

Abstract
Catalysis is at the heart of many manufacturing processes and underpins provision of the goods and infrastructure necessary for the effective wellbeing of society; catalysis continues to play a key role in the manufacture of chemical intermediates and final products. There is a continuing need to design new effective catalysts especially with the drive toward using sustainable resources. The identification that gold is an exceptionally effective catalyst has paved the way for a new class of active heterogeneous and homogeneous catalysts for a broad range of reactions. As a heterogeneous catalyst gold is the most active catalyst for the oxidation of carbon monoxide at ambient temperature. It is also the most effective catalyst for the synthesis of vinyl chloride by acetylene hydrochlorination, and a gold catalyst has recently been commercialized in China for this reaction. In this outlook the nature of the active gold species for these two reactions will be explored.
Short abstract
Heterogeneous gold catalysts display many active centers including cations, clusters of atoms, and nanoparticles all of which can exhibit activity depending on the reaction.
Introduction
Catalysis is a topic of immense general importance. Without catalysis society would not have access to food and medicines, and indeed most of materials that are manufactured require a catalyst as some stage to make them. Catalysis is therefore ubiquitous, but often the central importance of catalysis to everyday life is overlooked. There is always a quest for new more effective catalysts, and in this Outlook the recent discovery of catalysis by gold will be described.
Gold has fascinated people for millennia; its bright lustrous yellow color has been molded in great works of art, and it has been prized because it is the most noble of the metals and is considered immutable. Gold colloids have also been used to color glass for centuries, and the most vivid example is perhaps the Lycurgus Cup which dates to the fourth century AD and is a very early example of dichroic glass which changes from green to a translucent glowing red when light is shone through due to the presence of colloidal gold nanoparticles.1 It was Faraday who presented the first scientific paper on the properties and preparation of gold colloids and demonstrated these vividly colored nanoscale structures at the Royal Institution in 1847.2 Indeed, these gold sols are still stable and remain in the Royal Institution in London to this day.3 Perhaps it was the perception of the immutability of gold that hampered the development of the chemistry of gold. Until about 40 years ago gold was thought of as one of the least interesting elements in the periodic table with very few pages devoted to it in textbooks of the day. However, now gold is known to have an exceptionally rich and exciting chemistry, and gold nanoparticles have been finding efficacy in many applications especially in the medical arena for cancer treatment.4 The topic of catalysis by gold is now a very well-studied topic as both homogeneous5,6 and heterogeneous catalysts.7 The new advent in the interest in the chemistry of gold has its origins in two discoveries in the 1980s when gold was found to be the best heterogeneous catalyst for both the oxidation of carbon monoxide at ambient temperature8 and the synthesis of vinyl chloride by acetylene hydrochlorination,9 and the gold catalyst has recently been commercialized for this process in China.10 The use of gold complexes as homogeneous catalysts is now well-advanced after the initial discovery;11 however, although heterogeneous gold catalysts are now finding commercial application the nature of the active gold species in these catalysts has until recently been an intensely debated topic. In this Outlook the recent advances in understanding the nature of the active site for the two reactions for which gold is, without doubt, an exceptional catalyst, namely, carbon monoxide oxidation and acetylene hydrochlorination, are discussed.
The Complexity of Heterogeneous Gold Catalysts
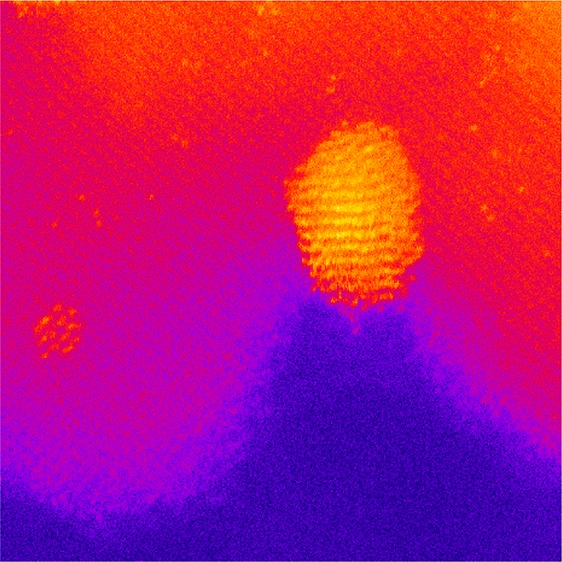
Ever since the discovery that gold is a very effective heterogeneous catalyst the quest has been to determine the origin of the active species. Until the advent of aberration-corrected scanning transmission electron microscopy (AC-STEM), most studies focused attention on the activity of 2–5 nm nanoparticles, which are readily detectable by the conventional microscopy techniques. However, the introduction of AC-STEM revealed the complexity of the gold species present in catalysts prepared by deposition–precipitation on TiO212 (Figure 1a) and on iron oxide13 (Figure 1b). In both cases it is clear that a whole range of gold species are present including individual atoms or cations, small clusters of a few atoms, small monolayer cluster, bilayer clusters, and small nanoparticles containing hundreds to thousands of atoms. In his Spiers lecture in 2011 Haruta14 introduced the concept of a hierarchy of activity for the gold species, and subsequently together we explored this proposal for low-temperature CO oxidation.
Figure 1.

(a) High-magnification Z-contrast micrographs showing 10 wt % loaded Au on anatase. In the oxidized precursor state following deposition–precipitation of Au (reproduced from ref (12)). (b) AC-STEM micrograph of a 5 wt % Au on iron oxide prepared by deposition–precipitation (reproduced from ref (13)). In both images a range of Au species from atoms clusters and nanoparticles are clearly visible.
Carbon Monoxide Oxidation Using Gold Catalysts
A key observation in heterogeneous catalysis is that the method of preparation can have a profound effect on the effectiveness of the catalyst. This is particularly true of heterogeneous gold catalysts. An example of this concerns two Au/FeOx catalysts prepared by two different coprecipitation methods.13 While the methods are quite similar, there are key differences in the sequence and rate of mixing the acidic and basic precursors. In one method (denoted CP-1), the acidic solution (Fe(NO3)3 + HAuCl4) was added very quickly (within 2 min) into the basic solution (Na2CO3), whereas in the second method (denoted CP-2), the basic solution (Na2CO3) was slowly added dropwise into the acidic solution (Fe(NO3)3 + HAuCl4) over 30 min. These subtle preparation differences have a dramatic effect on the catalytic behavior (Figure 2). The two materials were either dried at 120 °C or heated in air at 300 °C giving four materials that were tested for CO oxidation, and the catalyst activity was investigated as the reaction temperature was increased. Both the samples dried at 120 °C gave identical activity when tested in this way (Figure 2), but the samples heated at 300 °C behaved very differently with the CP-1-300 material showing increased activity as it was now effective for CO oxidation at subambient temperatures, whereas the CP-2-300 material was far less active. As we have four sets of materials and three distinct different manifestations of catalyst activity we reasoned that the differences were due to the different populations of gold species present in these catalysts or the nature of the iron oxide support. The two methods gave different support structures with the CP-1 method giving heamatite and the CP-2 method giving ferrihydrate. Of course, some of the observed effects could be the result of differences in the support that could originate from the two different methods. The effect of the support was ruled out by synthesizing the supports using the same methods but without the gold being present. The gold was then added to these supports by a deposition–precipitation (DP) method to give catalysts comprising 5% Au, and the catalysts were dried at 120 °C. When evaluated for CO oxidation these two dried DP catalysts exhibited very high activity similar to that of the dried catalysts that had been prepared by coprecipitation. Hence the structure of the support is not an important factor for the origin of the activity in the dried only catalysts. However, on heating to 300 °C the activity of the two DP catalysts decreased markedly, and hence the activation behavior observed with the CP-1 catalyst was not observed.
Figure 2.

Diverging catalyst behavior after heat treatment. (a) CO conversion at various temperatures (reproduced from ref (13)). Catalyst mass 150 mg, gas flow 50 mL min–1 1 vol % CO in air. (b) Arrhenius plots carried out at low conversion conditions. ○ (CP-1, dried, 6 wt % Au by ICP), ● (CP-1, calcined, 6 wt % Au by ICP), □ (CP-2, dried, 3.5 wt % Au by ICP), ■ (CP-2, calcined, 3.5 wt % Au by ICP). The arrows shown in part a represent the thermal activation behavior (black arrow) of the CP-1 catalyst and the thermal deactivation behavior (white arrow) of the CP-2 catalyst.
We then focused on the differences in the gold species present on the catalysts, but detailed analysis of the population densities of the species present in the catalysts showed that it was not possible to assign just one type of Au species as being solely active, while the others are inactive, to explain all three sets of data. In particular, the two dried catalysts CP-1-120 and CP-2-120 showed completely different population densities of Au species whereas if any particular species was the active species this would have been observed. To explain the behavior of these catalysts we suggested that a hierarchy of activity was present as had been first proposed by Haruta.14 Supported gold catalysts comprise a range of structures, including dispersed atoms, clusters, and nanoparticles. Hence, the final reported activities of the catalysts should be a weighted sum of the activity of each of the species present, combined with their relative population densities (i.e., total activity A = ∑iρiεi, where ρi and εi represent the population fraction and intrinsic activity for the ith active species). The relative activities εi of different Au species were estimated by Haruta.14 In the case of the iron oxide support we observed that the small clusters were more active than the 1–3 nm nanoparticles, and the atoms were inactive, which is consistent with an earlier AC-STEM we have carried out.15
In the coprecipitation method the CP-1 catalyst is the result of rapid mixing as compared with CP-2. This enhanced mixing probably leads to a more homogeneous precipitation process which leads to Au species being embedded within the oxide support, and this was verified experimentally.13 This then enabled us to explain the effects on activity that were observed when the CP catalysts were heated to 300 °C. As shown in Figure 3 CP1-300 increases in activity and during the heat treatment; the smallest Au species that are originally located on the surface will sinter into larger nanoparticles, which will be less active. However, the gold that is encapsulated in the iron oxide particles acts as a reservoir and diffuses to the surface thereby replenishing the active species. This is not possible for the CP2 sample as this is made by the slow coprecipitation and therefore has much lower levels of trapped gold within the iron oxide. So for CP2 the surface gold species sinter decreasing the activity, and no new surface gold species are formed. Support for this model of activation is provided in the elegant in situ electron microscopy experiments of Allard et al.16 The same is also true for the DP catalysts, where the Au was added after the support material was formed, and so no reservoir of trapped gold species can be available in these materials.
Figure 3.

Proposed mechanism for the thermal activation behavior of the CP-1 catalyst (reproduced from ref (13)). A series of schematic diagrams which illustrate the thermal evolution process of the (a) CP-1 (acid-into-base), (b) CP-2 (base-into-acid), and (c) DP-1 (acid-into-base) catalysts. The CP-1 catalyst (column a) has a much larger amount of atomic Au species buried inside the support material after only being dried as compared to the CP-2 and DP-1 catalysts (columns b and c, respectively). Therefore, after calcination at 300 °C, the loss of the more active smaller Au species (i.e., sub-nm clusters and 1–3 nm Au particles) due to agglomeration can be replenished in the CP-1 catalysts by the outward diffusion of the “trapped” internal Au species, which is not possible in the case of the CP-2 and DP-1 catalysts. As a result, the CP-1 catalyst after calcination at 300 °C can be even more active than the dried only stage (as highlighted by the dashed box). However, after prolonged calcination at higher temperatures (i.e., the 500 °C treatment) the Au reserves inside the CP-1 support particle eventually get depleted, and the catalytic activity decreases close to zero due to agglomeration of the surface Au species.
Why are the clusters and the small nanoparticles the active species in CO oxidation? This is related to the current view of the reaction mechanism.7 O2 is considered to be activated at the peripheral sites of the nanoparticles or clusters that are in contact with the surface of the support which carry a net positive charge which in the presence of water, an essential reactant, produces a hydroperoxyl species. CO is activated on Au(0) that is on the second layer of the cluster or the nanoparticles and this interacts with the oxygen species to form a hydroxy carbonate that then decomposes to release CO2. Hence bilayer clusters15 represent the optimum morphology if this reaction mechanism takes place, and so this is consistent with the experimental evidence.
Acetylene Hydrochlorination Using Gold Catalysis
Acetylene hydrochlorination is currently a major process operated commercially in China producing over 13 Mtpa of vinyl chloride. The industrial catalyst until recently has been mercuric chloride supported on carbon. Based on data for a wide range of carbon-supported metal chlorides that was presented by Shinoda17 and plotting the reactivity data against the standard electrode potential Hutchings observed9 a correlation could be observed, so that as the standard electrode potential of the metal became more positive so the catalyst activity increased (Figure 4a). This led to the prediction that Au would be an effective catalyst for this reaction as it had a higher standard electrode potential, a prediction that was subsequently demonstrated by the preparation of a catalyst from Au metal dissolved in aqua regia and impregnated onto activated carbon (Figure 4b).18 A gold catalyst has recently been commercialized for this reaction in China10 enabling the major use of mercury to be phased out, and as a result of this the Minimata Convention19 has now been passed into international law which will seek to eliminate the use of mercury in any application worldwide.
Figure 4.
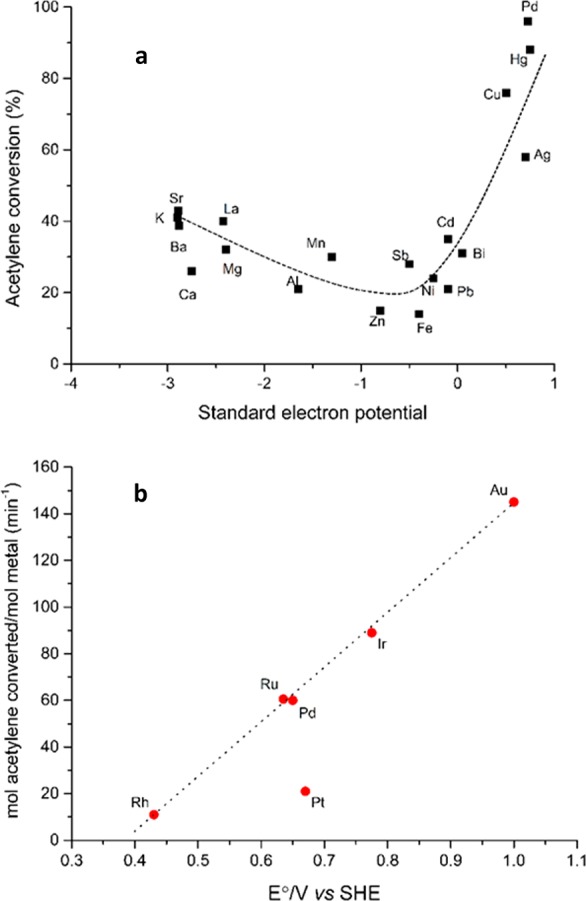
(a) Correlation of activity for acetylene hydrochlorination of carbon-supported metal chloride catalysts with the standard electrode potential (reproduced from ref (9)). (b) Correlation of initial acetylene hydrochlorination activity of supported metal chloride catalysts with the standard electrode potential of metal chloride salts (reproduced from ref (21)).
The nature of the active site in the gold catalyst for acetylene hydrochlorination has been studied over many years. However, until recently all of these studies had involved ex situ analysis. Using Mössbauer spectroscopy20 and X-ray photoelectron spectroscopy21 metallic gold was observed together with cationic gold, and transmission electron microscopy21 showed that gold nanoparticles were also present. The cationic gold was considered to be the active species, and this was confirmed by Mössbauer spectroscopy which showed that catalysts became deactivated when the cationic gold was depleted.20 Hence the model for the active site was proposed to be cationic Au at the peripheral sites of the gold nanoparticles in contact with the activated carbon support.
Recently the first in situ characterization of Au/C catalysts for the acetylene hydrochlorination reaction using X-ray absorption fine structure spectroscopy (XAFS) has been carried out under operating conditions, and this has been most instructive.22 XAFS has been widely applied to study heterogeneous and homogeneous catalysts and can be applied while working at realistic reaction condition through the design of suitable reactors. From the normalized Au L3-edge X-ray absorption near edge structure (XANES), information on Au speciation can be determined based on the white line, a sharp intense peak originating from electronic transitions before the adsorption edge that can be indicative of Au oxidation state by comparison with appropriate standards. In this study a series of gold catalysts supported on activated carbon were prepared using HAuCl4 with aqua regia, HNO3, or H2O as solvent. Also a Au/C catalyst prepared using a Au(I)-thiosulfate precursor analogous to the industrially validated catalyst10 was analyzed. The XAFS study of these catalysts under dilute acetylene hydrochlorination reaction conditions was performed while following the acetylene conversion and vinyl chloride monomer (VCM) production by mass spectrometry. This revealed the highly active catalysts (Au/C–aqua regia, Au/C–HNO3, and Au/C–S2O3) comprise almost entirely single site cationic Au entities with no evidence of Au nanoparticles of Au–Au scattering interactions categorically proving that the active form of these catalysts is cationic Au (Figure 5). The activity of the best performing catalysts correlates with the ratio of Au(I):Au(III) present, and all catalysts have both oxidation states present supporting a redox mechanism between Au(I)–Au(III) as proposed by Hutchings originally.9 Catalysts that comprised predominantly metallic-Au nanoparticles by XAFS (i.e., the catalyst prepared using water as solvent) were found to be almost inactive with no improvement with reaction time-online. Hence the in situ experiments showed that the gold catalyst was very different from that expected on the basis of ex situ analysis as no metallic Au nanoparticles are present on the catalyst under reaction conditions. We therefore revisited the microscopy of these catalysts. The STEM high-angle annular dark field (STEM-HAADF) imaging studies of the catalyst prepared using aqua regia as solvent or the Au/C–S2O3 catalyst prepared from an aqueous thiosulfate solution showed the presence of atomically dispersed Au on the C support (Figure 5).
Figure 5.

(a) k3 weighted XAFS data for 1 wt % Au/C–aqua regia and reference gold foil. (b) Representative STEM-HAADF image for 1 wt % Au/C–aqua regia showing isolated Au species. (c) k3 weighted XAFS data for 1 wt % Au/C–S2O3 and reference gold foil. (d) Representative STEM-HAADF image for 1 wt % Au/C–S2O3. (e) k3 weighted XAFS data for 1 wt % Au/C–aqua regia, 1 wt % Au/C–H2O, and reference gold foil. (f) Representative STEM-HAADF image for 1 wt % Au/C–aqua regia and 1 wt % Au/C–H2O showing the absence of isolated Au species (dated taken from ref (22)).
Further analysis of the Au/C catalyst prepared using aqua regia catalyst shows that, upon introduction of the reactant gases, an immediate and significant change in the Au L3-edge XANES spectrum occurred (Figure 6a). During the initial 180 min reaction the white-line intensity increases initially and then gradually decreases. The activity observed for the synthesis of vinyl chloride shows the opposite trend as the activity initially decreases and then gradually improves. The white-line height correlates with the oxidation state of the gold on the catalyst, and the observed trend shows that Au+ chloride type species initially present are oxidized to predominantly Au3+ chloride species by the reactants during the first 20 min of reaction. Over the subsequent 160 min of reaction the mean oxidation state of the Au species gradually reduces back toward that of Au+ before converging to a stable condition where both the Au+ and Au3+ species coexist. This measured change in white-line relative intensity during this process correlated strongly with the simultaneously recorded VCM productivity of the catalyst (Figure 6b), with higher productivity being observed with lower Au white-line relative intensity indicating that the most active catalysts have more Au(I) present than Au(III). Even after extended operation under these in situ conditions no gold nanoparticles were observed (Figure 6c).
Figure 6.

VCM productivity and in situ characterization of 1 wt % Au/C–AR catalyst as a function of time-online. (A) Catalytic performance as a function of time-online (black) and the change in normalized white-line intensity (blue) as a function of reaction time. (B) 3D profile plot of successive Au L3-edges from XANES spectra acquired in situ as a function of reaction time. (C) Representative STEM-HAADF image of Au/C–AR after use for 250 min showing the presence of atomically dispersed species and a few occasional sub-nm clusters (reproduced from ref (22)).
Figure 7 shows that a correlation can be made between the variation in the Au L3 white-line intensity and VCM production of the three active catalysts (Au/C–AR, Au/C–HNO3, and Au/C–S2O3). This correlation shows the highly dispersed Au(I) species are crucially important for this reaction together with a population of highly dispersed Au(III) like species strongly suggesting that the activity is related to a Au(I)–Au(III) redox couple. Analysis of the active catalysts after extended reaction times (e.g., as in Figure 6c) confirmed the prevalence of atomically dispersed Au species still predominantly composed of cationic AuClx. At no stage during these in situ reactions of these highly active catalysts were significant populations of Au–Au distances indicative of the formation of metallic Au nanoparticles observed. This combined with the observations that catalysts containing Au nanoparticles under in situ conditions were inactive shows that for heterogeneous gold catalysis it is essential to use in situ analysis if possible. Theoretical studies of acetylene activation mechanism revealed its initial binding to Au(I) and subsequent activation of HCl, and showed that there is no direct interaction between gold cations and HCl, which indicates that this gold catalyst may not be applicable to more general reactions involving HCl.22
Figure 7.
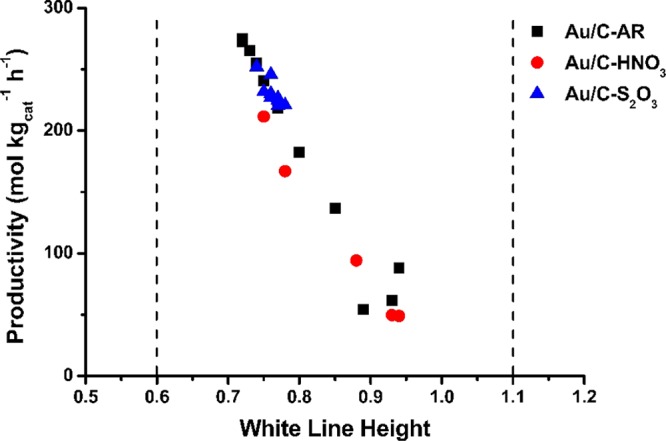
Correlation between VCM productivity and change in normalized white-line intensity of the 1 wt % Au/C–AR, 1 wt % Au/C–HNO3, and 1 wt % Au/C–S2O3 catalysts. Dashed lines represent the white-line intensities of the Au+ [AuCl2]− standard (value of 0.6) and the Au3+ KAuCl4 standard (value of 1.1). [AuCl2]− standard from difference spectra calculated in ref (24) (reproduced from ref (22)).
Concluding Comments
It is clear that supported gold species are highly active for both CO oxidation and acetylene hydrochlorination. In recent years much attention has been given to the study of bimetallic catalysts (e.g., AuPd) as these catalysts exhibit a synergistic enhancement in activity for a range of redox reactions.7 However, gold catalysts for CO oxidation and acetylene hydrochlorination do not exhibit any enhancement in activity when alloyed with Pd; rather addition of Pd leads to a less active catalyst, and the activity is correlated with the standard electrode potential which indicates that cationic gold is important in both these reactions. However, the active species of gold for these two reactions is different. For acetylene hydrochlorination it is highly dispersed Au(I) cations that are active, whereas in CO oxidation the gold atoms/cations are not active, and a hierarchy of active species exists with small clusters being the most active when iron oxide is used as support. Hence it appears that there is not a single active gold species, and this needs to be taken into account when designing heterogeneous gold catalysts for a new reaction; it should be born in mind that deposition–precipitation would be an effective preparation method to start any investigation since these catalysts comprise the full range of potential active species.
Acknowledgments
The author thanks Cardiff University for financial support.
The author declares no competing financial interest.
References
- British Museum—The Lycurgus Cup. https://britishmuseum.tumblr.com/post/120689869617/the-lycurgus-cup (June 4, 2015).
- Faraday M. The Bakerian Lecture: experimental relations of gold (and other metals) to light. Philos. Trans. R. Soc. London 1857, 147, 145–181. 10.1098/rstl.1857.0011. [DOI] [Google Scholar]
- Michael Faraday’s Gold Colloids. http://www.rigb.org/our-history/iconic-objects/iconic-objects-list/faraday-gold-colloids (2014).
- Huang X.; El-Sayed M. A. Gold nanoparticles: Optical properties and implementations in cancer diagnosis and photothermal therapy. J. Adv. Res. 2010, 1, 13–18. 10.1016/j.jare.2010.02.002. [DOI] [Google Scholar]
- Pflästerer D.; Hashmi A. S. K. Gold catalysis in total synthesis-recent achievements. Chem. Soc. Rev. 2016, 45, 1331–1367. 10.1039/C5CS00721F. [DOI] [PubMed] [Google Scholar]
- Echavarren A. M.; Hashmi A. S. K.; Toste F. D. Gold catalysis – steadily increasing in importance. Adv. Synth. Catal. 2016, 358, 1347. 10.1002/adsc.201600381. [DOI] [Google Scholar]
- Hashmi A. S. K.; Hutchings G. J. Gold catalysis. Angew. Chem., Int. Ed. 2006, 45, 7896–7936. 10.1002/anie.200602454. [DOI] [PubMed] [Google Scholar]
- Haruta M.; Kobayashi T.; Sano H.; Yamada N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. 10.1246/cl.1987.405. [DOI] [Google Scholar]
- Hutchings G. J. Vapour phase hydrochlorination of acetylene: Correlation of catalytic activity of supported metal chloride catalysts. J. Catal. 1985, 96, 292–295. 10.1016/0021-9517(85)90383-5. [DOI] [Google Scholar]
- Johnston P.; Carthey N.; Hutchings G. J. Discovery, Development and commercialisation of gold catalysts for acetylene hydrochlorination. J. Am. Chem. Soc. 2015, 137, 14548–14557. 10.1021/jacs.5b07752. [DOI] [PubMed] [Google Scholar]
- Teles J. H.; Brode S.; Chabanas M. Cationic gold(I) complexes: highly efficient catalysts for the addition of alcohols to alkynes. Angew. Chem., Int. Ed. 1998, 37, 1415–1418. . [DOI] [PubMed] [Google Scholar]
- Rashkeev S. N.; Lupini A. R.; Overbury S.H.; Pennycook S. J.; Pantelides S. T. Role of the nanoscale in catalytic CO oxidation by supported Au and Pt nanostructures. Phys. Rev. B: Condens. Matter Mater. Phys. 2007, 76, 035438. 10.1103/PhysRevB.76.035438. [DOI] [Google Scholar]
- He Q.; Freakley S. J.; Edwards J. K.; Carley A. F.; Borisevich A. Y.; Mineo Y.; Haruta M.; Hutchings G. J.; Kiely C. J. Population and hierarchy of active species in gold iron oxide catalysts for carbon monoxide oxidation. Nat. Commun. 2016, 7, 12905. 10.1038/ncomms12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruta M. Spiers Memorial Lecture : Role of perimeter interfaces in catalysis by gold nanoparticles. Faraday Discuss. 2011, 152, 11–32. 10.1039/c1fd00107h. [DOI] [PubMed] [Google Scholar]
- Herzing A. A.; Kiely C. J.; Carley A. F.; Landon P.; Hutchings Identification of active gold nanoclusters on iron oxide supports for CO oxidation. Science 2008, 321, 1331–1335. 10.1126/science.1159639. [DOI] [PubMed] [Google Scholar]
- Allard L. F.; Borisevich A. Y.; Deng W.; Si R.; Flytzani-Stephanopoulos M.; Overbury S. H. Behavior of Au species in Au/FeOx catalysts as a result of in-situ thermal treatments, characterized via aberration-corrected STEM imaging. Microsc. Microanal. 2009, 15, 1482. 10.1017/S1431927609097001. [DOI] [Google Scholar]
- Shinoda K. Vapor-phase hydrochlorination of acetylene over metal chlorides supported on activated carbon. Chem. Lett. 1975, 4, 219–220. 10.1246/cl.1975.219. [DOI] [Google Scholar]
- Nkosi B.; Coville N. J.; Hutchings G. J. Reactivation of a supported gold catalyst for acetylene hydrochlorination. J. Chem. Soc., Chem. Commun. 1988, 71–72. 10.1039/c39880000071. [DOI] [Google Scholar]
- Minamata Convention, Curbing Mercury Use, is Now Legally Binding. http://www.ipsnews.net/2017/08/minamata-convention-curbing-mercury-use-is-now-legally-binding/ (August 8, 2017).
- Nkosi B.; Coville N. J.; Hutchings G. J.; Adams M. D.; Friedl J.; Wagner F. E. Hydrochlorination of acetylene using gold catalysts: a study of catalyst deactivation. J. Catal. 1991, 128, 366–377. 10.1016/0021-9517(91)90295-F. [DOI] [Google Scholar]
- Liu X.; Conte M.; Elias D.; Lu L.; Morgan D. J.; Freakley S. J.; Johnston P.; Kiely C. J.; Hutchings G. J. Investigation of the active species in the carbon-supported gold catalyst for acetylene hydrochlorination. Catal. Sci. Technol. 2016, 6, 5144–5153. 10.1039/C6CY00090H. [DOI] [Google Scholar]
- Malta G.; Kondrat S. A.; Freakley S. J.; Davies C. J.; Lu L.; Dawson S.; Thetford A.; Gibson E. K.; Morgan D. J.; Jones W.; Wells P. P.; Johnston P.; Catlow C. R. A.; Kiely C. J.; Hutchings G. J. Identification of single-site gold catalysts in acetylene hydrochlorination. Science 2017, 355, 1399–1402. 10.1126/science.aal3439. [DOI] [PubMed] [Google Scholar]
- Malta G.; Freakley S. J.; Kondrat S. A.; Hutchings G. J. Acetylene hydrochlorination using Au/carbon: a journey towards single site catalysis. Chem. Commun. 2017, 53, 11733–11746. 10.1039/C7CC05986H. [DOI] [PubMed] [Google Scholar]
- Chang S.-Y.; Uehara A.; Booth S. G.; Ignatyev K.; Mosselmans J. F. W.; Dryfe R. A. W.; Schroeder S. L. M. Structure and bonding in Au(I) chloride species: a critical examination of X-ray absorption spectroscopy (XAS) data. RSC Adv. 2015, 5, 6912–6918. 10.1039/C4RA13087A. [DOI] [Google Scholar]