

Abstract
Background
In approximately 10% of all gastric cancer (GC) cases, a heritable cause is suspected. A subset of these cases have a causative germline CDH1 mutation; however, in most cases the cause remains unknown. Our objective was to assess to what extent these remaining cases may be explained by germline mutations in the novel candidate GC predisposing genes CTNNA1, MAP3K6 or MYD88.
Methods
We sequenced a large cohort of unexplained young and/or familial patients with GC (n=286) without a CDH1germline mutation for germline variants affecting CTNNA1, MAP3K6 and MYD88 using a targeted next-generation sequencing approach based on single-molecule molecular inversion probes.
Results
Predicted deleterious germline variants were not encountered in MYD88, but recurrently observed in CTNNA1 (n=2) and MAP3K6 (n=3) in our cohort of patients with GC. In contrast to deleterious variants in CTNNA1, deleterious variants in MAP3K6 also occur frequently in the general population.
Conclusions
Based on our results MAP3K6 should no longer be considered a GC predisposition gene, whereas deleterious CTNNA1 variants are confirmed as an infrequent cause of GC susceptibility. Biallelic MYD88 germline mutations are at most a very rare cause of GC susceptibility as no additional cases were identified.
Keywords: cancer: gastric, heritability, ctnna1 – map3k6 – myd88, next generation sequencing
Introduction
In familial or early-onset gastric cancer (GC) cases, a heritable germline aberration may underlie the development of GC. Elucidation of these germline defects is crucial to improve the clinical management of these patients and their relatives at risk. Pathogenic germline mutations in the CDH1 gene predispose to the development of hereditary diffuse gastric cancer (HDGC) with high GC risk of up to 70%,1 2 although the penetrance of CDH1 mutations identified in incident cases through gene panel testing for other reasons is not known. In more than 75% of the families that fulfil the CDH1 testing criteria,3 a causative germline mutation in CDH1 cannot be identified.2 4 5 In addition to these unexplained HDGC families, no heritable germline defects predisposing to the development of familial mixed-type and intestinal-type GC have been identified yet. Consequently, the putative causative germline aberration remains unknown for the majority of patients with GC.
The introduction of next-generation sequencing has revolutionised the identification of novel germline defects predisposing to (Mendelian) diseases, including heritable cancer syndromes.6–8 CTNNA1, MAP3K6 and MYD88 are considered novel candidate GC predisposing genes.9–11 Thus far, only for CTNNA1 the role in GC predisposition was confirmed in an independent study.2 Since there were only very few families reported with CTNNA1, MAP3K6 and MYD88 germline mutations, the histological characteristics of the GC associated with these mutations are not established yet. Here, we describe the sequencing of germline DNA of a large cohort of CDH1 mutation-negative patients with GC who were suspected for a hereditary predisposition based on age and/or family history (n=286) for variants affecting CTNNA1, MAP3K6 and MYD88 to further determine their potential role as GC predisposing genes.
Materials and methods
Patient selection
Our selected cohort was counselled in 16 different centres throughout Europe and contained 299 patients with GC from 297 families (the Netherlands (n=193), Poland (n=76), Germany (n=12), Italy (n=12), Portugal (n=5) and Norway (n=1)). All patients had early-onset GC (before the age of 50) and/or a family history of GC with first-degree and/or second-degree relatives with GC (for details see online supplementary file 1). Since germline variants in MAP3K6 have also been identified in patients with intestinal-type GC and the predisposing phenotypes of CTNNA1 and MYD88 germline mutations are uncertain, patients with all histological adenocarcinoma subtypes of GC were included. Of the 299 patients, 178 were diagnosed with diffuse-type, 62 with intestinal-type and 10 patients with mixed-type GC. In 49 patients the histological subtype was not specified. The absence of CDH1 germline mutations was confirmed in all 120 patients previously tested outside the Radboud university medical center using single-molecule molecular inversion probes (smMIP)-based targeted sequencing of CDH1 on germline DNA. For 13 of these patients samples were excluded, as the coverage did not fulfil our predetermined quality criteria (≥95% of the open reading frame covered with at least 15 unique reads). Subsequently, for the remaining 107 samples multiplex ligation-dependent probe amplification (MLPA) analysis was performed using MLPA Kit P083 (MRC-Holland, Amsterdam, The Netherlands) according to standard procedures, but no germline aberrations affecting CDH1 were detected (MLPA was unsuccessful for two of these samples). Therefore, our cohort contained 286 patients with GC without a causative pathogenic germline aberration affecting CDH1. Prior to this study whole-exome and targeted MYD88 sequencing was performed for 38 and 62 patients, respectively.11 12
jmedgenet-2017-104962supp001.docx (3.6MB, docx)
Targeted sequencing, data analysis and immunohistochemistry
To determine the germline status of CTNNA1, MAP3K6 and MYD88 in patients with GC, we applied targeted sequencing using smMIPs on blood-derived DNA of these patients according to previously published methods.13–16 Data were analysed using the SeqNext V.4.3.0 software package (JSI medical systems). The Exome Aggregation Consortium (ExAC) database, containing whole-exome sequencing data derived from 60 706 individuals, was used as a control data set to establish the variant allele frequency of variant calls in the general population (ExAC, Cambridge, Massachusetts, USA (http://exac.broadinstitute.org, accessed 01/04/2017)).17 For details, including the performed α-E-catenin immunohistochemistry, see online supplementary file 2.
Results
Targeted smMIP-based sequencing of CTNNA1, MAP3K6 and MYD88 was performed on germline DNA samples derived from 286 patients with GC from 284 families without a causative germline mutation affecting CDH1. Variant calling could not be performed for three of these samples due to a low amount of total aligned unique reads obtained for these samples (153, 190 and 3.695 unique reads, respectively) (see figure 1A). For the remaining 283 samples, representing 147 patients fulfilling HDGC criteria, 26 patients fulfilling familial intestinal gastric cancer criteria, 76 patients with familial GC and 34 patients diagnosed with GC between the age of 40 and 50, the total number of aligned unique reads (median number of aligned unique reads: 26.012 (range 6.373–145.043)), as well as the average coverage of the protein coding regions (including canonical splice sites) of CTNNA1, MAP3K6 and MYD88, was high (online supplementary file 3). In total 331 non-synonymous variants were called. After exclusion of common SNPs (minor allele frequency >0.01; n=303), 28 variants remained for subsequent interpretation (figure 1A and table 1).
Figure 1.
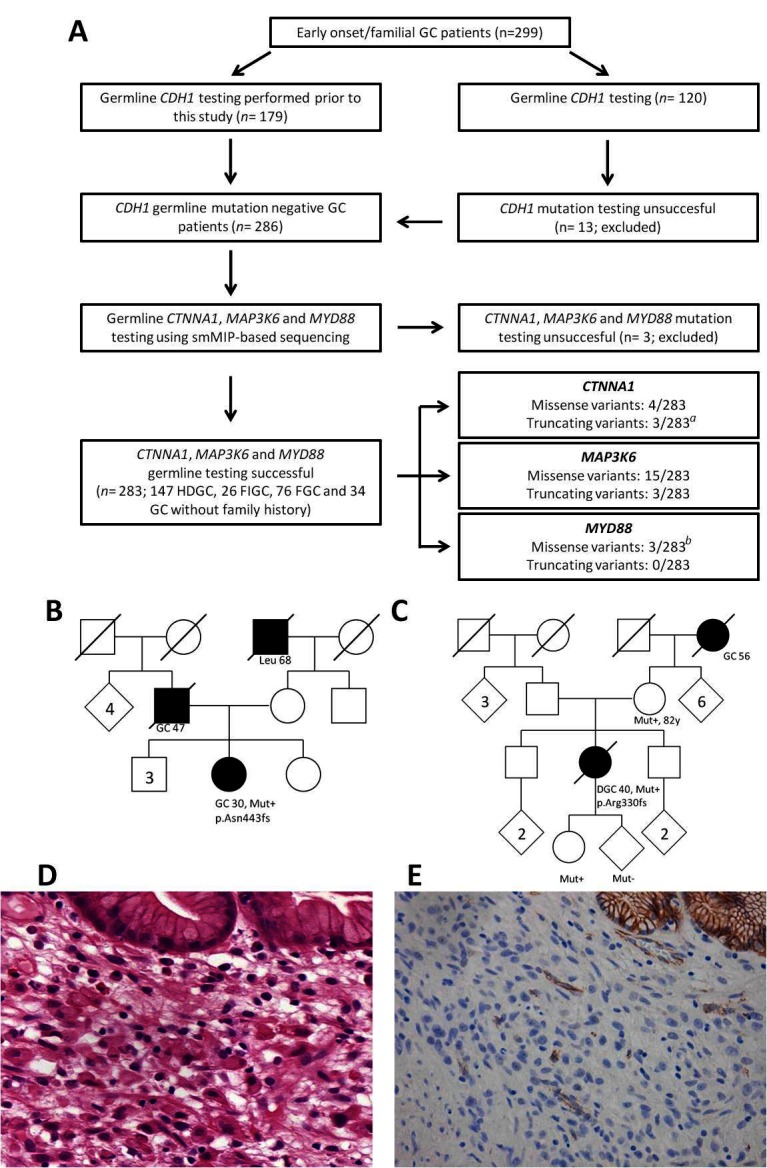
(A) Flow chart representing patient inclusion and subsequent germline analysis for CTNNA1, MAP3K6 and MYD88. aOne truncating germline variant in CTNNA1 was identified in a relative of a known CTNNA1 family.9 b One missense variant was identified in a homozygous state and previously reported.11 (B) Pedigree of the Polish patient with HDGC (746A) who carried a p.Asn443fs variant in CTNNA1. (C) Pedigree of the Dutch patient with HDGC (432A) who carried a p.Arg330fs variant in CTNNA1. (D) Microscopic image of DGC of patient 432A. Poorly cohesive polymorphic cells proliferate individually underneath normal gastric glands with foamy cytoplasm and sometimes signet ring cell morphology (H&E, 250×). (E) Immunohistochemistry for α-E-catenin shows total loss of protein expression in the poorly cohesive tumour cells, while the normal glands in the right upper corner and blood vessels in between show retained expression (magnification 200×). DGC, diffuse-type gastric cancer; FGC, familial gastric cancer; FIGC, familial intestinal gastric cancer; GC, gastric cancer; HDGC, hereditary diffuse gastric cancer; Leu, leukaemia; smMIP, single-molecule molecular inversion probe.
Table 1.
Variant calling of germline variants in CTNNA1, MAP3K6 and MYD88 in patients with GC
| Patient ID | HDGC/FIGC/FGC/ EOGC/other* |
Diagnosis index (age and gender) | GC in first-degree relatives, n (age) | GC in second-degree relatives, n (age) | Other cancer diagnoses in first-degree and second-degree relatives (age)† | Gene‡ | Nucleotide change | Amino acid change | MAF ExAC§ | PhyloP | Align GVGD¶ | SIFT** | PolyPhen†† |
| 270A | HDGC | DGC (62 M) | DGC: 1 (63); GC: 2 (54, 71) | GC: 4 (45, 51, 57, 72) | BRAT (49) | CTNNA1 | c.80_81del | p.Arg27fs§§ | 0 | N/A | N/A | N/A | N/A |
| 162A‡‡ | HDGC | DGC (33 M) | IGC: 1 (42) | ES (69) | CTNNA1 | c.536C>T | p.Ala179Val | 0.001179 | 4.16 | C0 | Tol. | Benign | |
| 528A | Other | GC (42 F) | DC (56) | CTNNA1 | c.536C>T | p.Ala179Val | 0.001179 | 4.16 | C0 | Tol. | Benign | ||
| 240A | HDGC | DGC (54 F) | IGC: 1 (80) | GC: 2 (73, 81) | PrCa (71); CRC (78) | CTNNA1 | c.618G>C | p.Gln206His | 0.004517 | 0.85 | C0 | Tol. | Pos. D. |
| 380A | HDGC | DGC (36 F) | Leu (37) | CTNNA1 | c.770A>G | p.Asn257Ser | 0.0005189 | 4.89 | C0 | Tol. | Benign | ||
| 432A | HDGC | DGC (40 F) | GC: 1 (56) | CTNNA1 | c.964_988dup | p.Arg330fs | 0 | N/A | N/A | N/A | N/A | ||
| 746A | HDGC | DGC (30 F) | GC: 1 (48) | Leu (?) | CTNNA1 | c.1328dup | p.Asn443fs | 0 | N/A | N/A | N/A | N/A | |
| 233A | HDGC | DGC (41 F) | GC: 1 | MAP3K6 | c.598G>T | p.Asp200Tyr | 0.002953 | 5.29 | C65 | Del. | Prob. D. | ||
| 250A | FGC | IGC (62 M) | IGC: 1 (54); GC: 1 (48) | GC: 1 (80) | MAP3K6 | c.598G>T | p.Asp200Tyr | 0.002953 | 5.29 | C65 | Del. | Prob. D. | |
| 183A | Other | GC (39 F) | MAP3K6 | c.1001C>T | p.Ala334Val | 0.00002531 | 5.21 | C65 | Del. | Pos. D. | |||
| 295A | FGC | IGC (62 M) | GC: 1 (61) | GC: 1 (78) | BRAT (85); CUP (60, 75); CRC (77, 84) | MAP3K6 | c.1256-2A>G | p.? | 0.003761 | N/A | N/A | N/A | N/A |
| 757A | HDGC | DGC (38 M) | MAP3K6 | c.1256-2A>G | p.? | 0.003761 | N/A | N/A | N/A | N/A | |||
| 709A | Other | GC (40 M) | MAP3K6 | c.1622T>C | p.Leu541Pro | 0 | 2.06 | C65 | Del. | Pos. D. | |||
| 167A‡‡ | HDGC | DGC (31 F) | BC (59); MM (60) | MAP3K6 | c.1772A>G | p.Tyr591Cys§§ | 0 | 2.71 | C65 | Del. | Benign | ||
| 737A | FGC | IGC (66 M) | GC: 1 (86) | GC: 1 (60) | MAP3K6 | c.2837C>T | p.Pro946Leu | 0.005296 | 3.35 | C65 | Del. | Prob. D. | |
| 770A | HDGC | DGC (48 M) | GC (51) | PrCa (71) | MAP3K6 | c.2837C>T | p.Pro946Leu | 0.005296 | 3.35 | C65 | Del. | Prob. D. | |
| 729A‡‡ | FIGC | IGC (48 F) | GC: 2 (40, 79) | MAP3K6 | c.2837C>T | p.Pro946Leu | 0.005296 | 3.35 | C65 | Del. | Prob. D. | ||
| 132A | HDGC | DGC (44 F) | DGC: 1 (34) | Leu (62) | MAP3K6 | c.2954C>T | p.Pro985Leu | 0.0009774 | 3.43 | C65 | Del. | Pos. D. | |
| 108A | FGC | IGC (68 M) | DGC: 1 (22); GC: 1 (78) | CRC (50, 59) | MAP3K6 | c.2954C>T | p.Pro985Leu | 0.0009774 | 3.43 | C65 | Del. | Pos. D. | |
| 210A | Other | DGC (40 F) | MAP3K6 | c.3070A>G | p.Lys1024Glu | 0.004362 | −0.6 | C0 | Tol. | Benign | |||
| 244A | HDGC | DGC (26 M) | LiC (55) | MAP3K6 | c.3070A>G | p.Lys1024Glu | 0.004362 | −0.6 | C0 | Tol. | Benign | ||
| 166A | HDGC | DGC (66 F) | GC: 1 (48) | GC: 1 (60) | BC (60); PC (48, 60); PrCa (71); LC (53, 55); CRC (?); RC (53) | MAP3K6 | c.3143A>G | p.His1048Arg | 0.0002113 | 1.09 | C25 | Del. | Benign |
| 183A | EOGC | GC (39 F) | MAP3K6 | c.3181G>A | p.Ala1061Thr | 0.0007999 | 1.34 | C0 | Tol. | Benign | |||
| 113B | FGC | IGC (72 F) | IGC: 1 (82); GC: 3 (63, 66, 82) | HL (72) | MAP3K6 | c.3481C>G | p.Pro1161Ala | 0.00001684 | 0.61 | C0 | Tol. | Benign | |
| 753A | HDGC | DGC (32 M) | MAP3K6 | c.3562C>T | p.Gln1188* | 0.00001682 | N/A | N/A | N/A | N/A | |||
| 270A | HDGC | DGC (62 M) | DGC: 1 (63); GC: 2 (54, 71) | GC: 4 (45, 51, 57, 72) | BRAT (49) | MYD88 | c.251C>T | p.Thr84Ile | 0 | 1.66 | C0 | Tol. | Prob. D. |
| 537A | Other | GC (41) | MYD88 | c.518G>A | p.Arg173His | 0.00002477 | −0.04 | C0 | Tol. | Benign | |||
| 036A‡‡ | HDGC | DGC (23) | BC (45, 45); EC (70) | MYD88 | c.712C>T | p.Arg238Cys§§ | 0 | 3.76 | C15 | Del. | Pos. D. |
N/A, not available; (?), age is not known.
*EOGC, early-onset gastric cancer; FGC, familial gastric cancer; FIGC, familial intestinal gastric cancer; HDGC, hereditary diffuse gastric cancer. For details on these categories, see online supplementary file 2.
†BC, breast cancer; BRAT, brain tumour; CRC, colorectal cancer; CUP, cancer of unknown primary; DC, duodenal cancer; DGC, diffuse-type gastric cancer; EC, endometrial cancer; ES, oesophageal cancer; GC, gastric cancer; HL, Hodgkin lymphoma; IGC, intestinal-type gastric cancer; LC, lung cancer; Leu, leukaemia; LiC, liver cancer; MM, malignant melanoma; PC, pancreatic cancer; PrCa, prostate cancer; RC, renal cancer.
‡NM_001903.2 (CTNNA1), NM_004672.4 (MAP3K6) and NM_001172567.1 (MYD88).
§Minor allele frequency (MAF) of the corresponding variant in the Exome Aggregation Consortium (ExAC) database (see Materials and methods).
¶ GVGD, Grantham Variation Grantham Difference score. Class score; C0 is considered benign.
** SIFT, Sorting Intolerant from Tolerant; Del., deleterious; Tol., tolerated.
††Pos. D., possibly damaging; Prob. D., probably damaging.
‡‡ Whole exome sequencing (WES) has been performed on germline DNA derived from these individuals prior to this study.12
§§Variant has previously been reported in other studies.9 11 12
In CTNNA1 a heterozygous germline variant was observed in seven patients (table 1). An individual with the frameshift mutation p.Arg27fs turned out to be a family member of the first reported CTNNA1 family,9 and this case was discarded from further analyses. Two frameshift variants, p.Asn443fs and p.Arg330fs, were not observed in the control data set and were considered deleterious. The variant p.Asn443fs was identified in a Polish patient with HDGC diagnosed with a poorly differentiated (not otherwise classified) gastric adenocarcinoma at age 30. Unfortunately, the gastric tumour specimen of this patient was not available for pathological review (family pedigree provided in figure 1B). The variant p.Arg330fs was encountered in a Dutch patient with HDGC diagnosed with a diffuse-type GC (poorly cohesive adenocarcinoma, with signet ring cell differentiation, according to the WHO classification) at age 40 (family pedigree provided in figure 1C). Subtotal gastrectomy revealed a tumour extending into the subserosal fat tissue with three lymph node metastases (classified as a stage IIIA, seventh edition,18 see microscopic image in figure 1D). There was a background of chronic inflammation with intestinal metaplasia. Immunohistochemistry for α-E-catenin showed loss of protein expression in the tumour cells, while normal gastric glands showed preserved expression (figure 1E). In control GC cases, α-E-catenin showed retained staining in tumour cells of most cases (online supplementary file 4). Segregation analysis showed the same variant in the patient’s mother (82 years without a diagnosis of cancer) and daughter (26 years without a diagnosis of cancer). Four missense variants were recurrently encountered in the general population and are likely benign SNVs based on in silico predictions (table 1).
In MAP3K6 a total of 18 heterozygous variants (12 unique variants) were observed in 16 patients (table 1). A non-sense variant (p.Gln1188*) and canonical splice site variant (c.1256-2A>G; identified in two unrelated patients) were recurrently encountered in the control data set at low frequency. Two missense variants, p.Asp200Tyr and p.Pro946Leu, which have previously been associated with familial GC, occur two and three times in our cohort, respectively.10 However, both variants are also frequently observed in the control data set. The variant frequencies in our GC cohort and the control data set are not significantly different (P=0.8 and P=1.0, respectively). Similarly, one highly conserved and predicted deleterious missense variant, p.Pro985Leu, was observed in two patients with GC, but was observed at similar frequencies in the control data set (P=0.2). Five other missense variants, representing four different SNVs, were weakly conserved (PhyloP <2) and/or predicted to be benign using in silico predictions (table 1). The three remaining missense variants, p.Ala334Val, p.Leu541Pro and p.Tyr591Cys, which were called in the germline of three different patients with GC, are moderately/highly conserved and predicted possibly deleterious or deleterious and nearly absent or absent in the control data set (table 1).
In MYD88 the homozygous p.Arg238Cys variant was detected in the patient already reported by us.11 Two unrelated patients with GC had a heterozygous missense variant, p.Thr84Ile and p.Arg173His. These variants affect poorly conserved amino acids and are likely benign based on in silico predictions (table 1).
To determine a potential enrichment of strong loss-of-function germline variants (ie, nonsense variants, frameshift variants and variants affecting a canonical splice site) in CTNNA1, MAP3K6 and MYD88 in our GC cohort compared with the general population, the frequency of such variants was established in 60 706 individuals with exome data in the public domain. The cumulative variant allele frequency (cVAF) of strong loss-of-function germline variants in CTNNA1 and MYD88 is low in the general population (cVAF: 0.000057879 and 0.000074822, respectively). In contrast, such variants in MAP3K6 are relatively common (cVAF: 0.004688221). Based on these numbers, the number of germline truncating alleles in CTNNA1 was highly significantly enriched in patients with GC compared with the general population (2/564 vs 7/121 412, P<0.0001), whereas this was not the case for strong loss-of-function alleles affecting MAP3K6 (3/566 vs 509/121 412, P=0.94) or MYD88 (0/566 vs 9/121 412, P=0.84) (for details see online supplementary file 2).
Discussion
To further establish the GC predisposing effect and prevalence of germline aberrations affecting CTNNA1, MAP3K6 and MYD88, we sequenced a large cohort of unexplained, CDH1-negative, familial and/or young patients with GC (n=283) using a targeted next-generation sequencing approach. These 283 patients had early-onset GC (before the age of 50) and/or a family history of GC; 147 patients fulfilled HDGC/CDH1 testing criteria.19 This cohort was significantly enriched for truncating germline variants in CTNNA1, confirming that loss-of-function mutations in CTNNA1 predispose to the development of (familial) HDGC.
The putative GC predisposing role of CTNNA1, encoding the protein α-E-catenin, was first established in a large Dutch HDGC family with multiple affected family members.9 α-E-catenin functions in the same junctional complex as E-cadherin, encoded by the main HDGC predisposing gene CDH1. Subsequent screening by Majewski et al9 of 25 other HDGC pedigrees did not reveal any additional loss-of-function germline variants affecting this gene. Hansford et al revealed two additional families with a germline mutation affecting CTNNA1 in 144 HDGC/FGC.2 In the current study, we detected two germline truncating mutations in CTNNA1 in two unrelated GC cases. Therefore, a total of five CTNNA1 mutation-positive GC families have now been identified (see online supplementary file 5). The clinical picture of these CTNNA1 families seems to be similar to that of CDH1 mutation-positive families, except that no lobular breast cancer or cleft lip/palate was reported thus far. The diffuse morphology could not be established for all affected relatives because not all original pathology reports or specimens were available. The age of onset of cancer in confirmed CTNNA1-mutation carriers with GC varies between 22 and 72 years.2 The incidence of heterozygous germline mutations in CTNNA1 is low in unexplained families with GC (~1%–2%). Due to the limited number of CTNNA1 families identified, proper estimation of disease penetrance and evidence-based advice on preventive measurements is not yet possible.
Heterozygous germline aberrations affecting MAP3K6 were reported in multiple unrelated individuals with familial GC.10 Initially, a heterozygous missense mutation (p.Pro946Leu) was identified that partly cosegregated with the development of GC in a large family. Remarkably, an 80-year-old homozygous family member was never diagnosed with cancer. Subsequent targeted screening on germline DNA derived from 115 unrelated unexplained GC cases with intestinal-type and/or diffuse-type GC revealed five additional heterozygous germline variants affecting MAP3K6 (ie, one frameshift and four missense variants).10 However, most of these variants are recurrently encountered in control cohorts as well, and in silico predictions to determine their pathogenicity reveal inconsistent results. In our study, we also found multiple variants in MAP3K6; however, these also occur frequently in the general population. In fact, strong loss-of-function germline variants affecting MAP3K6 are encountered in approximately 0.5% of the general population, clearly conflicting with a highly penetrant predisposing effect of, in Western populations, a rare phenotype like GC. Therefore, germline aberrations affecting MAP3K6 are unlikely to be involved in high-penetrant GC predisposition.
In contrast to the proposed dominant inheritance patterns of GC predisposition due to CTNNA1 and MAP3K6 mutations, we previously reported that germline mutations affecting MYD88 may predispose to the development of GC in a recessive manner.11 A homozygous missense variant (p.Arg238Cys) in MYD88 was identified in an individual suffering from recurrent candidiasis and diffuse-type GC at a young age (23 years old). This mutation results in an impaired immune response and, possibly, an increased GC risk.11 Identification of additional patients with GC with biallelic deleterious MYD88 variants would have further supported the role of MYD88 in GC predisposition; however, such variants were not detected in our cohort.
To evaluate the role of proposed cancer predisposing genes, it is crucial to test large cohorts in independent studies, as incorrect associations between germline aberrations and an increased lifetime risk to develop cancer will result in unnecessary screenings and inappropriate clinical management of carriers of such aberrations. In the current study, we confirm that inactivating mutations in CTNNA1 are an infrequent cause of DGC predisposition. However, based on our data it is unlikely that MAP3K6 is a GC predisposition gene. Biallelic MYD88 mutations as a cause of GC susceptibility are at most very rare. To substantiate its GC predisposing effect, it should be confirmed in other patients.
Acknowledgments
We would like to thank the Radboud Genomics Technology Center for performing the sequencing runs described in this manuscript and Nanne van Heumen for technical assistance.
Footnotes
RDAW and RSP contributed equally.
Contributors: RDAW, RSvdP, IPV, NH and MJLL were responsible for study concept and design. Pathology review was performed by RSvdP. All authors were responsible for data acquisition. Data analysis and interpretation were performed by RDAW, RSvdP and MJLL. RDAW, RSvdP, NH and MJLL wrote the manuscript, with assistance and final approval of all authors.
Funding: This study was funded by KWF Kankerbestrijding (grant number KUN 2013-5876, RSvdP).
Competing interests: None declared.
Patient consent: Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
Ethics approval: This study was approved by the medical ethics committee of the Radboud university medical center, reference number 2013/201.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature 1998;392:402–5. 10.1038/32918 [DOI] [PubMed] [Google Scholar]
- 2. Hansford S, Kaurah P, Li-Chang H, Woo M, Senz J, Pinheiro H, Schrader KA, Schaeffer DF, Shumansky K, Zogopoulos G, Santos TA, Claro I, Carvalho J, Nielsen C, Padilla S, Lum A, Talhouk A, Baker-Lange K, Richardson S, Lewis I, Lindor NM, Pennell E, MacMillan A, Fernandez B, Keller G, Lynch H, Shah SP, Guilford P, Gallinger S, Corso G, Roviello F, Caldas C, Oliveira C, Pharoah PD, Huntsman DG. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol 2015;1:23–32. 10.1001/jamaoncol.2014.168 [DOI] [PubMed] [Google Scholar]
- 3. Fitzgerald RC, Hardwick R, Huntsman D, Carneiro F, Guilford P, Blair V, Chung DC, Norton J, Ragunath K, Van Krieken JH, Dwerryhouse S, Caldas C; International Gastric Cancer Linkage Consortium. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet 2010;47:436–44. 10.1136/jmg.2009.074237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benusiglio PR, Malka D, Rouleau E, De Pauw A, Buecher B, Noguès C, Fourme E, Colas C, Coulet F, Warcoin M, Grandjouan S, Sezeur A, Laurent-Puig P, Molière D, Tlemsani C, Di Maria M, Byrde V, Delaloge S, Blayau M, Caron O. CDH1 germline mutations and the hereditary diffuse gastric and lobular breast cancer syndrome: a multicentre study. J Med Genet 2013;50:486–9. 10.1136/jmedgenet-2012-101472 [DOI] [PubMed] [Google Scholar]
- 5. van der Post RS, Vogelaar IP, Manders P, van der Kolk LE, Cats A, van Hest LP, Sijmons R, Aalfs CM, Ausems MG, Gómez García EB, Wagner A, Hes FJ, Arts N, Mensenkamp AR, van Krieken JH, Hoogerbrugge N, Ligtenberg MJ. Accuracy of hereditary diffuse gastric cancer testing criteria and outcomes in patients with a germline mutation in CDH1. Gastroenterology 2015;149:897–906. 10.1053/j.gastro.2015.06.003 [DOI] [PubMed] [Google Scholar]
- 6. Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genome Biol 2011;12:228 10.1186/gb-2011-12-9-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Guarino Almeida E, Salguero I, Sherborne A, Chubb D, Carvajal-Carmona LG, Ma Y, Kaur K, Dobbins S, Barclay E, Gorman M, Martin L, Kovac MB, Humphray S, Lucassen A, Holmes CC, Bentley D, Donnelly P, Taylor J, Petridis C, Roylance R, Sawyer EJ, Kerr DJ, Clark S, Grimes J, Kearsey SE, Thomas HJ, McVean G, Houlston RS, Tomlinson I; CORGI Consortium WGS500 Consortium. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013;45:136–44. 10.1038/ng.2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L, van Zelst-Stams WA, Jongmans MC, Gilissen C, Hehir-Kwa JY, Hoischen A, Shendure J, Boyle EA, Kamping EJ, Nagtegaal ID, Tops BB, Nagengast FM, Geurts van Kessel A, van Krieken JH, Kuiper RP, Hoogerbrugge N. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 2015;47:668–71. 10.1038/ng.3287 [DOI] [PubMed] [Google Scholar]
- 9. Majewski IJ, Kluijt I, Cats A, Scerri TS, de Jong D, Kluin RJ, Hansford S, Hogervorst FB, Bosma AJ, Hofland I, Winter M, Huntsman D, Jonkers J, Bahlo M, Bernards R. An α-E-catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. J Pathol 2013;229:621–9. 10.1002/path.4152 [DOI] [PubMed] [Google Scholar]
- 10. Gaston D, Hansford S, Oliveira C, Nightingale M, Pinheiro H, Macgillivray C, Kaurah P, Rideout AL, Steele P, Soares G, Huang WY, Whitehouse S, Blowers S, LeBlanc MA, Jiang H, Greer W, Samuels ME, Orr A, Fernandez CV, Majewski J, Ludman M, Dyack S, Penney LS, McMaster CR, Huntsman D, Bedard K. Germline mutations in MAP3K6 are associated with familial gastric cancer. PLoS Genet 2014;10:e1004669 10.1371/journal.pgen.1004669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vogelaar IP, Ligtenberg MJ, van der Post RS, de Voer RM, Kets CM, Jansen TJ, Jacobs L, Schreibelt G, de Vries IJ, Netea MG, Hoogerbrugge N; International Gastric Cancer Genetics Group. Recurrent candidiasis and early-onset gastric cancer in a patient with a genetically defined partial MYD88 defect. Fam Cancer 2016;15:289–96. 10.1007/s10689-015-9859-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vogelaar IP, van der Post RS, van Krieken JHJ, Spruijt L, van Zelst-Stams WA, Kets CM, Lubinski J, Jakubowska A, Teodorczyk U, Aalfs CM, van Hest LP, Pinheiro H, Oliveira C, Jhangiani SN, Muzny DM, Gibbs RA, Lupski JR, de Ligt J, Vissers L, Hoischen A, Gilissen C, van de Vorst M, Goeman JJ, Schackert HK, Ranzani GN, Molinaro V, Gómez García EB, Hes FJ, Holinski-Feder E, Genuardi M, Ausems M, Sijmons RH, Wagner A, van der Kolk LE, Bjørnevoll I, Høberg-Vetti H, van Kessel AG, Kuiper RP, Ligtenberg MJL, Hoogerbrugge N. Unraveling genetic predisposition to familial or early onset gastric cancer using germline whole-exome sequencing. Eur J Hum Genet 2017;25:1246–52. 10.1038/ejhg.2017.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boyle EA, O’Roak BJ, Martin BK, Kumar A, Shendure J. MIPgen: optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics 2014;30:2670–2. 10.1093/bioinformatics/btu353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hiatt JB, Pritchard CC, Salipante SJ, O’Roak BJ, Shendure J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res 2013;23:843–54. 10.1101/gr.147686.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O’Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012;338:1619–22. 10.1126/science.1227764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weren RD, Mensenkamp AR, Simons M, Eijkelenboom A, Sie AS, Ouchene H, van Asseldonk M, Gomez-Garcia EB, Blok MJ, de Hullu JA, Nelen MR, Hoischen A, Bulten J, Tops BB, Hoogerbrugge N, Ligtenberg MJ. Novel BRCA1 and BRCA2 Tumor Test as Basis for Treatment Decisions and Referral for Genetic Counselling of Patients with Ovarian Carcinomas. Hum Mutat 2017;38:226–35. 10.1002/humu.23137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bosman FT; World Health Organization, International Agency for Research on Cancer. WHO classification of tumours of the digestive system. 4th ed Lyon: International Agency for Research on Cancer, 2010. [Google Scholar]
- 19. van der Post RS, Vogelaar IP, Carneiro F, Guilford P, Huntsman D, Hoogerbrugge N, Caldas C, Schreiber KE, Hardwick RH, Ausems MG, Bardram L, Benusiglio PR, Bisseling TM, Blair V, Bleiker E, Boussioutas A, Cats A, Coit D, DeGregorio L, Figueiredo J, Ford JM, Heijkoop E, Hermens R, Humar B, Kaurah P, Keller G, Lai J, Ligtenberg MJ, O’Donovan M, Oliveira C, Pinheiro H, Ragunath K, Rasenberg E, Richardson S, Roviello F, Schackert H, Seruca R, Taylor A, Ter Huurne A, Tischkowitz M, Joe ST, van Dijck B, van Grieken NC, van Hillegersberg R, van Sandick JW, Vehof R, van Krieken JH, Fitzgerald RC. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74. 10.1136/jmedgenet-2015-103094 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmedgenet-2017-104962supp001.docx (3.6MB, docx)