Studya.

Note: T* denotes the Thr-O-GalNAc moiety.
Figure 1.

Biophysical characterization of GalNAc-T4. (a) Peptide glycosylation kinetics of GalNAc-T4 against (glyco)peptides 1–6 (see also Figure 4a). Michaelis–Menten kinetic values, Km, Vmax, and catalytic efficiency (Vmax/Km) for glycopeptides 3–6 were obtained from the nonlinear least-squares fit to the initial rate data, obtained as described in the Methods section and given in Table 2. Peptide substrates 1 and 2 are largely unglycosylated by GalNAc-T4.14 (b) Left panel: SPR sensogram for binding of glycopeptide 6 to GalNAc-T4. Right panel: Fitting of the SPR binding data giving a Kd of 70 ± 15 μM. (c) Mapping of substrate binding epitopes by saturation transfer difference (STD) NMR. The size of the colored spheres represents the normalized STD-NMR intensity (i.e., binding) observed for the indicated protons/residues. For sake of clarity, the STD response given for the indicated amino acid residues corresponds to the average of STD for all of the protons in the residue that could be accuracy measured. See Figures S4–S6 for the detailed STD-NMR enhancements of the identified residues/protons. Note that in addition to the GalNAc protons amino acid protons in the -T*TPGP- sequence also gave STD-NMR enhancements. (d) Representative 600 MHz 1H NMR spectra of glycopeptide 6 (-T*--T*T-) at 877 μM in the presence of 13.5 μM GalNAc-T4, 75 μM UDP, and 75 μM MnCl2 obtained at 298 K. The off resonance reference spectrum (labeled Off res) is displayed in blue, and the on resonance STD spectrum (labeled STD) is in red. Key proton resonances are labeled in the STD spectrum. Note the different STD responses for the identified GalNAc H2 protons of the glycosylated Thr3 and Thr11 found between 4.0 and 4.1 ppm of the STD spectrum.
To test the rationale above, detailed enzyme kinetics of GalNAc-T4 against the (glyco)peptides listed in Table 1 was performed using UDP-3H-GalNAc as donor (see the Methods section for details). Plots of initial rates versus substrate concentration are given in Figure 1a, while the obtained kinetic constants are listed in Table 2. Note that the results for peptides 3, 4, and 5 could be fitted to a standard Michaelis–Menten model, while peptide 6 was best fitted to a model which included apparent substrate inhibition and will be discussed below. As can be readily seen from Figure 1a, glycopeptides 3 (-T*--TT-) and 6 (-T*--T*T-) show the largest activity, which is ∼10-fold higher than that for glycopeptides 4 (-T*T) and 5 (-T*T--T*-). As previously reported, peptides 1 (-TT-) and 2 (-TT--T*-) were imperceptibly glycosylated.14 Note also that only the second Thr is glycosylated in those peptides containing the -TTPGP- sequence.14 These results are consistent with our previous findings that GalNAc-T4 displays a directly neighboring N-terminal prior glycosylation preference, as well as a long-range N-terminal prior glycosylation activity, while showing poor activity against the naked peptide or glycopeptides containing only a C-terminal remote prior glycosylation.10 Thus, for glycosylation to take place in glycopeptide 4 (-T*T), it can be concluded that the neighboring glycosylated Thr must directly bind to the catalytic domain as the acceptor Thr is contiguous to this site. The similar plots and kinetic constants obtained for diglycopeptide 5 (-T*T--T*-) and monoglycopeptide 4 (-T*T-), (Figure 1a and Table 2) suggest that the glycosylation of diglycopeptide 5 may be dominated by -T*T- binding at the catalytic domain rather than binding of the remote C-terminal T* at the lectin domain. This agrees with the imperceptibly low activity of glycopeptide 2 (-TT--T*-), which we take as evidence that the C-terminal glycosite either did not to bind the lectin domain, or if bound, it failed to correctly orient the N-terminal acceptor region of the substrate into the catalytic domain.14 However, as discussed below, our kinetic studies of the GalNAc-T4 lectin mutant suggest that the lectin domain may play some role in the observed activity of diglycopeptide 5.
Table 2. Kinetic Parameters for the Wild-Type and Mutant GalNAc-T4.

Kinetic values and R2 obtained from the GraphPad Michaelis–Menten fit of the plots in Figures 1a and 4.
Kinetic values and R2 obtained from the GraphPad Michaelis–Menten fitting with substrate inhibition of the plots in Figure 4.
Cat Eff, catalytic efficiency obtained from the Vmax/Km ratio.
UDP-GalNAc hydrolysis obtained from Sephadex G10 chromatography summarized in Figure S8.
The comparison of the two most active monoglycopeptides 3 (-T*--TT-) and 4 (-T*T-) reveals the dominant influence of the remote N-terminal prior glycosylation on enzyme activity, where peptide 3 has a ∼8-fold higher Vmax, ∼12-fold lower Km, and a ∼100-fold higher catalytic efficiency (Vmax/Km) compared to peptide 4 (Table 2). This influence of the lectin domain is also observed for diglycopeptide 6 (-T*--T*T-), which possesses both remote and neighboring glycosites N-terminal of the acceptor site (Figure 1a). However, the kinetic plot for diglycopeptide 6 was best fitted to a Michaelis–Menten model with substrate inhibition (Figure 1a, Table 2). The Km of peptide 6 (-T*--T*T-) is ∼1.3-fold lower than that for peptide 3 (-T*--TT-) (∼39 vs ∼51 μM) suggesting a weak synergistic effect of the two glycosites in peptide 6, each binding separately to the lectin and catalytic domains of the enzyme. This fact implies that substrate binding to the enzyme is slightly stronger when both the lectin and catalytic domains bind GalNAc residues with the appropriate N-terminal placement, as found for the diglycosylated substrate. This synergistic effect is clearly observed in the substrate Kd values obtained by surface plasmon resonance (SPR) in the presence of excess of UDP and MnCl2 (see the Methods section). While the Kd values for both monoglycopeptides 4 (-T*T-) and 3 (-T*--TT-) were relatively weak (in the mM range; see Figure S1 and ref (14)), the Kd value for diglycopeptide 6 (-T*--T*T-) was 70 ± 15 μM (Figure 1b). Interestingly, the observed Km values for these three glycopeptides are lower than their Kd values, especially for glycopeptides 3 and 6. The discrepancies between GalNAc-T4’s higher-affinity Km values obtained from our kinetics studies (Table 2) and the Kd values obtained from direct binding studies, particularly for peptide 3, can be attributed to the fact that the UDP-GalNAc donor was present in the enzyme kinetics, but absent in the SPR binding studies.14 UDP-GalNAc stabilizes the so-called flexible loop of the catalytic domain active site in a “closed” conformation, which completes the formation of the peptide-binding groove and leads to an active transferase.13,14,18 Thus, our SPR studies in the absence of UDP-GalNAc likely represent the weak binding of peptide substrate to the “open” flexible loop conformation of the enzyme. However, this discrepancy is not as large for peptide 6, where Kd is only ∼2-fold higher than the Km observed from the kinetic analysis. This suggests that the high binding affinity of peptide 6 (due to GalNAc binding at both domains) might further stabilize/drive the GalNAc-T4’s catalytic domain flexible loop in a closed conformation in the presence of either UDP-GalNAc or UDP (as confirmed by the crystal structure discussed below). The synergistic effects observed in the kinetic and Kd values obtained for diglycopeptide 6 (-T*--T*T-) compared to monoglycopeptides 3 (-T*--TT-) and 4 (-T*T) are due to the divalency of peptide 6, which may simultaneously or consecutively bind to both the lectin and catalytic domains of the transferase. Such synergy is common to multivalent carbohydrate–lectin binding systems.23,24 In summary, the initial catalytic efficiency (Vmax/Km) of peptide 6 (-T*--T*T-) is ∼1.5- and ∼150-fold higher than the catalytic efficiency of peptides 3 (-T*--TT-) and 4 (-T*T-), respectively, showing again that the remote T* binding to the lectin domain dominates overall catalytic efficiency. It is also worth noting that the much higher catalytic efficiencies of glycopeptides 3 and 6 compared to those deduced for glycopeptides 4 and 5 correlate with their extent of nonproductive hydrolysis of UDP-GalNAc by the transferase, where glycopeptides 3 and 6 display only ∼5% hydrolysis, while glycopeptides 4 and 5 display ∼20% hydrolysis (Table 2 and the Methods section). Likewise, peptides 1 and 2, with extremely low catalytic efficiencies, display even higher rates of hydrolysis of ∼40–50% (Figure S8).14
As mentioned above, the kinetic plot for diglycopeptide 6 (-T*--T*T-) is best fitted to a Michaelis–Menten kinetic model that includes substrate inhibition (Figure 1a and Table 2). The appearance of substrate inhibition at high diglycopeptide 6 concentration could be due to the nonproductive binding of the neighboring GalNAc in the -T*TPGP- sequence at the lectin domain. Such a binding event would compete with the “productive” binding of the remote GalNAc (-T*-) at the lectin domain and would lead to the observed decrease in activity at high substrate concentration. This explanation implies that the binding affinity of the -T*TPGP- epitope at the lectin domain is weaker than that of the remote T* GalNAc of glycopeptide 6. In this case, the glycosylation rate at high substrate concentration, would ideally be ∼1/2 of the initial maximum rate, as indeed observed (Figure 1a).
GalNAc-T4–Substrate Interactions with (Glyco)peptide Substrates by STD-NMR
To further characterize the different modes of glycopeptide substrate binding to GalNAc-T4, we performed saturation transfer difference (STD) NMR experiments on the substrates in Table 1, in the presence of GalNAc-T4, UDP, and MnCl2, as described in the Methods section. Our previous STD-NMR studies with peptides 1–3 (-TT-, -TT--T*-, -T*--TT-)14 have revealed different features: (1) No significant STD enhancements were observed for peptide 1 (-TT-), suggesting that it poorly binds GalNAc-T4, in agreement with its very low glycosylation rate. (2) Poor recognition of the peptide residues was revealed in glycopeptides 2 and 3 (-TT--T*-, -T*--TT-). (3) Significant, but different, STD intensity patterns for the GalNAc protons of glycopeptides 2 and 3, suggesting the existence of different binding modes, were probably related to their different rates of glycosylation14 (Figure S2). These STD-NMR results were further supported by the crystal structure of the complex of GalNAc-T4-UDP-Mn2+-glycopeptide 3, which shows the GalNAc moiety of glycopeptide 3 bound to the GalNAc binding site at the lectin domain.14
Here, we continued these STD-NMR studies with the new glycopeptides 4, 5, and 6 (-T*T-, -T*T–T*-, and –T*--T*T-). In contrast to monoglycopeptides 2 and 3, glycopeptides 4, 5, and 6 show significant STD-NMR enhancements for the protons of the Thr and Pro residues comprising the -T*TPGP- peptide sequence (Figure 1c, Figures S2–S6 and Tables S1–S3). The GalNAc moieties in 4, 5, and 6 also gave clear STD-NMR enhancements for the methyl protons of the GalNAc N-acetyl group (NHAc) (>75%, Figures S2–S6 and Tables S1–S3). However, differences in the relative STD-NMR enhancements of the GalNAc ring protons are clearly observed between glycopeptides 3, 4, 5, and 6. When comparing monoglycopeptides 3 and 4, (-T*--TT- and -T*T-) the highest STD-NMR responses are observed for the H2, H4, and NHAc methyl protons in 3, while H3 and NHAc methyl protons give the largest STD-NMR enhancements in 4 (Figure 1c and Figure S4). These differences in the STD-NMR intensities between glycopeptides 3 and 4 suggest different recognition modes of the GalNAc moieties by the transferase, fully consistent with their proposed binding at the lectin and catalytic domains, respectively. The observed STD-NMR enhancements for the protons of the Thr, Pro, and GalNAc moieties in the -T*TPGP- acceptor sequence of monoglycopeptide 4 are consistent with the GalNAc residues interacting at the catalytic domain rather than at the lectin domain (see below).
For diglycopeptides 5 and 6 (-T*T--T*- and -T*--T*T-), given the observed extensive overlapping, most of the observed STD-NMR enhancements in the GalNAc residue represent the combined STD from both GalNAc residues. Nevertheless, the individual NHAc methyl resonances from the different GalNAc residues in 5 and 6 were well-resolved, showing high STD-NMR enhancements (Figures S5 and S6), which suggests that both NHAc groups are bound. In contrast, the STD-NMR response for the resolved H2 protons of the two GalNAc residues in 6 are rather different (Figure 1c and Figures S5 and S6). The H2 proton of the GalNAc at Thr3 (i.e., the remote lectin bound GalNAc) shows a much stronger STD (90%) than that (50%) at Thr11 (i.e., the catalytic domain bound neighboring GalNAc-). This low STD response for the H2 proton of the GalNAc at Thr11 in 6 (-T*--T*T-) is similar to that observed for the monoglycopeptide 4 (-T*T-) and strongly suggests that the neighboring GalNAc moiety in 6 also preferentially binds to the catalytic domain of GalNAc-T4. This is again supported by the observation of STDs for the protons of the peptide residues of the -T*TPGP- acceptor sequence in both 4 and 6 (Figure 1c and Figures S4 and S6).
On the other hand, there are no STD enhancements for the peptide backbone of glycopeptides 2 and 3, which lack the neighboring GalNAc-Thr residue in the acceptor sequence, -TTPGP-,14 while STD enhancements are clearly present in glycopeptides 4, 5, and 6, whose acceptor sequences do contain it. This, taken together with the different STD-NMR patterns for the GalNAc protons and the higher activities of the -T*T- containing peptides compared to the -TT- analogues (particularly 1 vs 4 and 2 vs 5), strongly suggests that the neighboring GalNAc in -T*T- and the flanking -PGP- motif together increase peptide substrate binding (in the absence of a remote N-terminal prior glycosite). Furthermore, this binding event must specifically occur at the catalytic domain, since the GalNAc moiety is transferred to the free Thr in the -T*TPGP- acceptor sequence upon catalysis.
Glycopeptides 3 and 6 (-T*--TT- and -T*--T*T-), which show the highest Vmax and catalytic efficiency of all of the peptides (Table 2), nevertheless display dramatically different STD-NMR enhancements at their -TTPGP- and -T*TPGP- acceptor sequences. In fact, 6 provides high STDs, while 3 does not give any enhancements. This is consistent with a binding event between the remote N-terminal prior glycosite and the lectin domain in both glycopeptides, while only in 6 is there tight binding at the catalytic domain due to the presence of a neighboring glycosite in the acceptor T*TPGP sequence. This is in keeping with our earlier structure of 3 (-T*--TT-) bound to GalNAc-T4, which revealed no electron density for the peptide bound to the catalytic domain and the lack of STD enhancements for the acceptor -TTPGP- sequence. Therefore, the combination of kinetics and STD-NMR results provides strong evidence that a separate peptide GalNAc binding exists in the catalytic domain of GalNAc-T4 that accounts for its N-terminal short-range neighboring glycosylation preference.
Crystal Structure of GalNAc-T4 in Complex with UDP-Mn2+ and Glycopeptide 6 (-T*--T*T-)
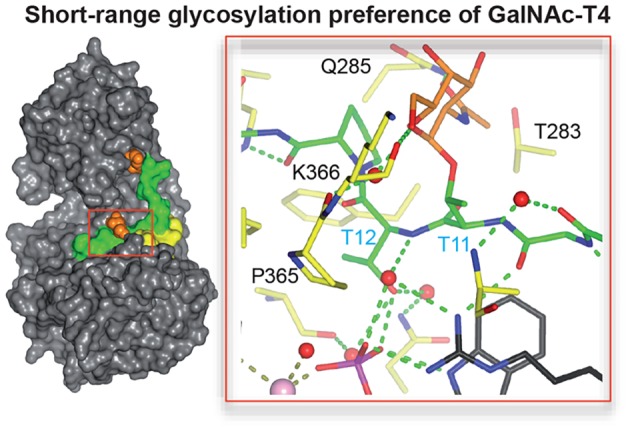
To identify the catalytic domain (neighboring) GalNAc-peptide binding site on GalNAc-T4 and to further understand how the transferase recognizes both donor and glycopeptide acceptors, we obtained triclinic crystals of GalNAc-T4 that were subsequently soaked with diglycopeptide 6, UDP, and MnCl2. The resulting crystal was solved giving a structure of the transferase/diglycopeptide complex at 1.80 Å resolution, containing two independent GalNAc-T4 molecules in the asymmetric unit (Table S4). The obtained structure shows a compact GT-A fold and a lectin domain located at the N- and C-terminal regions of the transferase, respectively. Moreover, a clear structure of diglycopeptide 6 is evident bound to both domains (Figure 2a). Additionally, the structure clearly shows the flexible linker connecting both domains and the flexible loop of the catalytic domain (Figure 2a), which account for the distinct long-range glycosylation preferences and for the inactivation/activation process of these enzymes, respectively.14,18 In our earlier crystal structure of GalNAc-T4 complexed with 3 (-T*--TT-), the flexible loop at the catalytic domain was disordered, and no density for glycopeptide 3 could be observed at the catalytic domain.14 In contrast, the present structure of GalNAc-T4-UDP/Mn2+ complexed with 6 (-T*--T*T-) shows both structural features fully ordered (Figure 2a). This is likely due to the stabilization provided by the binding of both UDP and the GalNAc-containing peptide to the catalytic domain (Figure 2a,b). Herein, a loop protruding from the lectin domain toward the catalytic domain glycopeptide binding site is also evident, which we have called the “lectin flexible loop” (Figure 2a residues 460–472; hereafter named LFL). This LFL motif has not been previously described for other GalNAc-T structures, and its significance will be discussed below.
Figure 2.

Crystal structure of GalNAc-T4 in complex with UDP-Mn+2 and glycopeptide 6. (a) Two different views of GalNAc-T4 in complex with 6. The catalytic and lectin domains are colored in gray, and the flexible linker and catalytic domain active site loop are depicted in red and yellow, respectively. The lectin domain flexible loop (LFL) is indicated by a black arrow. The GalNAc moiety of the Thr3-GalNAc and Thr11-GalNAc is shown as orange carbon atoms while the rest of the peptide is shown as green carbon atoms. The nucleotide is depicted as brown carbon atoms whereas the manganese atom is shown as a pink sphere. Inserted between the structures is a surface representation of GalNAc-T4 with the same orientation as the cartoon representation of the leftmost structure. (b) Electron density maps are FO–FC (blue) contoured at 2.0 σ for glycopeptide 6, UDP, and manganese ion. (c) Two different views of GalNAc-T2 in complex with MUC5AC-13 (PDB entry 5AJP(14)) again with an inserted surface representation between structures. Atom colors are the same as in part a above. (d) Close-up views of the GalNAc-T2-UDP-MUC5AC-13 and the GalNAc-T4-UDP-glycopeptide 6 complexes showing the bound glycopeptide and the catalytic domain active site flexible loop (in black). Note that flexible loop residues Trp331 in GalNAc-T2 and Trp334 in GalNAc-T4 adopt an “in” loop conformation.
Comparison between the GalNAc-T4 and T2 Structures Bound to Glycopeptides
The GalNAc-T4 structure complexed with 6 (-T*--T*T-) and UDP/Mn2+ was compared (Figure 2c) to that of GalNAc-T2 bound to with MUC5AC-13 (GTTPSPVPTTSTT*SAP) and UDP/Mn2+ (PDB entry 5AJP). The observed different orientations of the lectin domain account for the distinct long-range/remote prior glycosylation preferences of these transferases, i.e., for N-/C-terminal glycosites, respectively (compare left panels and space filled inserts of Figure 2a,c).14 In addition, in both crystal structures, the flexible loop of the catalytic domain (residues 363–374 in GalNAc-T4 and residues 360–372 in GalNAc-T2; see Figure S7) adopts the so-called “closed conformation”, indicating an active state structure. The active form of the GalNAc-T4-6 complex is further supported by the presence of the so-called “in conformation” of the catalytic Trp334 (Trp331 in GalNAc-T2, Figure 2d).13 On this basis, the crystal structure of GalNAc-T4 bound to UDP and 6 describes the active conformation.
Lectin Domain Substrate Binding
The remote GalNAc moiety (Thr3-GalNAc of 6) bound at the lectin domain is tethered by hydrogen bonds by the conserved residues Asp459/His478/Asn483 (Figure 3a) (for GalNAc-T2, Thr13-GalNAc of MUC5AC-13 interacts with Asp458/His474/Asn479, Figure 3b). In addition, CH−π interactions are also observed between the Thr3-GalNAc moiety and Phe475 of GalNAc-T4 (Tyr471 in GalNAc-T2) (Figure 3a,b). Out of the 16 residues of 6, ∼1/4 (i.e., Gly4-Ala7) are fully exposed to the solvent when bound to GalNAc-T4, while this is not observed for the homologous MUC5AC-13 bound to GalNAc-T2 (Figure 3a,b). This implies there may be a gap in substrate binding between the lectin domain GalNAc binding site and the catalytic domain (glyco)peptide binding site that has not been observed for GalNAc-T2 (see the surface representations in Figure 2a,b). Although we cannot rule out the role of the particular sequence of the peptide substrate sequence, this observation suggests that the two transferases may indeed differ in their binding mode of glycopeptide substrates in the region spanning the lectin and catalytic domain (Figure 3a,b).
Figure 3.

Structural features of peptide, UDP, and lectin-domain-binding sites of GalNAc-T4. (a) View of the complete sugar nucleotide, peptide, and lectin-domain-binding sites of the GalNAc-T4-UDP-glycopeptide 6 complex. Upper panel: close-up view of bound glycopeptide. Lower panel: close-up view of the manganese binding site. The residues forming sugar-nucleotide, peptide, and lectin-domain-binding sites are depicted as black, yellow, and gray carbon atoms, respectively. UDP and the glycopeptide are shown as brown and green carbon atoms, respectively. Mn2+ and GalNAc moiety are depicted as a pink sphere and orange carbon atoms, respectively. Hydrogen bond interactions are shown as dotted green lines. Water molecules are depicted as red spheres. Note that we only show water-mediated interactions in which only one water molecule acts as a bridge between the residues. (b) View of the sugar nucleotide, glycopeptide, and lectin-domain-binding sites of the GalNAc-T2-UDP-MUC5AC-13 complex (PDB entry 4D0T). Colors are the same as above. (c) Modeled structure for a GalNAc-T4-UDP-GalNAc-glycopeptide 6 complex. The coordinates of the UDP-GalNAc were obtained by superposing the structure of GalNAc-T2 containing UDP-GalNAc (PDB entry 4D0T) with the GalNAc-T4-UDP-GalNAc-glycopeptide 6 complex. The structure shows that the Thr12 acceptor of glycopeptide 6 is close to the anomeric carbon of UDP-GalNAc.
Catalytic Domain Substrate Binding
At the catalytic domain peptide-binding site of GalNAc-T4 bound to 6, most of the interactions with the peptide are through direct and water-mediated hydrogen bonds and to a lesser extent through hydrophobic interactions. Direct hydrogen bonds are observed with the Gly8/Ala9 and Pro13 peptide substrate backbone carbonyls and the side chain NH’s of Arg372 and Trp286, respectively, while a CH−π interaction takes place between Pro15 and Trp286. The acceptor Thr12 side chain hydroxyl group is hydrogen bonded to the UDP β-phosphate, showing that this residue is well-located to accept the GalNAc moiety of the UDP-GalNAc (Figure 3a,c). In addition, the Thr12 methyl group is located in a hydrophobic environment formed by the side chains of Phe284, Phe364, Trp286, and Ala310, as earlier described for GalNAc-T2 (residues Phe280, Trp282, Phe361, and Ala307) in complex with the mEA2 peptide (STCPA) (PDB entry 4D0Z).18 We proposed that this environment helps to correctly position the acceptor Thr hydroxyl group to accept the anomeric carbon of UDP-GalNAc, thus enhancing the turnover and further explaining why Thr residues are better acceptors than Ser residues.18 Water-mediated hydrogen bonds are observed through (1) Ala9/Gly10 and Gly16 backbone with Ala368/Arg372 backbone and Thr258 side chain hydroxyl, respectively; (2) Thr12 backbone with Arg 372 backbone; and (3) Thr12 side chain with Asn338 side chain and Ala310 backbone (Figure 3a).
In contrast to the observations for 6 bound to GalNAc-T4, most of the interactions of the MUC5AC-13 glycopeptide (GTTPSPVPTTSTT*SAP) bound to GalNAc-T2 take place through hydrophobic and water-mediated hydrogen bond interactions, with few direct hydrogen bond interactions (Figure 3b).13 Overall, our comparison of the GalNAc-T2 and -T4 peptide substrate bound structures demonstrates that both employ a well-balanced number of direct and water-mediated interactions that differ in type and number. While the complex of 6 with GalNAc-T4 mainly relies on direct hydrogen bonds, the binding of MUC5AC-13 to GalNAc-T2 is dominated by multiple hydrophobic interactions. These results may be due to both the difference between the residues forming the peptide binding pocket and the different peptide substrate sequences. However, both enzymes share nearly identical hydrophobic interactions that are used to recognize the -PXP- motif found in both peptides, Pro13 in 6 (GAT*GAGAGAGT*TPGPG, italicized), and Pro6 in MUC5AC-13 (GTTPSPVPTTSTT*SAP; Figure 3a,b). The analysis of both crystal structures permits us to infer that the relative promiscuity of both enzymes (outside the -PXP- motif) for recognizing multiple and dissimilar protein/peptide substrates relies on a combination of direct interactions of the peptide substrate amino acid residues with the transferase and with water-mediated hydrogen bond interactions. A similar mechanism of protein substrate recognition has been described for the promiscuous O-fucosyltransferase 2 (PoFUT2).25 Hence, this recognition event may be general for diverse peptide substrates binding to enzymes.
GalNAc-T4 Catalytic Domain Glycopeptide-GalNAc Binding Site
A putative GalNAc binding site in the catalytic domain of GalNAc-T4 proposed in our earlier random glycopeptide studies10 has been confirmed by the kinetics and STD-NMR experiments described above. Such a binding site is not present in GalNAc-T2.10 The structure of GalNAc-T4 bound to diglycopeptide 6 (-T*--T*T-) clearly reveals the GalNAc moiety on Thr11 (directly N-terminal of the acceptor Thr12) tethered to the surface of the catalytic domain (Figure 3a). This binding event is mediated by hydrogen bond interactions between GalNAc-O6 and the Lys336 backbone carbonyl and between the carbonyl group of GalNAc and the Gln285 side chain amide. In addition, water-mediated and CH3/CH3 hydrophobic interactions are shown between the GalNAc O6 and the Pro365 backbone carbonyl, and the methyl group of GalNAc and the Thr283 methyl, respectively (Figure 3a). As shown below, mutation of Thr283 and Gln285 significantly alters GalNAc-T4’s neighboring glycopeptide activity. All together, these interactions show how the specific GalNAc-Thr binding event at the catalytic domain directly presents the adjacent C-terminal Thr acceptor into the correct orientation for subsequent GalNAc transfer from the bound UDP-GalNAc. Thus, in addition to the remote lectin bound GalNAc residue, the presence of the neighboring substrate GalNAc moiety together provides the specific and essential contacts with the transferase that are required for optimal activity.
GalNAc-T4 Nucleotide-Sugar Binding Site
Both GalNAc-T2 and T4 share a very similar nucleotide-sugar binding sites with ∼60% identical residues (Figure 3a,b). The UDP pyrimidine moiety in GalNAc-T4 is sandwiched between Tyr144 and Leu207 (His145 and Leu204 in GalNAc-T2), while the Mn2+ ion is hexagonally coordinated by the UDP pyrophosphate group, His362, the D227XH229 motif (His359 and D224XH226 in GalNAc-T2), and a water molecule (Figure 3a). UDP is mostly tethered by conserved hydrogen bonds with Ala142, Asp175, Arg204, Cys228, Trp334, and Tyr370 that are also present in GalNAc-T2 (Thr143, Asp176, Arg201, Ser225, Trp331, and Tyr367 in GalNAc-T2). Both enzymes also share interactions between Tyr370 (Tyr367 in GalNAc-T2) with the pyrophosphate moiety. The complex of GalNAc-T4 shows a hydrogen bond between the Tyr144 backbone and the uridine ribose, which is not found in that of GalNAc-T2. However, most of the differences at this binding site between both enzymes arise from the nonconserved amino acids in their flexible loops and the differences in peptide substrates. For example, for GalNAc-T4, Arg372 and Pro365/Lys366 recognize the β–pyrophosphate/glycopeptide 6 and GalNAc on Thr11, respectively, while, for GalNAc-T2, His365 and Arg362 interact with the MUC5AC-13 substrate (Figure 3a,b). Overall, our results confirm that the recognition of UDP-GalNAc is highly conserved between GalNAc-Ts, whereas the nonconserved flexible loop and the peptide-binding groove are responsible for their different peptide substrate specificity.
Correlation between Crystal Structure and STD-NMR
The STD-derived epitope mapping reported in Figure 1c and Figures S2–S6 is consistent with the X-ray crystal structure of 6 bound to GalNAc-T4. For the bound -T*TPGP- fragment at the catalytic domain, the observed STDs for methyl group protons of GalNAc and both Thr residues, together with the CH2 protons of both Pro residues of compounds 4, 5, and 6, are readily explained by the X-ray structure since all these protons are in close contact to the transferase surface (Figure S9). The sugar moiety appears relatively far from the catalytic domain. However, the GalNAc moiety bound to the lectin domain fits into a cleft that displays multiple contacts, including the NHAc methyl group protons (Figure S9). These interactions account for the STD enhancements observed for the GalNAc H2, H3, and H4 and NHAc methyl protons in 3. All together, these results are fully consistent with a model in which the bound GalNAc at the catalytic domain shows poor contacts with the enzyme and therefore weak STDs, while that bound at the lectin domain displays important interactions with large STD enhancements.
GalNAc Binding Site Mutants of GalNAc-T4
To further assess the roles of the individual lectin and catalytic domains on the binding of GalNAc glycopeptide substrates, GalNAc binding mutants were expressed for kinetic analysis against the (glyco)peptide substrate library in Table 1. These were designed to (1) eliminate the remote GalNAc binding site at the lectin domain (lectin mutant); (2) attempt to eliminate the neighboring GalNAc binding on the catalytic domain (catalytic mutant); (3) understand the role of the lectin flexible loop (LFL mutant); (4) understand the combined role of the catalytic domain and LFL on GalNAc binding (catalytic/LFL mutant); and finally (5) understand the effect of eliminating both the lectin and catalytic domain GalNAc binding sites (lectin/catalytic mutant). The results of these kinetic studies in comparison to those carried out for wt GalNAc-T4 are given in Figure 4 and Table 2. To summarize, the results clearly show the expected effects of the different mutations. The remote lectin domain glycopeptide activity is eliminated in the lectin mutants (left panels Figure 4), while the neighboring glycopeptide activity is reduced but not completely eliminated in the catalytic domain and LFL mutants (right panels Figure 4). These findings show that it is possible to produce a transferase that lacks one or the other prior GalNAc binding activities or one that is nearly void of any prior glycosylation activity. We discuss the obtained findings for each mutant in more detail below.
Figure 4.
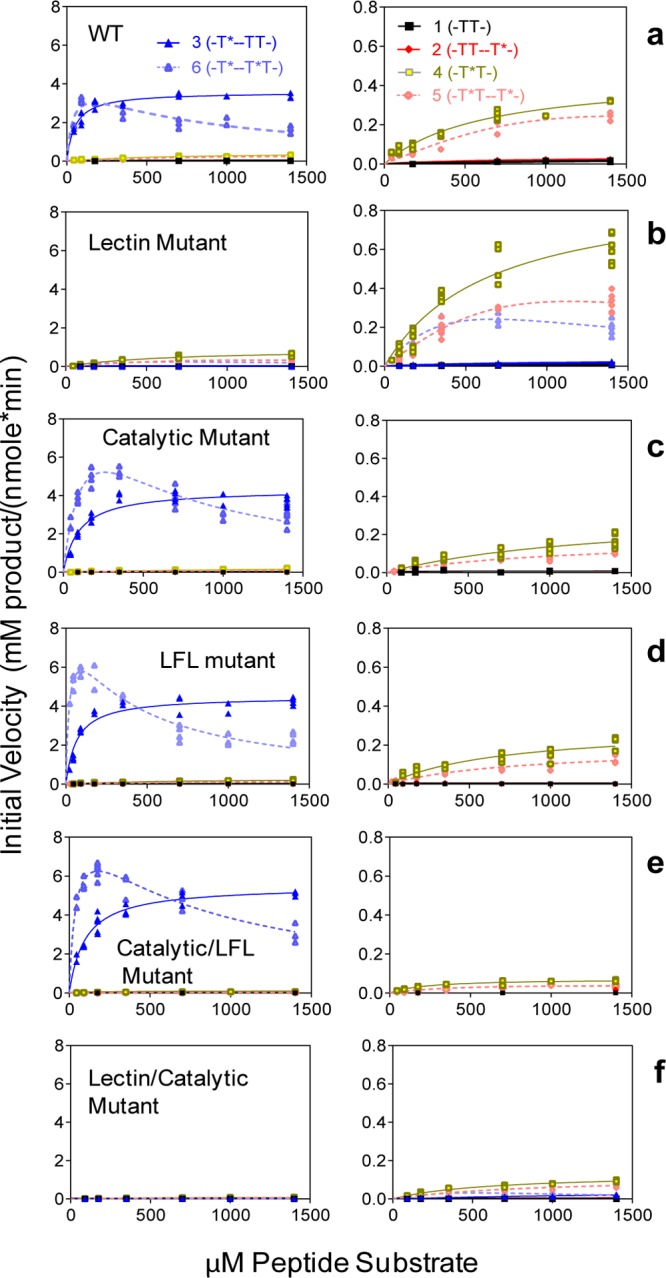
Enzyme kinetics of wt and mutant GalNAc-T4 against the (glyco)peptide substrates in Table 1. Note that the left and right panels are plotted with different initial activity scales. Kinetic constants obtained from the plots are given in Table 2. (a) Wild-type GalNAc-T4 showing both long-range (left panel) and short-range (right panel) glycopeptide activities. (b) Lectin mutant (D549H) showing the loss of its long-range glycopeptide activity. (c) Catalytic mutant (T283S, Q285A) showing the partial loss of GalANc-T4’s short-range prior glycopeptide activity. (d) Lectin flexible link (LFL) mutant (P463DNNP467 to GGG) showing a partial loss of the short-range glycopeptide activity while possessing an intact catalytic domain. (e) Catalytic/LFL combined mutant (T283S, Q285A, D464A) showing a more complete loss of the short-range glycopeptide activity. (f) Lectin/catalytic combined mutant (T283S, Q285A, D459H) showing the near complete loss of both the long-range and short-range glycopeptide activities.
Lectin Domain GalNAc Binding Mutant
To eliminate the long-range (i.e., -T*--T*T- and -T*--TT-) glycopeptide binding at the lectin domain, the critical Asp in the classical CysLeuAsp (CLD) GalNAc binding motif was mutated to His (D459H lectin mutant). Such Asp to His mutations in other GalNAc-Ts, including T4, have been shown to eliminate their lectin binding properties.11,26−29 Accordingly, the GalNAc-T4 D459H mutant shows a dramatic loss in its long-range prior glycosylation activity against glycopeptides 3 and 6 (-T*--TT- and -T*--T*T-) (Figure 4b, left panel), which can only be observed in the expanded scale plot at the right (Figure 4b). Thus, for 3, the Km increased ∼20-fold, and its Vmax decreased over 1000-fold, approaching the activity of the naked peptide 1.14 For 6, Km also increased ∼20-fold, but its apparent Vmax decreased only ∼5-fold, providing half of the activity of glycopeptide 4 (Figure 4b). In addition, the nonproductive UDP-GalNAc hydrolysis significantly increased to ∼70% and ∼20%, for 3 and 6, respectively, in the lectin mutant, while for the wt transferase hydrolysis is only ∼5% for both substrates (Table 2 and Figure S8). These results clearly demonstrate the role of the lectin domain in directing the long-range prior glycosylation activity of GalNAc-T4 and that this is the dominant activity of GalNAc-T4. Nevertheless, the mutant displays a residual activity for glycopeptide 3 (-T*--TT-) compared to peptide 1 (-TT-) and a slight substrate inhibition for 6 that suggests that a weak lectin domain binding activity remains in the mutant, probably due to weak binding at its β- or γ-lectin subdomains.10
Although the GalNAc binding site of the catalytic domain was unchanged, the glycosylation of 4, containing the neighboring glycosylation motif -T*T-, was significantly higher, giving a ∼2-fold higher Vmax and catalytic efficiency compared to the wt transferase. Moreover, the UDP-GalNAc hydrolysis decreased by ∼1/2 (Figure 4b right, Figure S8, and Table 2). We attribute this ∼2-fold increase in activity to the loss of the nonproductive binding of -T*TPGP- at the lectin domain in this mutant compared to the wt GalNAc-T4. Interestingly, both diglycopeptides 5 and 6 (-T*T--T*- and -T*--T*T-), which contain both the long-range and the neighboring prior GalNAc residues, show about half of the activity of glycopeptide 4 (-T*T-) (Figure 4b, right panel, Table 2). This lower activity can be rationalized by recognizing that diglycopeptides 5 and 6 contain two substrate GalNAc residues that compete for binding at the catalytic domain GalNAc binding site. This competition process leads to a net reduction in overall transfer, since only the bound -T*T- sequence can be glycosylated. Thus, the observed rates of glycosylation of the diglycopeptides should approach half of that of glycopeptide 4 (Figure 4b and Table 2). Hence, the kinetic parameters derived for monoglycopeptide 4 with this “simplified” mutant likely represent the intrinsic rates of GalNAc-T4 for glycosylating the -T*TPGP- sequence.
Catalytic Domain GalNAc Binding Mutant
Based on the structure of the bound diglycopeptide 6 (-T*---T*T-), the side chains of Thr283 and Gln285 of the catalytic domain are the only potentially mutable residues involved in direct interactions with the neighboring peptide GalNAc residue (i.e., -T*T-) of the glycopeptide substrate. This catalytic mutant (T283S/Q285A) was therefore expressed to disrupt these interactions. As expected, the catalytic activities (Vmax) against glycopeptide substrates 4 and 5 (-T*T-, -T*T--T*-), were reduced to ∼1/3 of the wt GalNAc-T4 activity (Figure 4c right, Table 2). This incomplete inactivation suggests that the neighboring GalNAc binding site is only partially abolished in this mutant, presumably due to the remaining GalNAc-peptide backbone binding interactions shown in the crystal structure (i.e., Lys336 and Pro365, Figure 3), which are not eliminated by this mutation. Furthermore, the Km values for glycopeptides 4 and 5 were ∼2-fold higher than those of the wt and lectin mutant GalNAc-T4. Finally, the small increase in nonproductive hydrolysis of UDP-GalNAc observed with these glycopeptides correlates with the lower activity of glycopeptides 4 and 5 compared to the wt enzyme (Table 2 and Figure S8). All together, these observations strongly suggest that the -PGP- motif remains a significant contributor to the binding of substrate in the catalytic mutant.
To further confirm that the catalytic mutant has reduced or eliminated the −1 neighboring glycopeptide activity (i.e., for the -T*T- motif), a random glycopeptide GPIID (GAGAXXXXXT*XXXXXAGAG, where X= GARPNEYV and T) was glycosylated by the catalytic mutant and Edman amino sequenced to determine the sites of 3H-GalNAc incorporation.10 As shown in Figure S10, the catalytic mutant clearly showed a reduction of incorporation of 3H-GalNAc at the Thr acceptor contiguous to the T* at the −1 site, compared to the wt GalNAc-T4, further confirming, in a nonambiguous manner, the partial loss of the neighboring glycosylation activity in this mutant.
As expected, glycosylation of 3 and 6 (-T*--TT- and -T*--T*T-) by the catalytic mutant is dominated by their remote prior glycosylation activity, due to GalNAc binding to the lectin domain. Thus, the obtained kinetic plots are similar to those for the wt GalNAc-T4 (Figure 4a,c), including the presence of substrate inhibition for diglycopeptide 6. However, 6 displays a ∼4-fold higher Km and a ∼6-fold higher Kd by SPR (Figure S11) and a ∼2-fold higher initial Vmax compared to the wt transferase, resulting in about 1/2 the catalytic efficiency, Vmax/Km (see Table 2). These higher Km and Kd values in the catalytic mutant are consistent with a significant loss of GalNAc binding at the catalytic domain and the subsequent loss of divalent binding to the enzyme, as compared to the native GalNAc-T4. Although merely speculative, the observed elevation in apparent Vmax for 6 could be explained by the loss of nonproductive binding at the catalytic domain for the remote -T*- substrate GalNAc residue, along with the weaker binding of the lectin domain to the -T*TPGP- sequence (Figure 4c and Table 2). Nevertheless, 6 still displays substrate inhibition at high concentrations, presumably due to the onset of nonproductive binding of the -T*TPGP- sequence at the lectin domain competing with the productive binding of the remote -T*-. In the catalytic mutant, monoglycopeptide 3 (-T*--TT-) does not show any significant increase in rate compared to the wt transferase, likely due to the fact that the catalytic domain GalNAc binding site has not been fully inactivated in this mutation. As shown below, when the catalytic domain GalNAc binding is more fully reduced, the Vmax for 3 indeed increases. Finally, it should be mentioned that the extent of nonproductive UDP-GalNAc hydrolysis for glycopeptides 3 and 6 for the catalytic mutant are very low, as observed for the wt GalNAc-T4, again consistent with their high activities and lectin domain binding (Table 2 and Figure S8).
Lectin Domain Flexible Loop Mutant
As mentioned above, the structure of GalNAc-T4 bound to glycopeptide 6 displays a flexible loop (LFL) protruding from the lectin domain toward the catalytic domain bound GalNAc in the -T*TPGP- sequence. The superposition of GalNAc-T4 structures in the apo form with those in complex with 3 and 6 revealed conformational changes in the LFL (see below and Figure S12). Thus, a truncated mutant was generated, where the 463PDNNP467 sequence located at the tip of the loop was replaced by -GGG-. Surprisingly, this mutant gave nearly identical reductions in catalytic efficiency against glycopeptides 4 and 5 as those obtained for the catalytic mutant (i.e., ∼50% reductions compared to wt GalNAc-T4, Figure 4d and Table 2). These decreases were driven by Vmax and not by changes in the Km values (Table 2). These results confirm that this loop plays a significant role in the binding of the -T*TPGP- sequence at the catalytic domain. It is also worth noting that the neighboring glycosylation activity of the LFL mutant against the random glycopeptide acceptor GPIID shows the identical −1 glycosylation preference as the wt GalNAc-T4 (see Figure S10). Therefore, the loss of the lectin flexible loop does not alter its intrinsic glycopeptide specificity, which is consistent with the LFL mutant having an intact catalytic domain.
Further evidence that the LFL stabilizes both glycopeptide and peptide substrate binding was deduced by the observation that the nonproductive hydrolysis of UDP-GalNAc for glycopeptides 4 and 5 increased to ∼50%. For peptides 1 and 2 (-TT- and -TT--T*), hydrolysis nearly doubled to ∼90%, compared to the wt transferase (Table 2 and Figure S8). These significant decreases in activity and increases in UDP-GalNAc hydrolysis with an intact catalytic domain strongly suggest that the LFL must play a significant role in transient substrate binding or recognition of both glycosylated (-T*TPGP-) and nonglycosylated (-TTPGP-) substrates. This is the first example demonstrating that the lectin domain of a GalNAc-T can directly modulate transferase catalytic activity through its interactions with the substrate bound to the catalytic domain.
As expected, the kinetic plots of the LFL mutant against glycopeptides 3 and 6 (-T*--TT-, -T*--T*T-) were very similar to those observed for the catalytic mutant and the wt GalNAc-T4 enzymes, as they all contain an intact lectin domain GalNAc binding site. For 3, the Km and Vmax values are nearly the same with all three enzymes, except for a ∼2-fold higher Km in the catalytic mutant (Table 2). For 6, a nearly 2-fold higher apparent Vmax is observed in the LFL mutant as compared to the wt GalNAc-T4. This number is similar to that observed for the catalytic mutant. Interestingly, the Km value for glycopeptide 6 in the LFL mutant appears slightly lower, although within experimental error of the wt transferase, while for the catalytic mutant the Km value is ∼3-fold higher (Table 2). Thus, the apparent catalytic efficiency, Vmax/Km, of glycopeptide 6 is increased compared to the wt and catalytic mutant GalNAc-T4. Since the LFL loop deletion/mutation is located only 4 residues C-terminal away of the critical Asp459 of the lectin domain GalNAc binding CLD motif, it is therefore possible that the LFL mutant’s GalNAc binding properties are altered such that the binding of 6 is enhanced.
To further examine the effects of the LFL mutation on the structure of the LFL, molecular dynamics (MD) simulations were performed. The wild-type GalNAc-T4 and its LFL mutant, in complex with UDP-Mn+2 and peptide 6, were employed as starting geometries for 0.5 μs MD simulations on each (see MD simulations protocol for details). Both complexes were stable through the complete MD trajectory. Markedly, in the LFL mutant the modified loop (-GGG-) provided an open-like structure and was rather flexible. In contrast, the wild-type enzyme showed a well-defined loop (-PDNNP-), with a closed-like structure (Figure S13). This closed structure was stabilized by the formation of transient hydrogen bonds between O3 of the Thr11 GalNAc residue bound to the catalytic domain and the Asp464 side chain of the lectin loop (Figure S14). These observations provide further support to the hypothesis that the lectin flexible loop, and particularly Asp464, stabilizes the binding of neighboring prior glycosylated peptide substrates containing the -T*TPGP- motif. The MD simulations were further complemented with a combination of steered molecular dynamics and umbrella sampling simulations (see computational binding simulations) that consisted of pulling out the glycopeptide 6 from the catalytic domain of wt GalNAc-T4 (Figure S15). During this process, the hydrogen bond between Asp464 and the Thr11-GalNAc residue of 6 was lost and replaced with a transient hydrogen bonding with the acceptor hydroxyl of Thr12 (Figure S15). These alternative simulations offer further support for a key role for the LFL and particularly Asp464 in substrate binding.
Combined Catalytic Domain and LFL Mutant
To access the combined roles of the catalytic domain GalNAc binding residues, T283 and Q285, and the LFL key D464, the T283S, Q285A, D464A triple mutant was expressed and purified (named as the catalytic/LFL mutant). It was expected that this mutant would show further reduced activity against the -T*TPGP- motif while retaining its long-range N-terminal -T*- preferences. Indeed, the apparent activity and Vmax values of 4 and 5 were reduced to ∼1/4 of the values for the catalytic and LFL mutants (Figure 4e, right panel, Table 2). Interestingly, their Km values were also reduced compared to the wt and catalytic and LFL mutants; however, this may be an artifact of the low activity of these substrates resulting in an inadequate dynamic range for accurate data fitting. As expected, 3 and 6 displayed similar kinetic plots as the individual mutants with intact lectin domain GalNAc binding sites. However, both glycopeptides gave elevated apparent Vmax values compared to the wt transferase. These findings are consistent with the previous observation of higher rates of glycosylation for the catalytic domain mutant. Note however that the Km value for the diglycopeptide 6 in the catalytic/LFL mutant was identical to that for the wt transferase and ∼4-fold lower than that for the catalytic mutant. In contrast, the Km for monoglycopeptide 3 was ∼2-fold higher than that for the wt, but the same as that observed for the catalytic mutant (Table 2). Although a full explanation for all these observations remains elusive, the catalytic/LFL mutant may nevertheless be considered a kinetically simplified version of GalNAc-T4, with an intact N-terminal long-range prior glycosylation preference, while nearly lacking its neighboring preference for GalNAc. Finally, it is worth noting that the patterns of UDP-GalNAc hydrolysis for both the LFL and catalytic/LFL mutants differ from the catalytic mutant, as shown in Figure S8. For both LFL-containing mutants, the degree of hydrolysis in the presence of 1 (-TT-), 2 (-TT--T*-), 4 (-T*T-), and 5 (-T*T--T*-) is much higher than that with the catalytic mutant. The fact that hydrolysis is doubled in the presence of both the -TT- and -T*T- containing substrates that lack the N-terminal prior -T* again suggests that the lectin flexible loop plays an important role in productive -TTPGP- and -T*TPGP- substrate binding. Based on our previous experience with the GalNAc-Ts, hydrolysis tends to correlate with poorer substrate activity, which can be considered to represent incomplete or poor binding of substrate. In fact, in the absence of any (glyco)peptide substrate, hydrolysis is typically very low.10
Combined Lectin Domain and Catalytic Domain Mutant
Next, an attempt to remove both the long- and short-range prior glycosylation preferences of GalNAc-T4 was carried out, by combining the lectin and catalytic domain mutants. Thus, a new triple mutant (T283S, Q285A, D459H) was generated, which will be called the lectin/catalytic mutant. As shown in Figure 4f and Table 2, the target was largely achieved, although a residual activity for those glycopeptides containing the -T*TPGP- motif still remained. As discussed above, this residual activity may be due to the remaining GalNAc binding residues in the catalytic domain and/or to the presence of Asp464 at the LFL lectin domain. Moreover, UDP-GalNAc hydrolysis for all the substrates, except for glycopeptides 4 and 5, were significantly increased compared to wt GalNAc-T4 (Figure S8), although hydrolysis in the presence of (glyco)peptides 1, 2, 4, and 5 with this lectin/catalytic mutant were still lower that those observed for both LFL mutants. Together, these results further support the role of the LFL assisting in binding substrates at the catalytic domain and the dominance of the lectin domain binding remote N-terminal glycosylated substrates.
Peptide Substrate Preferences of GalNAc-T4 and Its Catalytic Domain Mutant
We have previously developed a series of random peptide substrates, as GAGAXXXXXTXXXXXAGAG, (where X = randomized amino acids), to characterize GalNAc-T peptide substrate preferences (see random peptide sequence motif determination in the Methods section).30,31 By using high enzyme and substrate concentrations along with long incubation times, these substrates were glycosylated by GalNAc-T4 and its catalytic mutant. Their substrate preferences were evaluated in terms of the obtained enhancement values (EVs) (Figures S16 and S17). As expected, both wt and catalytic mutant transferases revealed preferences for the TPXP motif, due to the presence of three highly conserved Phe and Trp residues in the catalytic domain of most GalNAc-T family members.10,13 GalNAc-T4 also revealed high preferences at the −1 position (relative to the site of glycosylation) for Val, Ile, and Met. On this basis, GalNAc-T4 shows peptide substrate preferences fairly close to that of GalNAc-T3.30 Regarding the question whether mutations in the catalytic domain GalNAc binding site would alter its “naked” peptide substrate specificity, particularly at the −1 position,10 no significant differences in the preferences were observed between the wt and mutant transferases (Figures S16 and S17). The observed preferences for Thr-O-GalNAc, Val, and Ile at the −1 position could be related to the presence of their β-branched methyl groups. Indeed, intense STDs are observed for the Thr methyl protons in 4, 5, and 6 in the presence of GalNAc-T4. The presence of methyl-containing residues at the −1 site are likely related to the reported conformational features provided by these residues.32
Thus, additional MD simulations were performed on GalNAc-T4 complexed with UDP-GalNAc and a naked peptide (GAGAGAGXTPGPG, where T is the acceptor Thr) in which the residue X8 (−1 with respect to the acceptor Thr) was replaced by either Val or Ala (Figure S18 and Movies S1 and S2). According to the MD calculations, the peptide containing the Val8 is stabilized by hydrophobic interaction between the methyl groups of Val8 and Ala368. In turn, this hydrophobic patch promotes the proximity between the hydroxyl group of Thr9 and the anomeric carbon of GalNAc. Conversely, the interactions between the methyl groups of Ala8 and Ala368 in the Ala8 substrate were negligible, presumably due to the longer distance between their hydrophobic side chains, prompting Thr9 to move away from the active site. These results satisfactorily explain why GalNAc-T4 prefers β-branched amino acids at position −1 for optimal glycosylation.
Conclusions
The GalNAc-Ts comprise a large family of evolutionary conserved glycosyltransferase isoforms that differentially exhibit substrate specificities for peptides and partially glycosylated GalNAc-glycopeptides. All isoforms use their unique C-terminal lectin domains to bind GalNAc-glycopeptides, and here for the first time we demonstrate that a subset of the GalNAc-T isoforms exemplified by GalNAc-T4 also contain a GalNAc binding site in the catalytic domain. We provide the first conclusive evidence for the direct interaction of the catalytic domain with a GalNAc residue immediately adjacent to the acceptor site explaining the observed GalNAc-glycopeptide substrate specificity of GalNAc-T4 and related isoforms. Unambiguous evidence for the two distinct GalNAc binding capabilities was obtained by the structure of GalNAc-T4 bound to a diglycopeptide (i.e., glycopeptide 6, -T*--T*T-). This structure revealed how the catalytic domain of GalNAc-T4 recognizes a glycopeptide substrate, and represents the first structure of a GalNAc-T with a glycopeptide GalNAc residue bound to its catalytic domain.
Interestingly, the key residues at the catalytic domain responsible for the binding of the neighboring GalNAc residue have been identified, showing that the binding process is dominated by rather weak interactions, as further supported by kinetic studies. When both long- and short-range prior glycosylation sites are combined in one substrate, i.e., diglycopeptide 6, apparent substrate inhibition is observed. Comparing the kinetics of the mutant and wt transferases suggests that the observed inhibition may be due to the weak nonproductive binding of the substrate -T*TGPG- sequence at the lectin domain. This competition with the binding of the remote GalNAc at high substrate concentrations then leads to a decrease in activity. This observation furthermore suggests the lectin domain of GalNAc-T4 may also possess unique peptide sequence preferences as found for GalNAc-T2.28
A unique aspect of this enzyme is the presence of a flexible loop (LFL) protruding from the lectin domain, which assists both GalNAc binding and release. We believe that with this finding we have identified a new structural feature, out of the few already described for GalNAc-Ts, that serves to modulate GalNAc-T4 activity and specificity. Thus, alterations outside the catalytic domain and the lectin domain GalNAc binding sites, that would not be predicted to be deleterious, can have profound effects on the catalytic activity of GalNAc-T4.
In addition, the generation of several GalNAc-T4 mutants has allowed us to individually characterize the kinetics and binding features of the different parts of the enzyme. Finally, the “naked” peptide substrate motif for GalNAc-T4 and its catalytic domain mutants reveals that GalNAc-T4 displays the expected TPXP preference and prefers the β-branched residues Val, Ile, and Met residues preceding its acceptor site.
In summary we have identified the molecular basis for GalNAc-T4’s long- and short-range prior GalNAc glycosylation preferences, demonstrating that the long-range specificity greatly dominates the activity/function of this isoform. The combination of long- and short-range GalNAc-glycopeptide substrate specificity makes GalNAc-T4 ideal for performing the proposed role as a follow-up isoenzyme that fills in unoccupied acceptor sites in densely O-glycosylated regions such as in the tandem repeats of mucins.11,21 This is consistent with GalNAc-T4’s high expression levels in mucin secreting tissues such as the colon, lung, and sublingual gland.1,33 Our newly acquired ability to selectively eliminate the long- and/or short-range glycopeptide activities of GalNAc-T4, based on the crystal structure of GalNAc-T4-UDP-glycopeptide 6 complex, will be an invaluable tool for understanding how GalNAc-T4 performs this important filling-in role.
Methods
Cloning, Site-Directed Mutagenesis, Expression, and Purification
The expression plasmid pPICZαAgalnact4 (36–578) was used as the template for introducing the following single and multiple amino-acid changes by site-directed mutagenesis as described:14 T283S-Q285A (catalytic mutant), D459H (lectin mutant), T283S-Q285A-D459H (lectin/catalytic mutant), and T283S-Q285A-D464A (catalytic/LFL mutant). For generating the lectin flexible loop deletion mutant (LFL mutant), the residues P463DNNP467 were removed and replaced by three Gly residues. Site-directed mutagenesis was performed by GenScript. Wild-type and mutant transferases were purified using the protocol developed for the wild-type enzyme.14
Synthesis of Glycopeptides
Glycopeptides were synthesized as described previously and confirmed by Edman amino acid sequencing on a Shimadzu PPSQ-53A peptide sequencer.14 See Table S5 for the HPLC and MS characterization of each glycopeptide.
Transferase Assays and Kinetics
GalNAc-T glycosylation reactions against (glyco)peptides 1–6 were performed as described.14 Briefly, reactions consisted of 75 mM sodium cacodylate, pH 6.5, 1 mM 2-mercaptoethanol, 10 mM MnCl2, 0.25 mM [3H]-radiolabeled UDP-GalNAc (∼6 × 108 DPM/μmole, American Radiolabeled Chemicals Inc.), and 0.004–1.4 mM of glycopeptide substrate and variable concentration of transferase (0.02–0.2 μM) in a final reaction volume of 20 μL. After incubating at 37 °C reactions were quenched by the addition of 20 μL of 250 mM EDTA. UDP and nonhydrolyzed UDP-GalNAc were removed by passage over a small Dowex 1 × 8 anion exchange resin. Total UDP-[3H]-GalNAc utilization (transfer to peptide substrate plus transfer to water, i.e., UDP-[3H]-GalNAc hydrolysis) was determined by scintillation counting (Beckman LS5801 scintillation counter) aliquots before and after passage over Dowex. The actual [3H]-GalNAc transfer to peptide and the extent of hydrolysis were determined by gel filtration analysis on Sephadex G10, as described.19 Example gel filtration chromatograms are given in Figure S8. In all cases the reported transferase activity has been corrected (reduced) for the presence of the nonproductive UDP-[3H]-GalNAc hydrolysis which varied with the glycopeptide substrate and with the transferase mutant. For detailed kinetics studies incubation times (typically 10–180 min) were chosen such that no more than 30% of the UDP-GalNAc donor was depleted while typically giving less than 10% (glyco)peptide glycosylation. Initial velocities/activities were determined using 1 or 3 reaction time points for each substrate concentration and were repeated 2–5 times. Typically 24–30 individual specific activity values were obtained over the entire substrate concentration range. These individual data points were used to calculate the kinetic constants of Km, Vmax, and Ki using the nonlinear Michaelis–Menten and the Michaelis–Menten with substrate inhibition fitting programs found in GraphPad Prism 7.03.
Random Peptide Sequence Motif Determination
Peptide sequence motifs for GalNAc-T4 and its mutants were obtained as described for other GalNAc-Ts.31,34,35 Briefly overnight transferase reactions (0.3 μM enzyme) were performed with each of the 3 random peptides PVI, PVII, and PVIII (GAGAXXXXXTXXXXXAGAGK, where X = G,A,R,P,E,H,Q,Y,V,L (PVI), G,A,P,R,D,F,I,M,K,N (PVII), and G,A,P,R,E,Y,V,K,N,S (PVIII))30 at ∼6.6 mM in the presence of 200 mM sodium cacodylate, pH 6.9, 1 mM 2-mercaptoethanol, 10 mM MnCl2, 0.1% TritonX100, and 2 mM [3H]-radiolabeled UDP-GalNAc (∼6 × 108 DPM/μmole, American Radiolabeled Chemicals Inc.) in a final volume of 200 μL. Reactions were quenched with 100 μL of 250 mM EDTA, passed over Dowex 1 × 8 anion exchange resin and peptide and glycopeptide products isolated by Sephadex G10 gel filtration. Glycopeptide product was isolated by lectin chromatography on a mixed bed lectin column containing immobilized lectins (SJA (Sophora japonica), SBA (Glycine max), HPA (Helix pomatia), and VVA (Vicia villosa)) as described.31 After final purification on Sephadex G10 chromatography the glycopeptide product was Edman amino acid sequenced on a Shimadzu PPSQ-53A peptide sequencer to determine the compositions of the X positions of the random peptides. Enhancement values (EVs) were obtained from the ratio of the mole fractions of each amino acid residue in the product glycopeptide to that in the starting random peptide.31 Thus, EVs greater than one indicate an enrichment in the glycopeptide while EVs lower than 1 indicate a depletion in the glycopeptide. On this basis the EVs reflect the transferase’s preference for a particular amino acid residue at each X position.30,31 EVs were obtained from triplicate determinations on each random peptide; thus for each amino acid residue there were between 3 and 9 individual EV determinations at each X position depending on their presence in the three different random peptides. The obtained averaged EVs are plotted and compared in Figures S16 and S17 for wt and catalytic mutant GalNAc-T4.
Determination of Site of Glycosylation
Substrate glycosylation sites were determined by Edman amino acid sequencing on a Shimadzu PPS-Q53A protein sequencer. Briefly, G10 isolated [3H]-GalNAc glycosylated substrates were spotted on a Polybrene precycled glass fiber disk (GFD) and sequenced using a modified GFD program. The glycosylated PTH-Thr derivatives (eluting between 2.85 and 3.5 min using the standard PPSQ HPLC buffers and flow rate) were collected directly into scintillation vials on a Shimadzu FRC-10A fraction collector and scintillation counted for [3H]-GalNAc content (Beckman LS5801 scintillation counter). Note that [3H]-GalNAc lag is commonly observed after a peak of [3H]-GalNAc incorporation in these determinations. This is due to the poor extraction of the glycosylated-PTH residues from the glass fiber disks compared with the nonglycosylated amino acid PTH derivatives.10,19
Surface Plasmon Resonance Experiments
The SPR experiments for peptides 4 and 6 were performed as described for glycopeptide 3.14 As found for peptide 3, binding saturation was not achieved for glycopeptide 4; thus its Kd could not be determined. However, saturation was reached for glycopeptide 6 allowing an accurate Kd determination.
NMR Experiments
All NMR experiments were recorded on a Bruker Avance 600 MHz spectrometer equipped with a triple channel cryoprobe head. The 1H NMR resonances of the glycopeptides 4, 5, and 6 were completely assigned through standard 2D-TOCSY (30 and 80 ms mixing time) and 2D-NOESY experiments (400 ms mixing time) obtained at 278 K. Glycopeptides were 1–3 mM in 25 mM perdeuterated tris-d11 (uncorrected pH meter reading 7.4) in H2O/D2O (90:10) with 7.5 mM NaCl and 1 mM DTT. The resonance of 2,2,3,3-tetradeutero-3-trimethylsilylpropionic acid (TSP) was used as a chemical shift reference (δ TSP = 0 ppm) in the 1H NMR experiments. Peak lists for the 2D-TOCSY and 2D-NOESY spectra were generated by interactive peak picking using CARA software. STD-NMR experiments were performed at 298 K in deuterated water in the presence of 25 mM perdeuterated tris-d11 (uncorrected pH meter reading 7.4), 7.5 mM NaCl, and 1 mM DTT, using ∼877 μM glycopeptide, 13.5 μM GalNAc-T4, 75 μM UDP, and 75 μM MnCl2. STD-NMR spectra were acquired and the data analyzed as described.14
Crystallization
Crystals of GalNAc-T4 were grown as described before.14 The crystals were soaked for 30 min with a mix containing 20 mM glycopeptide 6 and 20 mM UDP in 25 mM Tris pH 7.5 and 2 mM MnCl2. The crystals were subsequently cryoprotected with 25% ethylene glycol, 18% PEG3350, and 0.1 M ammonium nitrate, and frozen in a nitrogen gas stream cooled to 100 K.
Structure Determination and Refinement
Diffraction data were collected on the synchrotron beamline I03 of the Diamond Light Source (Harwell Science and Innovation Campus, Oxfordshire, UK) at a wavelength of 0.97 Å and temperature of 100 K. Data were processed and scaled using the XDS package36 and CCP437,38 software. Relevant statistics are given in Table S4. The crystal structure was solved by molecular replacement with Phaser37,38 using the PDB entry 5NQA of human GalNAc-T4 as template. Initial phases were improved by several cycles of manual model building in Coot39 and further refined using REFMAC5.40 The final model of GalNAc-T4 soaked with glycopeptide 6 and UDP was validated with PROCHECK where model statistics are given in Table S4. The asymmetric unit of the triclinic crystal contained 2 molecules of GalNAc-T4 while only one of the monomers contained UDP, Mn+2, and glycopeptide 6. The Ramachandran plot shows that 95.14%, 3.69%, and 1.17% of the amino acids are in most favored, allowed, and disallowed regions, respectively.
MD Simulations with Peptide and Glycopeptide Substrates
The wild-type and the LFL mutant of GalNAc-T4, both in complex with UDP-Mn+2 and the glycopeptide 6, were subjected to 500 ns of MD simulation as described previously.14 Similarly, the wild-type GalNAc-T4 in complex with UDP-GalNAc and a naked peptide (GAGAGAGXTPGPG where X denotes either Val or Ala and T as the acceptor Thr) was subjected to 200 ns of MD simulations. In all cases mutants were generated using PyMol. The starting coordinates of the UPD-GalNAc in GalNAc-T4 were taken from the X-ray structure of GalNAc-T2 previously reported by our group (PDB ID: 4D0T).
Glycopeptide Pull-Out Computational Details
The initial structure for the simulations was taken from the GalNAc-T4-UDP-glycopeptide 6 complex. The UDP substrate was completed adding the GalNAc sugar from the PDB entry 4D0T by superimposition. The protonation states and hydrogen atom positions of all amino acid residues were determined by visual inspection according to protein environment. The system was solvated with a box of 15 Å around the protein surface (31.595 water molecules), and the global charge was neutralized by the addition of 1 sodium ion, leading to a total of 103 442 atoms. Molecular dynamics (MD) simulations using Amber11 software were performed. The protein was modeled with the FF99SB force field, and the carbohydrate substrate and water molecules were described with the GLYCAM06 and TIP3P force fields, respectively. The MD simulation was carried out in several steps. First, the system was minimized, holding the protein and substrate fixed. Then, the entire system was allowed to relax. To gradually reach the desired temperature, weak spatial constraints were initially added to the protein and substrate, while water molecules and sodium ions were allowed to move freely at 100 K. The constraints were then removed, and the working temperature of 300 K was reached after two more 100 K heatings in the NVT ensemble. Afterward, the density was converged up to water density at 300 K in the NPT ensemble, and the simulation was extended to 50 ns in the NVT ensemble. Steered molecular dynamics41 (SMD) and umbrella sampling42 (US) simulations were performed to pull out the neighboring glycan from the catalytic domain. The first method was used to generate the initial pathway from which the last method explored the phase-space. One collective variable (CV) was used for the pull-out, defined as the distance between the α-carbon of Asn224 (buried in the binding pocket of the catalytic domain) and the α-carbon of Thr12 (the acceptor threonine of diglycopeptide 6). A total of 20 trajectories with random velocities were taken from the reference structure from the equilibration MD, allowing them to relax for 1 ns. Subsequently, a movable harmonic potential of 50 kcal/(mol A2) was used to drive the CV 40 Å apart during 2 ns, with a pulling velocity of 20 Å/ns. The trajectory with the lower energy was taken for the US simulations, and the pathway was divided in 81 windows with a regular separation of 0.5 Å between them. Force constants of 10 kcal/(mol A2) were used for the harmonic potentials. Every window was sampled during 10 ns, leading to a total of ∼0.8 μs of simulation data. The firsts 2 ns of each window were considered as an equilibration step. Analysis of the trajectories was carried out using standard tools of Amber and VMD.43 Particularly, the hydrogen bond analysis was performed using the cpptraj utility from Amber14, taking into account all the interactions between the substrate (diglycopeptide) and the receptor (GalNAc-T4 bound to UDP-GalNAc), with a distance cutoff of 3.0 Å between heteroatoms and 135° for the angle that defines the hydrogen bond.
Acknowledgments
We thank synchrotron radiation sources DLS (Oxford) and in particular beamline I03 (experiment number MX10121-15). We thank ARAID, MEC (CTQ2013-44367-C2-2-P, BFU2016-75633-P, CTQ2015-67727-R, CTQ2015-70524-R, and CTQ2017-85496-P), AGAUR (SGR2017-1189), the National Institutes of Health (R01-GM113534, and instrument Grant GM113534-01S to T. A. Gerken), the Danish National Research Foundation (DNRF107), the FCT-Portugal [UID/Multi/04378/2013 cofinanced by the FEDER (POCI-01-0145-FEDER-007728)], and the DGA (E34_R17) for financial support. I. Compañón thanks Universidad de La Rioja for the FPI grant. F. Marcelo thanks FCT-Portugal for IF Investigator grant (IF/00780/2015) and PTNMR supported by Project 022161. E. Lira-Navarrete acknowledges her postdoctoral EMBO fellowship ALTF 1553-2015 cofunded by the European Commission (LTFCOFUND2013, GA-2013-609409) and Marie Curie Actions. H. Coelho and J. Jiménez-Barbero thank EU for the TOLLerant project. The research leading to these results has also received funding from the FP7 (2007–2013) under BioStruct-X (Grant agreement 283570 and BIOSTRUCTX_5186). We would also like to acknowledge the assistance of Juwan Lee in obtaining the GalNAc-T4 random peptide motifs.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00488.
Accession Codes
The coordinate and structure factor has been deposited in the Worldwide Protein Data Bank (wwPDB) with the PDB code 6H0B (see Table S4).
Author Contributions
▰ M. de las Rivas and E. J. Paul Daniel contributed equally to this work. R. Hurtado-Guerrero designed the crystallization construct and solved the crystal structure. E. Lira-Navarrete, M. de las Rivas, and R. Hurtado-Guerrero purified the enzymes, crystallized the complex, and refined the crystal structure. I. Compañón, and F. Corzana synthesized the glycopeptides. F. Corzana, L. Raich, and C. Rovira performed the computational simulations. H. Coelho, A. Diniz, J. Jiménez-Barbero, and F. Marcelo performed the NMR experiments. T. A. Gerken and E. J. Paul Daniel performed the kinetic and peptide substrate motif studies together with the Edman amino acid sequencing. R. Hurtado-Guerrero and T. A. Gerken wrote the article together with main contributions of F. Corzana, H. Clausen, J. Jiménez-Barbero, and F. Marcelo. All authors read and approved the final manuscript.
The authors declare no competing financial interest.
Notes
Safety statement: no unexpected safety hazards were encountered.
Supplementary Material
References
- Bennett E. P.; Mandel U.; Clausen H.; Gerken T. A.; Fritz T. A.; Tabak L. A. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22 (6), 736–756. 10.1093/glycob/cwr182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K.; Jeanneau C.; Tarp M. A.; Benet-Pages A.; Lorenz-Depiereux B.; Bennett E. P.; Mandel U.; Strom T. M.; Clausen H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 2006, 281 (27), 18370–18377. 10.1074/jbc.M602469200. [DOI] [PubMed] [Google Scholar]
- Khetarpal S. A.; Schjoldager K. T.; Christoffersen C.; Raghavan A.; Edmondson A. C.; Reutter H. M.; Ahmed B.; Ouazzani R.; Peloso G. M.; Vitali C.; Zhao W.; Somasundara A. V.; Millar J. S.; Park Y.; Fernando G.; Livanov V.; Choi S.; Noe E.; Patel P.; Ho S. P.; Myocardial Infarction Exome Sequencing S.; Kirchgessner T. G.; Wandall H. H.; Hansen L.; Bennett E. P.; Vakhrushev S. Y.; Saleheen D.; Kathiresan S.; Brown C. D.; Abou Jamra R.; LeGuern E.; Clausen H.; Rader D. J. Loss of Function of GALNT2 Lowers High-Density Lipoproteins in Humans, Nonhuman Primates, and Rodents. Cell Metab. 2016, 24 (2), 234–245. 10.1016/j.cmet.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen N. B.; Wang S.; Narimatsu Y.; Yang Z.; Halim A.; Schjoldager K. T.; Madsen T. D.; Seidah N. G.; Bennett E. P.; Levery S. B.; Clausen H. Low density lipoprotein receptor class A repeats are O-glycosylated in linker regions. J. Biol. Chem. 2014, 289 (25), 17312–17324. 10.1074/jbc.M113.545053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaman E. M.; Brooks S. A. The extended ppGalNAc-T family and their functional involvement in the metastatic cascade. Histol Histopathol 2014, 29 (3), 293–304. 10.14670/HH-29.293. [DOI] [PubMed] [Google Scholar]
- Nguyen A. T.; Chia J.; Ros M.; Hui K. M.; Saltel F.; Bard F. Organelle Specific O-Glycosylation Drives MMP14 Activation, Tumor Growth, and Metastasis. Cancer Cell 2017, 32 (5), 639–653e6. 10.1016/j.ccell.2017.10.001. [DOI] [PubMed] [Google Scholar]
- Tran D. T.; Ten Hagen K. G. Mucin-type O-glycosylation during development. J. Biol. Chem. 2013, 288 (10), 6921–6929. 10.1074/jbc.R112.418558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goth C. K.; Vakhrushev S. Y.; Joshi H. J.; Clausen H.; Schjoldager K. T. Fine-Tuning Limited Proteolysis: A Major Role for Regulated Site-Specific O-Glycosylation. Trends Biochem. Sci. 2018, 43 (4), 269–284. 10.1016/j.tibs.2018.02.005. [DOI] [PubMed] [Google Scholar]
- Goth C. K.; Halim A.; Khetarpal S. A.; Rader D. J.; Clausen H.; Schjoldager K. T. A systematic study of modulation of ADAM-mediated ectodomain shedding by site-specific O-glycosylation. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (47), 14623–14628. 10.1073/pnas.1511175112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revoredo L.; Wang S.; Bennett E. P.; Clausen H.; Moremen K. W.; Jarvis D. L.; Ten Hagen K. G.; Tabak L. A.; Gerken T. A. Mucin-type O-glycosylation is controlled by short- and long-range glycopeptide substrate recognition that varies among members of the polypeptide GalNAc transferase family. Glycobiology 2016, 26 (4), 360–376. 10.1093/glycob/cwv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan H.; Reis C. A.; Bennett E. P.; Mirgorodskaya E.; Roepstorff P.; Hollingsworth M. A.; Burchell J.; Taylor-Papadimitriou J.; Clausen H. The lectin domain of UDP-N-acetyl-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase-T4 directs its glycopeptide specificities. J. Biol. Chem. 2000, 275 (49), 38197–38205. 10.1074/jbc.M005783200. [DOI] [PubMed] [Google Scholar]
- Raman J.; Fritz T. A.; Gerken T. A.; Jamison O.; Live D.; Liu M.; Tabak L. A. The catalytic and lectin domains of UDP-GalNAc:polypeptide alpha-N-Acetylgalactosaminyltransferase function in concert to direct glycosylation site selection. J. Biol. Chem. 2008, 283 (34), 22942–22951. 10.1074/jbc.M803387200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira-Navarrete E.; de Las Rivas M.; Companon I.; Pallares M. C.; Kong Y.; Iglesias-Fernandez J.; Bernardes G. J.; Peregrina J. M.; Rovira C.; Bernado P.; Bruscolini P.; Clausen H.; Lostao A.; Corzana F.; Hurtado-Guerrero R. Dynamic interplay between catalytic and lectin domains of GalNAc-transferases modulates protein O-glycosylation. Nat. Commun. 2015, 6, 6937. 10.1038/ncomms7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de las Rivas M.; Lira-Navarrete E.; Daniel E. J. P.; Companon I.; Coelho H.; Diniz A.; Jimenez-Barbero J.; Peregrina J. M.; Clausen H.; Corzana F.; Marcelo F.; Jimenez-Oses G.; Gerken T. A.; Hurtado-Guerrero R. The interdomain flexible linker of the polypeptide GalNAc transferases dictates their long-range glycosylation preferences. Nat. Commun. 2017, 8 (1), 1959. 10.1038/s41467-017-02006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrine C. L.; Ganguli A.; Wu P.; Bertozzi C. R.; Fritz T. A.; Raman J.; Tabak L. A.; Gerken T. A. Glycopeptide-preferring polypeptide GalNAc transferase 10 (ppGalNAc T10), involved in mucin-type O-glycosylation, has a unique GalNAc-O-Ser/Thr-binding site in its catalytic domain not found in ppGalNAc T1 or T2. J. Biol. Chem. 2009, 284 (30), 20387–20397. 10.1074/jbc.M109.017236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz T. A.; Raman J.; Tabak L. A. Dynamic association between the catalytic and lectin domains of human UDP-GalNAc:polypeptide alpha-N-acetylgalactosaminyltransferase-2. J. Biol. Chem. 2006, 281 (13), 8613–8619. 10.1074/jbc.M513590200. [DOI] [PubMed] [Google Scholar]
- Song L.; Linstedt A. D., Inhibitor of ppGalNAc-T3-mediated O-glycosylation blocks cancer cell invasiveness and lowers FGF23 levels. eLife 2017, 6, 24051. 10.7554/eLife.24051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira-Navarrete E.; Iglesias-Fernandez J.; Zandberg W. F.; Companon I.; Kong Y.; Corzana F.; Pinto B. M.; Clausen H.; Peregrina J. M.; Vocadlo D. J.; Rovira C.; Hurtado-Guerrero R. Substrate-guided front-face reaction revealed by combined structural snapshots and metadynamics for the polypeptide N-acetylgalactosaminyltransferase 2. Angew. Chem., Int. Ed. 2014, 53 (31), 8206–8210. 10.1002/anie.201402781. [DOI] [PubMed] [Google Scholar]
- Gerken T. A.; Revoredo L.; Thome J. J.; Tabak L. A.; Vester-Christensen M. B.; Clausen H.; Gahlay G. K.; Jarvis D. L.; Johnson R. W.; Moniz H. A.; Moremen K. The lectin domain of the polypeptide GalNAc transferase family of glycosyltransferases (ppGalNAc Ts) acts as a switch directing glycopeptide substrate glycosylation in an N- or C-terminal direction, further controlling mucin type O-glycosylation. J. Biol. Chem. 2013, 288 (27), 19900–19914. 10.1074/jbc.M113.477877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey A. D. Jr.; Schwab R. D.; Boesteanu A. C.; Steentoft C.; Mandel U.; Engels B.; Stone J. D.; Madsen T. D.; Schreiber K.; Haines K. M.; Cogdill A. P.; Chen T. J.; Song D.; Scholler J.; Kranz D. M.; Feldman M. D.; Young R.; Keith B.; Schreiber H.; Clausen H.; Johnson L. A.; June C. H. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44 (6), 1444–1454. 10.1016/j.immuni.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett E. P.; Hassan H.; Mandel U.; Mirgorodskaya E.; Roepstorff P.; Burchell J.; Taylor-Papadimitriou J.; Hollingsworth M. A.; Merkx G.; van Kessel A. G.; Eiberg H.; Steffensen R.; Clausen H. Cloning of a human UDP-N-acetyl-alpha-D-Galactosamine:polypeptide N-acetylgalactosaminyltransferase that complements other GalNAc-transferases in complete O-glycosylation of the MUC1 tandem repeat. J. Biol. Chem. 1998, 273 (46), 30472–30481. 10.1074/jbc.273.46.30472. [DOI] [PubMed] [Google Scholar]
- Bermejo I. A.; Usabiaga I.; Companon I.; Castro-Lopez J.; Insausti A.; Fernandez J. A.; Avenoza A.; Busto J. H.; Jimenez-Barbero J.; Asensio J. L.; Peregrina J. M.; Jimenez-Oses G.; Hurtado-Guerrero R.; Cocinero E. J.; Corzana F. Water Sculpts the Distinctive Shapes and Dynamics of the Tumor-Associated Carbohydrate Tn Antigens: Implications for Their Molecular Recognition. J. Am. Chem. Soc. 2018, 140 (31), 9952–9960. 10.1021/jacs.8b04801. [DOI] [PubMed] [Google Scholar]
- Dam T. K.; Gerken T. A.; Brewer C. F. Thermodynamics of multivalent carbohydrate-lectin cross-linking interactions: importance of entropy in the bind and jump mechanism. Biochemistry 2009, 48 (18), 3822–3827. 10.1021/bi9002919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam T. K.; Brewer C. F. Lectins as pattern recognition molecules: the effects of epitope density in innate immunity. Glycobiology 2010, 20 (3), 270–279. 10.1093/glycob/cwp186. [DOI] [PubMed] [Google Scholar]
- Valero-Gonzalez J.; Leonhard-Melief C.; Lira-Navarrete E.; Jimenez-Oses G.; Hernandez-Ruiz C.; Pallares M. C.; Yruela I.; Vasudevan D.; Lostao A.; Corzana F.; Takeuchi H.; Haltiwanger R. S.; Hurtado-Guerrero R. A proactive role of water molecules in acceptor recognition by protein O-fucosyltransferase 2. Nat. Chem. Biol. 2016, 12 (4), 240–246. 10.1038/nchembio.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz T. A.; Hurley J. H.; Trinh L. B.; Shiloach J.; Tabak L. A. The beginnings of mucin biosynthesis: the crystal structure of UDP-GalNAc:polypeptide alpha-N-acetylgalactosaminyltransferase-T1. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (43), 15307–15312. 10.1073/pnas.0405657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T.; Shiba T.; Sugioka S.; Furukawa S.; Sawaki H.; Kato R.; Wakatsuki S.; Narimatsu H. Structural basis of carbohydrate transfer activity by human UDP-GalNAc: polypeptide alpha-N-acetylgalactosaminyltransferase (pp-GalNAc-T10). J. Mol. Biol. 2006, 359 (3), 708–727. 10.1016/j.jmb.2006.03.061. [DOI] [PubMed] [Google Scholar]
- Pedersen J. W.; Bennett E. P.; Schjoldager K. T.; Meldal M.; Holmer A. P.; Blixt O.; Clo E.; Levery S. B.; Clausen H.; Wandall H. H. Lectin domains of polypeptide GalNAc transferases exhibit glycopeptide binding specificity. J. Biol. Chem. 2011, 286 (37), 32684–32696. 10.1074/jbc.M111.273722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandall H. H.; Irazoqui F.; Tarp M. A.; Bennett E. P.; Mandel U.; Takeuchi H.; Kato K.; Irimura T.; Suryanarayanan G.; Hollingsworth M. A.; Clausen H. The lectin domains of polypeptide GalNAc-transferases exhibit carbohydrate-binding specificity for GalNAc: lectin binding to GalNAc-glycopeptide substrates is required for high density GalNAc-O-glycosylation. Glycobiology 2007, 17 (4), 374–387. 10.1093/glycob/cwl082. [DOI] [PubMed] [Google Scholar]
- Gerken T. A.; Jamison O.; Perrine C. L.; Collette J. C.; Moinova H.; Ravi L.; Markowitz S. D.; Shen W.; Patel H.; Tabak L. A. Emerging paradigms for the initiation of mucin-type protein O-glycosylation by the polypeptide GalNAc transferase family of glycosyltransferases. J. Biol. Chem. 2011, 286 (16), 14493–14507. 10.1074/jbc.M111.218701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerken T. A.; Raman J.; Fritz T. A.; Jamison O. Identification of common and unique peptide substrate preferences for the UDP-GalNAc:polypeptide alpha-N-acetylgalactosaminyltransferases T1 and T2 derived from oriented random peptide substrates. J. Biol. Chem. 2006, 281 (43), 32403–32416. 10.1074/jbc.M605149200. [DOI] [PubMed] [Google Scholar]
- Corzana F.; Busto J. H.; Jimenez-Oses G.; Garcia de Luis M.; Asensio J. L.; Jimenez-Barbero J.; Peregrina J. M.; Avenoza A. Serine versus threonine glycosylation: the methyl group causes a drastic alteration on the carbohydrate orientation and on the surrounding water shell. J. Am. Chem. Soc. 2007, 129 (30), 9458–9467. 10.1021/ja072181b. [DOI] [PubMed] [Google Scholar]
- Young W. W. Jr.; Holcomb D. R.; Ten Hagen K. G.; Tabak L. A. Expression of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase isoforms in murine tissues determined by real-time PCR: a new view of a large family. Glycobiology 2003, 13 (7), 549–557. 10.1093/glycob/cwg062. [DOI] [PubMed] [Google Scholar]
- Perrine C.; Ju T.; Cummings R. D.; Gerken T. A. Systematic determination of the peptide acceptor preferences for the human UDP-Gal:glycoprotein-alpha-GalNAc beta 3 galactosyltransferase (T-synthase). Glycobiology 2009, 19 (3), 321–328. 10.1093/glycob/cwn143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerken T. A.; Ten Hagen K. G.; Jamison O. Conservation of peptide acceptor preferences between Drosophila and mammalian polypeptide-GalNAc transferase ortholog pairs. Glycobiology 2008, 18 (11), 861–870. 10.1093/glycob/cwn073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760–763. 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isralewitz B.; Gao M.; Schulten K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001, 11 (2), 224–230. 10.1016/S0959-440X(00)00194-9. [DOI] [PubMed] [Google Scholar]
- Park S.; Im W. Theory of Adaptive Optimization for Umbrella Sampling. J. Chem. Theory Comput. 2014, 10 (7), 2719–2728. 10.1021/ct500504g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.