

Summary
The genome of kiwifruit (Actinidia chinensis) was sequenced previously, the first in the Actinidiaceae family. It was shown to have been affected by polyploidization events, the nature of which has been elusive. Here, we performed a reanalysis of the genome and found clear evidence of 2 tetraploidization events, with one occurring ∼50–57 million years ago (Mya) and the other ∼18–20 Mya. Two subgenomes produced by each event have been under balanced fractionation. Moreover, genes were revealed to express in a balanced way between duplicated copies of chromosomes. Besides, lowered evolutionary rates of kiwifruit genes were observed. These findings could be explained by the likely auto-tetraploidization nature of the polyploidization events. Besides, we found that polyploidy contributed to the expansion of key functional genes, e.g., vitamin C biosynthesis genes. The present work also provided an important comparative genomics resource in the Actinidiaceae and related families.
Subject Areas: Genetics, Evolutionary Biology, Plant Biology, Omics
Graphical Abstract
Highlights
-
•
Two independent paleo-tetraploidization events may have occurred in Actinidiaceae
-
•
The tetraploidization events are likely autotetraploid in nature
-
•
These events contribute to the expansion of key trait genes
-
•
Hierarchical deconvolution allowed analysis of the kiwifruit genome interweaving homology
Genetics; Evolutionary Biology; Plant Biology; Omics
Introduction
Kiwifruit (Actinidia chinensis) is known as “the king of fruits,” with a remarkably high vitamin C content and a balanced nutritional composition of minerals, fiber, and some health-beneficial metabolites, which are essential for human health (Seddon et al., 1994, Fraser and Bramley, 2004). Kiwifruit is a member of the family Actinidiaceae, consisting of 3 genera and around 360 species (Anderberg et al., 2002), most of which are economically and nutritionally important crops. Actinidiaceae is commonly known as the Chinese gooseberry family and is the basal family in the order Ericales, which is one of the earliest orders to diverge from the asterids and shares the core-eudicot common hexaploidization (ECH).
Polyploidy plays an important role in the evolution of land plants, contributing to their origin and diversification (Paterson et al., 2004, Soltis et al., 2008, Jiao et al., 2011). Because of recursive polyploidization and genome repatterning, plant genomes are often considerably complex, hindering the efforts to understand their formation and evolution, and explore functional innovations of genes. The kiwifruit genome has a large number of collinear regions, which often are shared with other plant genomes. It was proposed that 2 whole-genome duplication events occurred during kiwifruit genome evolution (Tao et al., 2010, Huang et al., 2013).
However, the ancestral ploidy of kiwifruit has not been very clear because of the lack of deep exploration. Here, by using our newly proposed pipeline to unravel hierarchical genomic homology reconstruction, we performed a comprehensive analysis of the kiwifruit genome with grape and coffee genomes as outgroup references, produced a list of orthologous and paralogous genes, and explored the outcomes of genomic fractionation. Besides, the present effort provided an important comparative genomics resource for further biological exploration in the Actinidiaceae and related families.
Results
Intra- and Intergenomic Gene Collinearity
Gene collinearity, showing the preservation of ancestral genome structure in the modern genome, is an important means of unveiling the cryptic nature of genomic evolution. We inferred collinear genes within the kiwifruit, coffee, and grape genomes, and between them, via ColinearScan (Wang et al., 2006), which is an effective way to evaluate genomic blocks of collinear genes. The grape and coffee genomes were used as outgroup references because these genomes are relatively simple and have been affected only by ECH. Thus, we also inferred collinear genes within each of these genomes and all 3 genomes (Tables S1 and S2).
Kiwifruit has a much more highly preserved intragenomic homology than either grape or coffee genomes (Tables S1 and S2). We revealed 964 homologous blocks with ≥4 collinear genes, containing 9,998 collinear gene pairs in total. At the same parameter setting, we found 214 and 279 homologous blocks in grape and coffee genomes, containing 2,099 and 2,502 collinear gene pairs, respectively. The number of homologous genes in kiwifruit was about 4–5 times as many as those in grape and coffee, supporting the fact that the kiwifruit genome has undergone additional polyploidy events.
As to intergenomic gene collinearity, 1,631 blocks containing 18,626 collinear gene pairs were revealed between kiwifruit and grape and 1,869 blocks containing 18,869 collinear gene pairs were revealed between kiwifruit and coffee, showing similar collinearity between kiwifruit and the 2 referenced plants (Tables S1 and S2).
Evidence for Two Paleo-Tetraploidization Events
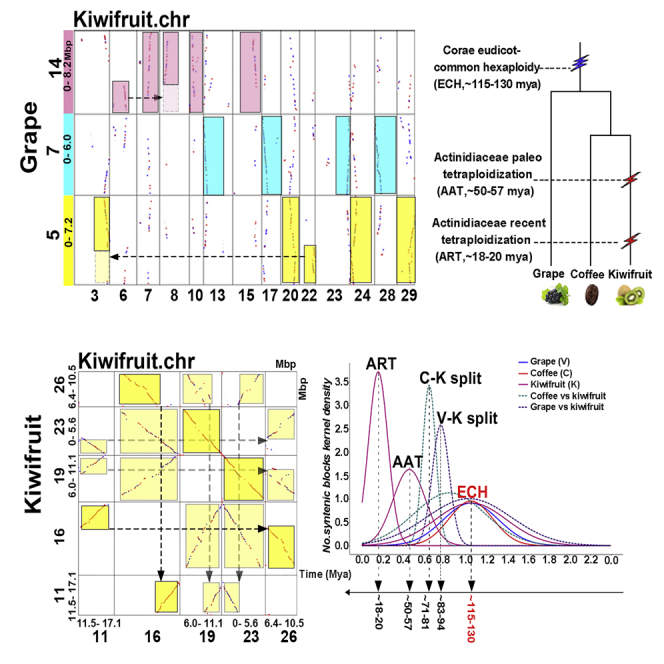
Our inference of ancient polyploidization features integration of sequence divergence and homologous gene dotploting. We first characterized the synonymous nucleotide substitutions on synonymous substitution sites (Ks) between the collinear genes inferred above. If all the Ks of collinear genes are pooled together, clustering analysis may result in absorbing effects. That is, a smaller cluster produced by one event (often an ancient one) would be covered by a bigger cluster produced by another event (often a recent one). Therefore, we calculated the median (relatively more stable than the mean) value of each block and used them to perform clustering analysis. The kiwifruit collinear blocks clearly produced 3 peaks (Figure 1A), and a curve-fitting process located them at 0.164 (+/−0.093), 0.462 (+/−0.156), and 1.55 (+/−0.297), showing 3 events. Then by mapping gene sequence onto whole-genome-based similarity inferred by BLASTN, we constructed the homologous gene dotplot within kiwifruit genome (Figure S1). We also mapped block median Ks values onto the dotplot. As to Ks and checking the dotplot, we could divide most of the blocks produced by 3 events, especially the longer blocks. In fact, if allowing the longer blocks to absorb their nearby smaller blocks, which could have been produced by genomic fractionation, we would distinguish all blocks by 3 events. Blocks produced by the most recent event covered 33.34% of the genomic DNA and 17.36% of total genes, and a covered region has a 1:1 correspondence, showing that it is tetraploidization. We called it as an Actinidiaceae recent tetraploidization (ART). Blocks produced by the mid-aged event covered 47.92% of the genomic DNA and 10.34% of total genes, and a covered region has a 1:1 correspondence, showing that it also is a tetraploidization event. We called it as an Actinidiaceae ancient tetraploidization (AAT). In contrast, we found that the blocks surely produced by the ECH covered 40.76% of the genomic DNA and only 6.96% of total genes.
Figure 1.

Original and Corrected Synonymous Nucleotide Substitutions between Collinear Genes (Ks)
ART, Actinidiaceae recent tetraploidization; AAT, Actinidiaceae ancient tetraploidization; ECH, core-eudicot-common hexaploidization; Mya, million years ago. Continuous curves are used to show Ks distribution in a genome, and broken ones are between genomes. (A) Density fitted by using original Ks values; (B) Inferred means; (C) Density fitted by using corrected Ks values; (D) Inferred evolutionary dates.
To verify our inference of the 3 events ART, AAT, and ECH, we used grape as a reference to decipher kiwifruit genome, a rosid eudicot preserving the ECH-triplicated genome structure of a core-eudicot common ancestor. Besides, we used an asteroid plant, coffee, as another reference, which is more closely related to kiwifruit than grape and has not been affected by other polyploidizations after the ECH. Homologous gene dotplots generated from the BLAST results between genomes allowed us to locate the corresponding homologous regions and were used to distinguish orthologous regions, established due to the grape-kiwifruit split, and outparalogous regions, established due to different duplications (Figures 2A, S2, and S3). We mapped the inferred median Ks values of each collinear block onto the dotplots (Figure S1). Integrating the aforementioned 2 lines of information helps distinguish orthologous and outparalogous blocks. The grape chromosomes were denoted with blocks in 7 colors, corresponding to the 7 ancestral eudicot chromosomes before the ECH, therefore a grape genomic region mostly has 2 paralogous regions in the same color (Jiao et al., 2012).
Figure 2.

Examples of Homologous Gene Dotplots between Kiwifruit, Coffee, and Grape
Chromosome numbers and regions (in Mbp) were shown. Best-hit genes make red dots, secondary hits make blue dots, and the others are shown in gray. Highlights show the best matched chromosomal regions. Arrows show complement correspondence produced by chromosome breakages during evolution.
(A) Homologous dotplot between selected grape and kiwifruit chromosomes.
(B) Homologous dotplot between selected coffee and kiwifruit chromosomes.
(C) Homologous dotplot within selected kiwifruit chromosomes.
As to the Ks of grape-kiwifruit collinear gene pairs, there is a bimodal distribution, and therefore can be decomposed into 2 distinct distributions that have means (0.764 ± 0.0802) and (1.034 ± 0.405) (Figure 1A), corresponding to the orthologous genes (originated through their split) and outparalogous genes (through the common ECH), respectively. According to the median Ks value of each block, it was not difficult to distinguish the orthologous and the outparalogous blocks, especially for the long ones with ≥10 collinear genes. Allowing the long blocks to absorb the nearby short ones, whose Ks often change much, we inferred orthology and outparalogy for all the predicted collinear blocks (Figures S2 and S3). With the orthologous blocks, by relating them to the AAT and ART blocks in kiwifruit dotplot, as to their locations on kiwifruit chromosomes, we separated the orthologous regions produced by different tetraploidization. For certain outparalogous regions lacking collinear genes, due to widespread and complementary gene losses (Maere et al., 2005), we transitively used the paralogy between grape chromosomes and orthology between grape and kiwifruit chromosomes to locate the regions where outparalogy should exist. The coffee-kiwifruit gene dotplot and Ks analysis revealed orthology and outparalogy between the 2 genomes (Figures 1, 2B, and S3). As expected, each grape or coffee genomic region often has 4 orthologous regions, 2 corresponding to the AAT and the other 2 to ART (Figure 3). These intergenomic analyses agree with the above inference of 2 sequential tetraploidizations in kiwifruit lineage.
Figure 3.

Species and Gene Phylogenetic Trees
(A) Phylogenetic tree of kiwifruit (K), coffee (C), and grape (V). The core-eudicot common hexaploidy (ECH) is denoted by a blue flash, and the two kiwifruit paleo-tetraploidizations are denoted by red flashes.
(B) Gene phylogeny: three paralogous genes in the grape and coffee genomes are denoted by V1, V2, and V3, and C1, C2, and C3, respectively, produced by the ECH, and each has 4 orthologs and 8 outparalogs in the kiwifruit genome (e.g., V1 has 4 orthologs, K111, K112, K121, and K122 and 8 outparalogs, K211, K212, K221, K222, K311, K312, K321, and K322 in kiwifruit). The species tree is produced based on our present analysis of homologous genes.
Reconstructed phylogeny of homologous genes provided further support to 2 tetraploidizations in kiwifruit. We used 426 homologous groups, including a grape gene, its ortholog coffee gene, and at least 3 orthologous kiwifruit genes, to reconstruct gene trees. Actually, we successfully constructed gene trees for 311 (73.00%) homologous gene groups (Figure S4), and the other groups were discarded due to insufficient sequence similarity. Checking the topology of these 311 trees, we found that 33 (10.61%) of them each involved 4 kiwifruit genes and were separated into 2 subgroups with each subgroup having 2 genes. This is just what is expected if 2sequential tetraploidizations occurred in the kiwifruit lineage.
Multiple Genome Alignment
The above-mentioned homologous gene dotplotting and Ks analysis distinguished orthology from outparalogy between genomes and the ART-, AAT-, and ECH-produced paralogous regions in kiwifruit. We retrieved detailed information of orthologous and (out)paralogous regions from the dotplots (Tables S3–S5), and counted duplicated genes produced by different events. We found that the ECH event produced 1,505 collinear paralogous pairs, containing 2,150 genes in 188 regions; the AAT event produced 1,983 collinear paralogous pairs, containing 3,196 genes in 153 regions; and the ART event produced 2,753 collinear paralogous pairs, containing 5,366 genes in 95 regions. The number of ECH-related kiwifruit paralog genes was much smaller than the 3,866 genes found in grape. Although the ECH-related paralogs were much less in number, there were more than twice as many, but much shorter, ECH-related regions in kiwifruit as in grape, showing the occurrence of severe kiwifruit genomic fractionation (Table 1).
Table 1.
Number of Duplicated Genes within the Kiwifruit Genome Related to Recursive Polyploidization Events
| Species | aECH Related | bAAT Related | cART Related |
|---|---|---|---|
| Grape | d87/2,432/3,866 | – | – |
| Coffee | 54/1,189/2,095 | – | – |
| Kiwifruit | 188/1,505/2,150 | 153/1,983/3,196 | 95/2,753/5,366 |
Core-eudicot common hexaploidization.
Actinidiaceae ancient tetraploidization.
Actinidiaceae recent tetraploidization.
Numbers of blocks, gene pairs, and gene numbers are separated by a solidus.
Intergenomic homology is much better than intragenomic homology. For example, there were 9,322 (30.16%) kiwifruit genes (9,508 pairs in 309 blocks) having coffee orthologs, and 2,885 (9.33%) genes (3,259 pairs in 261 blocks) having coffee outparalogs; meanwhile, 8,923 (28.87%) kiwifruit genes (9,125 pairs in 402 blocks) had grape orthologs, and 4,324 (14.07%) genes (3,681 pairs in 367 blocks) had grape outparalogs. Similar findings appeared in the grape and coffee alignment. More information can be found in Tables S3–S5.
With the grape genome as reference and by entering collinear gene IDs into a table, we constructed a hierarchical and event-related multiple-genome alignment (grape, coffee, kiwifruit), producing a table of homologous genes (Figures 4 and S5; Table S6). The table related paralogous genes in each genome and outparalogs between genomes to each polyploidization, and orthologs between genomes to their ancestral speciation (Figure 3). The table was translated to a whole alignment of the 3 genomes graphically (Figure 4), and a slice of the genome-level alignment can be illustrated in a linear format to give a close view of genomic fractionation (Figure 5). Moreover, to accommodate kiwifruit-specific genes with no grape collinearity, we constructed another homology table with coffee as reference (Figure S6; Table S7), which further proved the paleo-tetraploidizations in kiwifruit (for details see Transparent Methods).
Figure 4.

Homologous Alignments of Kiwifruit, Coffee, and Grape Genomes
With grape as a reference genome, genomic paralogy, orthology, and outparalogy information within grape and with coffee and kiwifruit are displayed in 18 circles. The curved lines within the inner circle, colored according to the 7 ancestral eudicot chromosomes (Jaillon et al., 2007), link paralogous pairs on the 19 grape chromosomes produced by the ECH. The short lines forming the innermost circle represent all predicted genes in grape, which have 2 sets of paralogous regions, forming another 2 circles. Each of the 3 sets of grape paralogous chromosomal regions has 1 orthologous copy in coffee and 4 orthologous copies in kiwifruit. Therefore, the 3 genomes result in 18 circles in the figure. Circles are respectively denoted by V, C, and K, corresponding to source plants in Figure 3. Homologous genes are denoted by short lines standing on a chromosome circle and are colored according to the chromosome number in the source plant shown in the inset legend.
Figure 5.

Local Alignment of the Kiwifruit Genome
Species are shown in different colors; homologous genes between adjacent chromosomes are ligated with Bezier curves.
Genomic Fractionation
As described in the previous sections, we mapped the kiwifruit genes, e.g., the AAT- and ART-produced paralogs, onto the grape genome, making it possible to check possible gene retention or loss (Figure S7A). Notably, we found that after 2 rounds of tetraploidizations, kiwifruit preserved a high fraction of genes collinear to grape orthologs. Actually, different grape chromosomes had collinear gene retention rate of 74%–95% in each of their 4 sets of orthologous regions (Table 2). With regard to 2 sets of AAT paralogous regions, grape gene retention rates are very similar. For 12 of 19 grape chromosomes, the difference of their gene retention rates in 2 AAT paralogous regions is ≤0.05. This means that there exist 2 AAT subgenomes retaining similar numbers of ancestral genes. Actually, the retention rate difference between the subgenomes, noted as AAT-1 and AAT-2, with arbitrarily assigned paralogous regions is 4.3% (Table 2). What is more interesting is that the gene retention rates between 2 sets of ART paralogous regions are just like the AAT regions. For 12 of 19 grape chromosomes, the ART paralogous regions have retention rate difference ≤0.05 (Table 2), suggesting 2 subgenomes with balanced gene retention. The ART paralogous regions are therefore arbitrarily assigned to ART subgenome, noted as ART-1 and ART-2, displaying a whole-genome difference ∼2.2%.
Table 2.
Gene Retention Rates in Subgenomes as to the Grape Genome
| Grape |
Kiwifruit Homoelogous Subgenomes |
||||||
|---|---|---|---|---|---|---|---|
| Chromosome # | Gene # | AAT- 1 | AAT-2 | AAT 1–2 Difference | ART-1 | ART-2 | ART 1–2 Difference |
| 1 | 1,327 | 0.86 | 0.88 | 0.02 | 0.80 | 0.74 | 0.06 |
| 2 | 1,237 | 0.88 | 0.87 | 0.01 | 0.80 | 0.78 | 0.02 |
| 3 | 1,000 | 0.79 | 0.86 | 0.07 | 0.80 | 0.90 | 0.10 |
| 4 | 1,638 | 0.85 | 0.81 | 0.04 | 0.86 | 0.81 | 0.05 |
| 5 | 1,748 | 0.86 | 0.89 | 0.03 | 0.84 | 0.75 | 0.09 |
| 6 | 1,779 | 0.95 | 0.89 | 0.06 | 0.76 | 0.79 | 0.03 |
| 7 | 1,409 | 0.88 | 0.82 | 0.06 | 0.83 | 0.83 | 0.00 |
| 8 | 1,867 | 0.80 | 0.79 | 0.01 | 0.83 | 0.95 | 0.12 |
| 9 | 1,221 | 0.85 | 0.87 | 0.02 | 0.92 | 0.90 | 0.02 |
| 10 | 632 | 0.81 | 0.87 | 0.06 | 0.77 | 0.75 | 0.02 |
| 11 | 1,107 | 0.88 | 0.74 | 0.14 | 0.79 | 0.83 | 0.04 |
| 12 | 1,481 | 0.84 | 0.84 | 0.00 | 0.84 | 0.90 | 0.06 |
| 13 | 1,329 | 0.87 | 0.79 | 0.08 | 0.89 | 0.80 | 0.09 |
| 14 | 1,729 | 0.84 | 0.86 | 0.02 | 0.85 | 0.81 | 0.04 |
| 15 | 561 | 0.88 | 0.78 | 0.10 | 0.89 | 0.90 | 0.01 |
| 16 | 647 | 0.92 | 0.95 | 0.03 | 0.78 | 0.87 | 0.09 |
| 17 | 1,168 | 0.82 | 0.81 | 0.01 | 0.85 | 0.86 | 0.01 |
| 18 | 1,886 | 0.74 | 0.73 | 0.01 | 0.87 | 0.92 | 0.05 |
| 19 | 1,135 | 0.95 | 0.80 | 0.05 | 0.85 | 0.86 | 0.01 |
| Total/Average | 24,901 | 0.86 | 0.83 | 0.04 | 0.83 | 0.84 | 0.05 |
We performed a scrutiny analysis of gene retention/loss using a sliding window along chromosomes. We found that, in nearly all local regions, with the exception of large patches of DNA losses in one copy of the duplicated chromosomes, genomic retention rates were often ≤0.05 (Figures S8 and S9). Actually, the difference observed at the chromosome level should have been caused by large patches of alternative segmental DNA losses due to genomic instability (Figures S8 and S9). With the coffee genome as the reference, we obtained similar observation (Table S8).
Modeling Genomic Fractionation
To explore the mechanism underlying genomic fractionation, we characterized the runs of continual gene deletions in kiwifruit when compared with the reference genomes (Wang et al., 2015). Although there were patches of chromosomal segmental losses (Figure S7), most of the runs of gene deletions were of 15 continual genes or fewer. A statistical fitness regression showed that deletion patterns followed a near geometric distribution (Figure S7). With grape and coffee genomes as reference, kiwifruit had a gene deletion pattern following similar distributions (geometric parameter p = 0.252–0.299, the probability of deleting one gene at a time, and goodness of fit F-test p value 0.89 to accept the fitness). This showed that 38%–42% of genes were deleted containing one or two genes, suggesting a mechanism of fractionation removing short DNA segment about 5–10 kb DNA in length. It seems that short deletion runs accounted for the majority initially, and then recursive deletion runs overlapping previous deletions elongated the observed length of runs, as revealed by a much-diverged reference genome.
Evolutionary Rate Divergence and Dating
By checking Ks, we managed to estimate the occurrence times of the AAT, ART, and other evolutionary events, e.g., speciation. As distribution of Ks between paralogs or orthologs related to a specific event, fitting by using normal distribution function, and the locations of the means (or peaks) and their variances were determined statistically (Figure 1A; Table S9). As to the divergent locations of the common ECH in different distributions, we found that the evolutionary rates of kiwifruit and grape were similar and the slowest among the 3 genomes, with evolution of the coffee genome faster by 47.20%.
Divergent evolutionary rates among plants affect dating ancestral events. Here, based on a modified version of an approach that we previously developed (Wang et al., 2015, Wang et al., 2018), we performed evolutionary rate correction by aligning the peaks in different genomes corresponding to the ECH event to the same locations (see Transparent Methods for details) (Figure 1B; Table S10). The present correction aligned the ECH peaks to the same location, correcting rate differences that have accumulated after the ECH event between rosids and asterids. Supposing that the ECH occurred at ∼115–130 million years ago (Mya) (Jiao et al., 2012, Vekemans et al., 2012), adopted by previous publications (Jaillon et al., 2007, Potato Genome Sequencing Consortium et al., 2011, Paterson et al., 2012), we inferred that the ART event occurred at ∼18–20 Mya and the AAT event occurred at ∼50–57 Mya. In addition, we inferred that kiwifruit (asterids) and grape (rosids) ancestors split at ∼83–94 Mya, and then kiwifruit (campanulids) and coffee (lamiids) ancestors split at ∼71–81 Mya. Therefore, the tetraploidization events might be shared by Actinidiaceae plants after the separation of Actinidiaceae and Roridulaceae sisters at 84.8 Mya (Magallón et al., 2015).
Balanced Gene Expression between Duplicated Chromosomes
Balanced gene expression was observed between duplicated copies of chromosomes produced in AAT and ART (Tables S11A–S11D). In AAT, between duplicated copies of chromosomes, 38.1% duplicated genes show no difference in gene expression, and the significantly diverged-expressed genes are distributed without significant difference. Between the ART duplicated copies of chromosomes, 33.7% and 40.1% of duplicated genes show no difference in expression and the significantly diverged-expressed genes are distributed without significant difference.
Ascorbic Acid (Vitamin C) Biosynthesis and Recycling Pathway
To check whether recursive polyploidization contributed to the expansion of key traits, such as the ascorbic acid (VC) biosynthesis and recycling pathway in kiwifruit, we used 18 VC-related previously reported gene families as seeds (Huang et al., 2013) and detected their homologs in grape, coffee, and kiwifruit at BLASTP E < 1e-10 and a score greater than 150 (Table S12). Kiwifruit had the most VC-related genes (123), 3.5–6.5 times more than in coffee and grape. Notably, 72.22%, 66.67%, and 61.11% of VC-related genes could be located at paralogous chromosomal locations related to the 3 polyploidization events (ART, AAT, and ECH), respectively. The gene copy numbers increased by 56 (44.53%) through the ECH, 35 (28.46%) through the AAT, and 52 (42.28%) through the ART. Moreover, among 18 VC-related families, 11, 12, and 13 of them exhibited an expansion after the ECH, AAT, and ART events, respectively. Specifically, for 22 kiwifruit GalUR-related genes, we found that 36.36%, 45.45%, and 27.27% of the copies were located in paralogous chromosomal regions related to the 3 polyploidization events (ART, AAT, and ECH), respectively. Comparatively, there are only 11 and 3 genes in grape and coffee, respectively.
Discussion
About Deciphering Complex Genomes
Plants often have complex genomes, due to recursive polyploidization and genome repatterning (Soltis et al., 2014, Soltis et al., 2015). This makes it difficult to deconvolute their genome structures, understand their formation, and explore their gene functional evolution. Here, we used a recently proposed pipeline to reanalyze the kiwifruit genome and showed that the kiwifruit genome was affected by 2 tetraploidization events. The pipeline features of integrative use of homologous gene dotplots and sequence divergence analysis make it possible to distinguish orthologous regions and (out)paraologous regions while comparing the genome under study with a well-characterized reference genome, or to distinguish paralogous regions in the studied genome resulting by different events. Therefore, this would help find the orthology (and outparalogy) ratio between genomes.
The produced event-related list of homologous genes among genomes is often very helpful in that it provided information about the origin and expansion of genes. Although it may not be complete as it does not include all genes produced by these events due to widespread fractionation over time, the list tells how and when a pair of homologs were produced and diverged, and whether there has been likely gene loss after certain events. Therefore, it can be useful information to reveal the evolutionary and function-innovation trajectories of genes, gene families, regulatory pathways, and economically and agriculturally important traits. As an example, we showed that genes related to VC biosynthesis and recycling pathway were expanded through each polyploidization event. By integrating more sophisticated phylogenetic and evolutionary analyses, many valuable genes could be re-checked at certain phylogenetic nodes to clarify specific evolutionary changes correlated with their functional innovation.
Two Likely Auto-Tetraploidization Events
A large number of genome sequencing analyses have shown that almost all plants on the earth have a polyploid ancestor. It has been proposed that polyploidization has contributed to the origin, divergence, and success of seed and flowering plants (Jiao et al., 2011) and their domestication (Kellogg, 2016).
The above-mentioned findings of balanced gene loss and balanced gene expression between the subgenomes in each tetraploidization event suggest little if any dominance between members of homologous chromosome pairs, which raises the likelihood of the 2 events, AAT and ART, being autotetroploid in nature.
Here we observed much lower evolutionary rates in kiwifruit genes when compared with the coffee homologs. This may be explained by the prolonged generation time of kiwifruit, which might further relate to auto-tetraploidization, as discussed below. Often, elevated evolutionary rates of genes are observed after polyploidization (Wang et al., 2016, Paterson et al., 2012). This may result from homoeologous recombination between duplicated chromosomal regions. Theoretically, the existence of a duplicated copy would buffer mutations in a gene. Therefore, allo-polyploidization would increase evolutionary rates due to homoeologous recombination and the existence of extra gene copies. However, (genetic) auto-tetraploidization results in multiple copies of homologous chromosomes, causing multisomic inheritance (Doyle and Egan, 2010). Multisomic inheritance, with multiple homologous chromosomes to interact and pair with one another in a reticular manner, may prolong the cell cycle time and therefore the generation time. In contrast, allopolyploids, although having doubled chromosomes, execute diploid inheritance and do not have prolonged cell cycle time or generation time. Actually, the effect in evolutionary rates may be a key difference between auto- and allopolyploids.
Autopolyploids may suffer from reduced fertility, whereas allopolyploids are thought to have advantages during the establishment phase owing to their potential for heterosis. These thoughts are consistent with the observation (Barker et al., 2016) that more crops are allopolyploid (e.g., wheat, cotton, tobacco, strawberry, and oilseed rape) than autopolyploid (e.g., potato, sugarcane, and banana). Based on information from sequenced genomes, maize, wheat, and the common ancestor of grasses were proposed to result from allopolyploidy, with only the most recent duplication in sugarcane proposed to be autopolyploidy (Schnable et al., 2011, Chalhoub et al., 2014, Mayer et al., 2014). Recently, we provided genomic evidence that soybean and Salicaceae plants may have autopolyploid origin (Wang et al., 2017, Liu et al., 2017). Here, the observation of balanced fractionation throughout the subgenomes after each of the AAT and ART provided evidence of their likely auto-tetraploidization nature. This is a singular observation so far in all sequenced genomes. However, we have to note that widespread genomic fractionation over millions of years does not allow the credible inference of auto- or allopolyploidy (Doyle and Egan, 2010) (Woodhouse et al., 2010).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We appreciate financial support from the Ministry of Science and Technology of the People's Republic of China (2016YFD0101001), China National Science Foundation (31510333 to J.W. and 3117022 to X.W.), and Natural Science Foundation of Hebei Province (C2015209069 to J.W.). We thank the helpful discussion with researchers at the iGeno Co. Ltd., China.
Author Contributions
Conceptualization: X.W. and J.-P.W.; Formal Analysis: J.-P.W., J.-G.Y., J.L., P.-C.S., L.W., F.-B.M., J.-Q.Y., S.-R.S., Y.-X.L., Y.-X.P., W.-N.G., Z.-Y.W., S.-Q.S., L.Z., X.-M.S., C.L., X.-Q.D., Y.-Q.X., Y.H., J.Z., and J.-Y.W.; Methodology: J.-P.W., and L.W.; Software: P.-C.S., T.-Y.L. and L.Z.; Data Curation: J.-G.Y. and J.L.; Writing – Original Draft: J.-P.W., J.-G.Y., and J.L.; Writing –Review & Editing: X.W., J.-P.W.; Supervision: X.W.
Declaration of Interests
The authors declare no conflict of interest.
Published: September 20, 2018
Footnotes
Supplemental Information includes Transparent Methods, 9 figures, and 13 tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.08.003.
Supplemental Information
References
- Anderberg A.A., Rydin C., Källersjö M. Phylogenetic relationships in the order Ericales SL: analyses of molecular data from five genes from the plastid and mitochondrial genomes. Am. J. Bot. 2002;89:677–687. doi: 10.3732/ajb.89.4.677. [DOI] [PubMed] [Google Scholar]
- Barker M.S., Husband B.C., Pires J.C. Spreading wings and flying high: the evolutionary importance of polyploidy after a century of study. Am. J. Bot. 2016;103:1139. doi: 10.3732/ajb.1600272. [DOI] [PubMed] [Google Scholar]
- Chalhoub B., Denoeud F., Liu S., Parkin I.A., Tang H., Wang X., Chiquet J., Belcram H., Tong C., Samans B. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345:950–953. doi: 10.1126/science.1253435. [DOI] [PubMed] [Google Scholar]
- Doyle J.J., Egan A.N. Dating the origins of polyploidy events. New Phytol. 2010;186:73–85. doi: 10.1111/j.1469-8137.2009.03118.x. [DOI] [PubMed] [Google Scholar]
- Fraser P.D., Bramley P.M. The biosynthesis and nutritional uses of carotenoids. Prog. Lipid Res. 2004;43:228–265. doi: 10.1016/j.plipres.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Huang S., Ding J., Deng D., Tang W., Sun H., Liu D., Zhang L., Niu X., Zhang X., Meng M. Draft genome of the kiwifruit Actinidia chinensis. Nat. Commun. 2013;4:2640. doi: 10.1038/ncomms3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O., Aury J.M., Noel B., Policriti A., Clepet C., Casagrande A., Choisne N., Aubourg S., Vitulo N., Jubin C. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- Jiao Y., Wickett N.J., Ayyampalayam S., Chanderbali A.S., Landherr L., Ralph P.E., Tomsho L.P., Hu Y., Liang H., Soltis P.S. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473:97–100. doi: 10.1038/nature09916. [DOI] [PubMed] [Google Scholar]
- Jiao Y., Leebens-Mack J., Ayyampalayam S., Bowers J.E., McKain M.R., McNeal J., Rolf M., Ruzicka D.R., Wafula E., Wickett N.J. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012;13:R3. doi: 10.1186/gb-2012-13-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg E.A. Has the connection between polyploidy and diversification actually been tested? Curr. Opin. Plant Biol. 2016;30:25–32. doi: 10.1016/j.pbi.2016.01.002. [DOI] [PubMed] [Google Scholar]
- Liu Y., Wang J., Ge W., Wang Z., Li Y., Yang N., Sun S., Zhang L., Wang X. Two highly similar poplar paleo-subgenomes suggest an autotetraploid ancestor of salicaceae plants. Front. Plant Sci. 2017;8:571. doi: 10.3389/fpls.2017.00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S., De Bodt S., Raes J., Casneuf T., Van Montagu M., Kuiper M., Van de Peer Y. Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. USA. 2005;102:5454. doi: 10.1073/pnas.0501102102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magallón S., Gómez-Acevedo S., Sánchez-Reyes L.L., Hernández-Hernández T. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytol. 2015;207:437. doi: 10.1111/nph.13264. [DOI] [PubMed] [Google Scholar]
- Mayer K.F.X., Rogers J., Doležel J., Pozniak C., Eversole K., Feuillet C., Gill B., Friebe B., Lukaszewski A.J., Sourdille P. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345:1251788. doi: 10.1126/science.1251788. [DOI] [PubMed] [Google Scholar]
- Paterson A.H., Bowers J.E., Chapman B.A. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. USA. 2004;101:9903. doi: 10.1073/pnas.0307901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson A.H., Wendel J.F., Gundlach H., Guo H., Jenkins J., Jin D., Llewellyn D., Showmaker K.C., Shu S., Udall J. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012;492:423. doi: 10.1038/nature11798. [DOI] [PubMed] [Google Scholar]
- Potato Genome Sequencing Consortium. Xu X., Pan S., Cheng S., Zhang B., Mu D., Ni P., Zhang G., Yang S., Li R. Genome sequence and analysis of the tuber crop potato. Nature. 2011;475:189. doi: 10.1038/nature10158. [DOI] [PubMed] [Google Scholar]
- Schnable J.C., Springer N.M., Freeling M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. USA. 2011;108:4069–4074. doi: 10.1073/pnas.1101368108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon J.M., Ajani U.A., Sperduto R.D., Hiller R., Blair N., Burton T.C., Farber M.D., Gragoudas E.S., Haller J., Miller D.T. Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. JAMA. 1994;272:1413–1420. [PubMed] [Google Scholar]
- Soltis D.E., Bell C.D., Kim S., Soltis P.S. Origin and early evolution of angiosperms. Ann. New York Acad. Sci. 2008;1133:3. doi: 10.1196/annals.1438.005. [DOI] [PubMed] [Google Scholar]
- Soltis D.E., Visger C.J., Soltis P.S. The polyploidy revolution then…and now: stebbins revisited. Am. J. Bot. 2014;101:1057–1078. doi: 10.3732/ajb.1400178. [DOI] [PubMed] [Google Scholar]
- Soltis P.S., Marchant D.B., Van de Peer Y., Soltis D.E. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 2015;35:119–125. doi: 10.1016/j.gde.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Tao S., HongWen H., Barker M.S., HeslopHarrison J.S.P. Ancient genome duplications during the evolution of kiwifruit (Actinidia) and related Ericales. Ann. Bot. 2010;106:497–504. doi: 10.1093/aob/mcq129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekemans D., Proost S., Vanneste K., Coenen H., Viaene T., Ruelens P., Maere S., Van de Peer Y., Geuten K. Gamma paleohexaploidy in the stem lineage of core eudicots: significance for MADS-box gene and species diversification. Mol. Biol. Evol. 2012;29:3793. doi: 10.1093/molbev/mss183. [DOI] [PubMed] [Google Scholar]
- Wang X., Shi X., Li Z., Zhu Q., Kong L., Tang W., Ge S., Luo J. Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice. BMC Bioinformatics. 2006;7:447. doi: 10.1186/1471-2105-7-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Wang J., Jin D., Guo H., Lee T.H., Liu T., Paterson A.H. Genome alignment spanning major Poaceae lineages reveals heterogeneous evolutionary rates and alters inferred dates for key evolutionary events. Mol. Plant. 2015;8:885–898. doi: 10.1016/j.molp.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Wang X., Guo H., Wang J., Lei T., Liu T., Wang Z., Li Y., Lee T.H., Li J., Tang H. Comparative genomic de-convolution of the cotton genome revealed a decaploid ancestor and widespread chromosomal fractionation. New Phytol. 2016;209:1252. doi: 10.1111/nph.13689. [DOI] [PubMed] [Google Scholar]
- Wang J., Sun P., Li Y., Liu Y., Yu J., Ma X., Sun S., Yang N., Xia R., Lei T. Hierarchically aligning 10 legume genomes establishes a family-level genomics platform. Plant Physiol. 2017;174:284–300. doi: 10.1104/pp.16.01981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Sun P., Li Y., Liu Y., Yang N., Yu J., Ma X., Sun S., Xia R., Liu X. An overlooked paleo-tetraploidization in Cucurbitaceae. Mol. Biol. Evol. 2018;35:16–26. doi: 10.1093/molbev/msx242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse M.R., Schnable J.C., Pedersen B.S., Lyons E., Lisch D., Subramaniam S., Freeling M. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol. 2010;8:e1000409. doi: 10.1371/journal.pbio.1000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.