

ABSTRACT
Prokaryotic genomes show a high level of information compaction often with different molecules transcribed from the same locus. Although antisense RNAs have been relatively well studied, RNAs in the same strand, internal RNAs (intraRNAs), are still poorly understood. The question of how common is the translation of overlapping reading frames remains open. We address this question in the model archaeon Halobacterium salinarum. In the present work we used differential RNA-seq (dRNA-seq) in H. salinarum NRC-1 to locate intraRNA signals in subsets of internal transcription start sites (iTSS) and establish the open reading frames associated to them (intraORFs). Using C-terminally flagged proteins, we experimentally observed isoforms accurately predicted by intraRNA translation for kef1, acs3 and orc4 genes. We also recovered from the literature and mass spectrometry databases several instances of protein isoforms consistent with intraRNA translation such as the gas vesicle protein gene gvpC1. We found evidence for intraRNAs in horizontally transferred genes such as the chaperone dnaK and the aerobic respiration related cydA in both H. salinarum and Escherichia coli. Also, intraRNA translation evidence in H. salinarum, E. coli and yeast of a universal elongation factor (aEF-2, fusA and eEF-2) suggests that this is an ancient phenomenon present in all domains of life.
KEYWORDS: IntraRNA, internal RNA, isoform, overlapping coding RNA, dRNAseq, dRNA-seq, differential RNA-seq, protein isoforms, alternative transcript, alternative protein, Halobacterium salinarum, archaea, intraORFeome, kef1, fusA, aEF-2, dnaK, cydA, gvpC, orc4, acs3, ancient phenomenon, LUCA, intraORF, uORF
Introduction
Prokaryotic genomes are compact and considered less complex than eukaryotic genomes due to their small size, lack of introns, and shorter intergenic non-coding and regulatory regions. However, this simplicity has been challenged by the widespread use of high-resolution transcriptome mapping technologies [1,2]. Identification of overlapping genomic elements revealed modular operon organization that allows for conditional co-modulation of genes in bacteria and archaea [3,4]. Abundant transcript signals established pervasive transcription as a general phenomenon, expanding our knowledge on the universe of non-coding RNAs [5]. Genome-wide mapping of transcription start sites (TSS) in diverse prokaryotes has confirmed that many of these signals are not artifacts but bona fide transcripts [6–8].
Most studies that mapped TSS throughout prokaryotic genomes have used differential RNA-seq technology (dRNA-seq), where 5ʹ triphosphorylated RNAs are enriched relative to 5ʹ monophosphorylated RNAs using TEX (Terminator 5ʹ phosphate dependent exonuclease) [9]. The use of dRNA-seq has allowed precise TSS mapping for most annotated coding sequences (CDS), antisense RNAs (asRNAs) and new intergenic RNAs in bacteria [7,9] and archaea [8,10]. Interestingly, many TSS inside coding regions (internal TSS, iTSS) have been mapped, but to date remain mostly uncharacterized. RNA molecules that have their transcription starting within CDS regions have been labeled intraRNAs [11,12].
Many iTSS have been associated with misannotation of genes, superimposed adjacent CDS sequences, non-coding RNAs overlapping CDS or transcription noise [13]. Medium and large scale comparative transcriptomics revealed enormous variability in the number of detected iTSS and although a modest conservation of iTSS was observed, it is still higher than antisense TSS [13,14]. Comparative transcriptomics revealed a modest ~ 30% conservation rate in eight different Shewanella species. However, analysis of transposon mutant fitness and transcription factor binding sequence motifs indicated that overlapping transcript production is probably significant to the proper functioning of the organism [13]. Nevertheless, the actual production of proteins derived from intraRNAs has been little explored. Coding potential of intraRNAs have been explicitly suggested in internal open reading frames (intraORFs) and detection of peptides attributed to them has been observed in Caulobacter crescentus [15], Bradyrhizobium japonicum [16] and Shigella flexneri [17].
Archaeal translation of intraRNAs was not yet reported. Moreover, recent findings that prokaryotes in general have high fractions of translated leaderless mRNAs [18,19] and sORFs (small open reading frames) [20,21] make the realization of intraRNAs coding potential worth investigating. Pervasive translation of intraRNAs would include prokaryotes into the meaningfulness of protein isoforms debate [22,23]. To investigate intraRNA translation in archaea we used Halobacterium salinarum NRC-1 as model organism.
H. salinarum NRC-1 is a halophilic archaeon that thrives in 4.3 M NaCl environments and presents a salt-in strategy for osmoregulation, as well as a highly acidic proteome [24]. Global gene regulatory networks have been studied in this organism using diverse environmental and genetic perturbations [25], ranking H. salinarum as having one of the most extensively studied archaeal transcriptomes.
In this work, we performed a global mapping of TSS and hereby strongly suggest that sense overlapping internal RNAs can drive the production of protein isoforms. In the next sections we: (i) define H. salinarum intraRNAs using iTSS; (ii) define intraORFs using intraRNAs; (iii) establish intraRNA translation; (iv) explore intraORFs overall sequence and expression characteristics; (v) suggest a link between asRNA and intraRNA that informs translation; and (vi) establish that intraRNA translation is present in highly conserved genes using Escherichia coli and yeast data. We end by discussing an alternative biogenesis hypothesis for protein isoform production in prokaryotes based on intraRNA translation.
Results
Internal transcription start sites reveal intraRNA molecules
TSS mapping was performed using dRNA-seq experiments with RNA samples from four different conditions to increase dRNA-seq sampling power: three time points in H. salinarum NRC-1 growth curve and a reference condition (Fig. S1). From all 2782 annotated CDS, a primary genuine TSS (gTSS) located at most 250 bp upstream of their annotated translation initiation sites (TIS) could be mapped for 1307 CDS (47%) at the statistical significance level of p-value <10−15. From this CDS sample, 71% presented short (<10 bp) 5ʹ untranslated regions (UTR), confirming H. salinarum preference for leaderless transcripts [4]. Additionally, using the same stringent statistical significance cutoff, 680 TSS were found antisense to annotated CDS (aTSS) and 300 did not fit into any of the previous categories (oTSS, ‘orphan’) (Table S1). Gaggle Genome Browser [26] files were made available at http://labpib.fmrp.usp.br/~rvencio/intrarna/ facilitate browsing and data-mining.
In the present work we focus on a specific class of molecules, intraRNAs, therefore we narrow down our iTSS search to those found internally to annotated CDS loci. Not all iTSS point to clear intraRNA candidates since known confounding issues exist. Evident issues include iTSS that are in fact gTSS for adjacent sense downstream genes or 5ʹ end genomic misannotations. Non-trivial instances include potential false positive artifacts from dRNA-seq protocol or analysis pipeline [10]. Therefore, we refined our analysis narrowing down to 520 iTSS mapped into 392 H. salinarum genes (Tables S2 and S3). To do that, we applied filters excluding iTSS which: (i) do not pass stringent statistical significance cutoff of <10−15, (ii) are located too close to CDS’ edges (90 bp or 10% of CDS length margin), or (iii) are upstream of sub-sequences prone to form structured molecules. The last filter is used since secondary structures in RNAs can prevent degradation by TEX and thus, generate dRNA-seq signal inside genes [10,27]. We computed the minimum free energy (MFE) of predicted structures on regions downstream of iTSS and compared them to the same procedure performed for the whole genome (Fig. S2). The majority of coding genes containing iTSS (312, 80%) share their locus with a single overlapping intraRNA and more than three intraRNAs per gene occurs rarely (Table S3).
Analysis of regions upstream of iTSS and gTSS revealed known regulatory region features (Fig. 1). The promoter regions of both TSS classes present an increase in the frequency of nucleotides from BRE/TATA regions [28]. As expected, gTSS show the highest degree of promoter sequence conservation, with a clear ATG start codon at position 0. Another noticeable feature is the frequency of pyrimidines (C or T) at −1 position, a signature found in other archaea that seems to be related to the recognition of the TSS [29,30].
Figure 1.

Sequence composition of regions upstream of genes and intraRNAs.
Logo representation of upstream regions of gTSS (A) and iTSS (B). Relative positions are shown positioning TSS as zero.
Internal open reading frames (intraORFs) defined by intraRNAs
To establish internal open reading frames, we sub-selected those intraRNAs with higher coding potential. We assume that intraRNAs are long enough to reach stop codons if they present at least one read among the top 5% longest (>173 nt, Fig. S3). An intraORF is, by definition, in the same frame as its cognate ORF, shares the same stop codon, and starts at the first start codon downstream of an intraRNA’s TSS. This stringent filter addresses the fact that many non-coding RNAs generated by pervasive transcription are generally short and unstable [31]. However, some intraORFs are short (<173 bp) and have intraRNA reads going full-length from iTSS to stop codons. These specific cases are considered potentially protein coding and are not excluded in spite of the fact that would not pass the aforementioned <173 nt RNA length filter.
Reading frames other than the cognate protein were not considered since (i) extensive searches in comprehensive H. salinarum NRC-1 mass spectrometry databases did not reveal peptides in different reading frames (Kusselbach & Moritz, personal communication) and (ii) searches in current NCBI’s NR protein database using predicted out-of-frame intraORFs returned few unreliable marginally significant hits.
Finding translation initiation sites (TIS) for usual genes is challenging but relatively well addressed computationally and experimentally, contrary to internal/alternative TIS [32,33]. Therefore, we adopted the simplest working definition: the TIS is located at the nearest ATG or GTG start codon downstream of the iTSS. Although archaea and bacteria can use other alternative start codons [34], these two are by far the most common so we restricted the putative internal coding sequences to them. Additional reliability filters were applied: (i) predicted short polypeptide sequences were excluded (<30 amino acid residues); (ii) 5ʹ intraUTRs longer than 173 nt were excluded; and (iii) if the 5ʹ intraUTRs are shorter than 10 nt, only ATG is considered as the start codon since leaderless transcripts were shown to be ATG-dependent in haloarchaea [35].
We were able to associate intraORF sequences to 274 (out of 520, 51%) intraRNAs (Table S4). Due to gene duplications and few genes with multiple iTSS, these intraRNAs define 220 putative protein isoforms.
Experimental detection of protein isoforms encoded by intraORFs in H. salinarum
In order to experimentally validate the production of alternative proteins from intraRNAs in vivo, we constructed two recombinant H. salinarum NRC-1 strains carrying the FLAG tag at the 3ʹ end of target genes: acs3, which encodes for a long-chain-fatty-acid CoA ligase; and kef1, which encodes for a sodium transporter (Table S11). There was no special selection criteria on signal strength, length, location or sequence characteristics. The only guideline used was the existence of a downstream PFAM domain.
As an adaptation to high salinity environments, the H. salinarum proteome is known to be unusually acidic, with an average pI of 5.1 [24,36]. Consequently, observed migration bands of acidic proteins in SDS-PAGE gels are well known to not correspond directly to their molecular weight (MW). Fortunately, a carefully devised correction equation was recently shown to address this long standing problem [37]. Our main line of argumentation relies on the observation of protein isoforms with sizes (inferred by MW) quantitatively consistent with translation products from intraRNAs.
A clear TEX+ > TEX- enrichment signal is detected inside acs3 (TSS_2633_3) along with two clear bands in western blot (Fig. 2A, Fig. S4). A northern blot assay confirms the existence of an intraRNA transcript reaching the stop codon (Fig. S5). The acs3’s intraRNA encodes for an 8.1 kDa isoform which, after DYKDDDDK FLAG-tag addition and acidic correction, would migrate as 12.1 kDa, consistent with the smaller band. Acs3 is a 57.0 kDa protein that migrates as 58.0 kDa. Our intraORF definition considers only ATG and GTG start codons by default but TTG is known to be rarely used and, if allowed in this case, would be the first in-frame start codon predicting a 9.4 kDa isoform which migrates as 13.9 kDa, also consistent with the experimental result.
Figure 2.
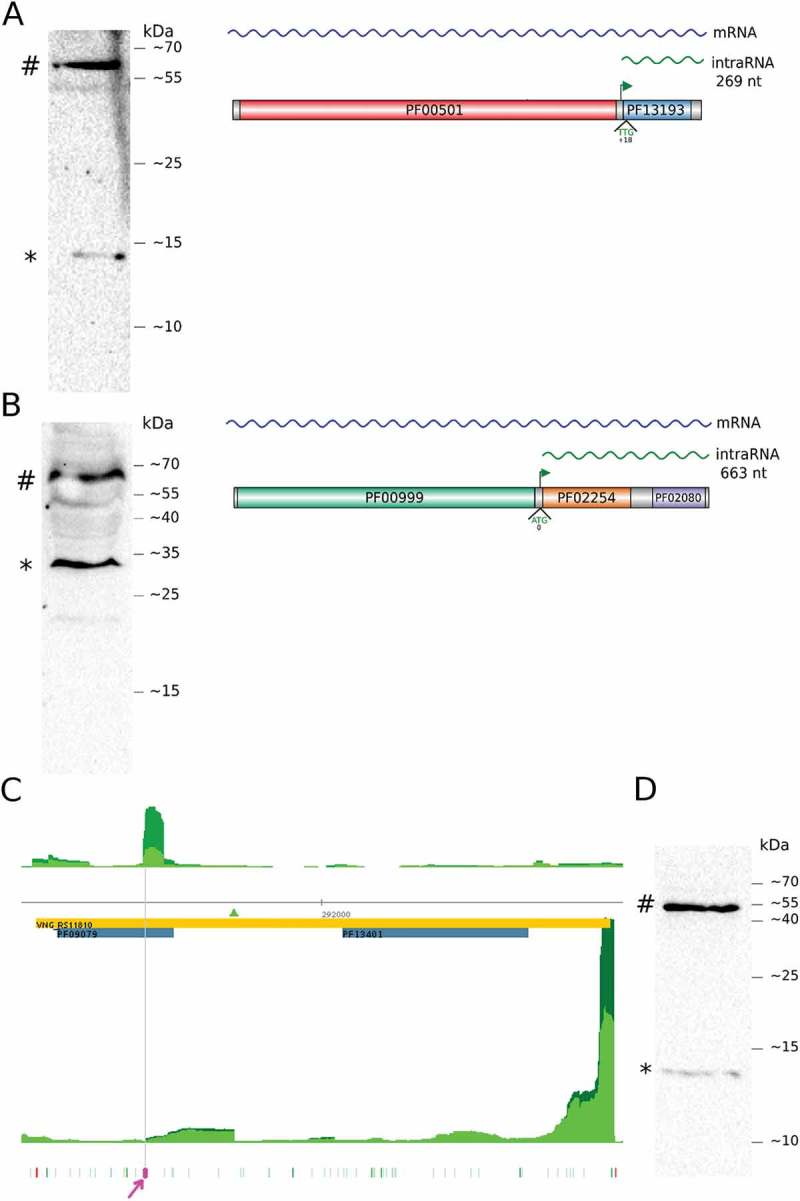
intraRNA translation validation.
Western blots for chromosomally tagged acs3 (A), kef1 (B) and orc4 (D). # indicates the full length protein and * the isoform translated from the intraRNA. Panels on the right (A and B) show the gene organization with PFAM domains, iTSS (green arrow) and the putative start codon for the protein isoform. (C) Aligned reads coverage along genomic coordinates for TEX+ (dark green) and TEX- (green) (arbitrarily scaled and normalized). Coding sequence is in reverse strand (orange rectangle, locus ID inside, 5ʹ→3ʹ direction is right to left). Forward and reverse coverage signals are shown above and below horizontal axis respectively. Domain annotation (blue rectangle), identified iTSS (green triangle), all possible archaeal start codons (green) and stop codon (red) are shown. Predicted TIS is highlighted (magenta). (D) Western blot of C-terminally FLAG-tagged Orc4 protein (#) and isoform (*).
The kef1 gene encodes for a 64.8 kDa protein and its leaderless intraRNA (TSS_9088_3) would produce a 23.4 kDa protein. The two most abundant bands are consistent with intraRNA translation and Kef1 corrected MW of 31.1 kDa and 67.1 kDa, respectively (Fig. 2B, Fig. S6).
Aiming robustness, we searched published mass spectrometry (MS) data for protein experiments that could provide additional intraRNA translation candidates using an orthogonal line of argumentation. Instead of relying on concordance between predicted and measured MW of translation products, we searched H. salinarum MS datasets for tryptic and semi-tryptic peptides since one would expect intraORF N-termini to generate different peptide sizes to the overlapping region of the full-length ORF due to the lack of a protease site at the N-terminus. We used two large-scale sources: PeptideAtlas database (www.peptideatlas.org [38]), which accumulated diverse H. salinarum NRC-1 proteomics data; and a more recent SWATH (Sequential Window Acquisition of all Theoretical Fragment Ion Spectra) high-depth data (PRIDE database accession PXD003667) [39] for H. salinarum R1.
Contrary to bacteria, archaeal prokaryotes do not start their proteins with N-Formylmethionine (fMet). Moreover, it is very common to observe the cleavage of initial methionine in H. salinarum [40]. Therefore we searched for confidently identified peptides matching the first semi-tryptic peptide of predicted isoforms, including or not the first amino acid. Also, the intraORF predicted isoform sequence could not be preceded by an R or K amino acid residue in its cognate CDS since it would turn it indistinguishable from a cognate’s tryptic peptide. We validated five intraORFs using this approach: ugpB, which encodes for a ABC-type transport system periplasmic substrate-binding protein; sdhB, which encodes for a succinate dehydrogenase subunit; ibp, which encodes for an iron ABC transporter substrate-binding protein; acs2, which encodes for an acetate CoA ligase; and noxA, which encodes for a NAD(P)/FAD-dependent oxidoreductase (Table S11).
The ugpB gene has an intraRNA (TSS_11337_3) that would encode for a protein which has MLWDSTSNLVSLVAGAK as its first semi-tryptic peptide. The PeptideAtlas database returned peptides LWDSTSNLVSLVAGAK (accession ID PAp00368925) and QAFLTEQAAMLWDSTSNLVSLVAGAK (PAp00372660) which can be interpreted as a methionine cleaved observation of the intraRNA translated isoform and a tryptic peptide obtained from the cognate protein, respectively (Fig. S7). The sdhB gene has an intraRNA (TSS_7794_3) that would translate a protein isoform consistent with semi-tryptic and tryptic peptides PAp00371526 and PAp02308521, respectively (Table S11). The ibp gene presents a putative intraORF (TSS_7012_3) consistent with semi-tryptic and tryptic peptides PAp00629266 and PAp02365987, respectively. The acs2 gene has two close iTSS (TSS_2031_3 and TSS_2032_3) that specify the same intraORF sequence and its translation is consistent with the semi-tryptic peptide PAp02365046. Finally, the noxA gene has an intraORF (TSS_3496_3) that would translate an isoform for which the peptide LADSGHDVEIEPFR was found by SWATH in H. salinarum R1 strain. Its cognate tryptic peptide PAp00366234 is present only in NRC-1 strain database.
The algorithmic inclusion criteria filtered out <30 amino acid long isoforms and treated ATG and GTG start codons equally although ATG is much more frequent. The data mining validation strategy pointed out potential false negative examples caused by the unavoidably imperfect intraORF inclusion criteria: korB, gap and arcD genes. The korB gene encodes for a pyruvate ferredoxin oxidoreductase subunit and shows an intraORF (TSS_7476_3) which would translate a 28 amino acid protein isoform consistent with peptide PAp02311112. The genes gap (TSS_397_3) and arcD (TSS_12846_3), which encode for glyceraldehyde-3-phosphate dehydrogenase and an arginine/ornithine antiporter, respectively, present peptides consistent with intraRNA translation if instead of the first start codon (GTG) we choose the second (ATG). Redefining gap’s intraORF turn it consistent with the semi-tryptic and overlapping peptides PAp02312588 and PAp00365226, respectively. Redefining arcD’s intraORF turn it consistent with PAp00368704 and PAp03629496, overlapping the tryptic peptide PAp05372640.
Taken together, our western blot data and published MS data present supporting evidence for a total of 10 intraRNA translation cases. The successful validation effort by western blot with no extensive trial-and-error candidate sampling is also considered as reinforcing circumstantial evidence. Therefore it is assumed for the purpose of the following descriptive statistics that all listed intraORF sequences have coding potential in spite of unavoidable false positives. The additional 220 proteins would represent a modest increase of 8% to the H. salinarum NRC-1 predicted proteome.
Sequence and expression characteristics of intraRNAs and associated intraORFs
Overall, only 20 (9%) intraRNAs are predicted to be leaderless transcripts (≤ 10 nt), less than the genome-wide estimate of 71%. The 5ʹ intraUTR length distribution is markedly different from the 5ʹ UTR sample for which we obtained gTSS evidence (Fig. S8). However, if leaderless transcripts are excluded from the CDS sample, the remaining distribution becomes similar to the 5ʹ intraUTR length distribution.
Shine-Dalgarno-like (SD-like) signatures GGTG or GGAG (16S ribosome tail sequence is GATCACCTCCTAA [41,42]) in 5ʹ intraUTRs were found within 20 nt from the TIS in 37 cases (14%) (Table S5). This figure is similar to the estimated rate of 20% in 5ʹ UTRs from annotated CDS [43,44] and above the 10% rate found for random 20-mer sequences inside CDS. Unsupervised search for novel motifs did not yield any significant pattern (data not shown).
Since intraORFs are subsequences of previously annotated ORFs, in silico functional annotation relies on the domain annotation of cognate predicted proteins. Overall, 16% (45 out of 274) of the predicted isoforms overlap hypothetical proteins, lower than the whole genome frequency of 33%. We found that 61 (22%) intraORFs encompass 41 known PFAM domains (Table S6). From these intraORFs, 15 are multidomain and 46 have a single annotated domain. The most frequent domains found are the transcription factor TFIIB repeat domain (PF00382) and the GHKL (Gyrase, Hsp90, Histidine Kinase, MutL) domain (PF02518). The most frequent biological process represented by domains found are signal transduction (GO:0007165) and oxidation-reduction (GO:0055114) by means of ATP binding (GO:0005524) and oxidoreductase activity (GO:0016491) molecular functions.
All 46 mono-domain isoforms putatively translated in H. salinarum can be found as mono-domain proteins in >10 other organisms, according to the PFAM domain architecture database [45]. Some mono- or bi-domain isoforms can be found also as standalone proteins in H. salinarum itself (Table S6). Examples include the RCK K+ conductance domain (PF02254 and PF02080 concatenated) which is found simultaneously in an intraRNA-translated isoform and in standalone H. salinarum proteins encoded from distinct loci (Fig. S9). This domain covers essentially the whole Kef1 protein isoform discussed in the validation section.
Conversely, some domains/motifs that are left out in isoforms could also implicate biological features. The main examples are proteins targeted by the Sec and TAT twin-arginine protein translocation pathways. Given that almost all secreted proteins in H. salinarum use these systems to be exported [46], isoforms that left them out would show very different qualitative properties. From 103 proteins predicted to contain such signal peptides [46], 10 had detected intraRNAs that would leave the export signals out in translated isoforms (Table S7).
In spite of being the best characterized archaea from the gene regulatory network point of view [25,47], there is only a small set of transcription factor (TfbB, TfbD, and TfbG) binding sites mapped using the ChIP-seq technology [48], which have the resolution necessary to investigate TF control of intraRNAs. Only 9 intraRNAs with valid intraORFs are immediately downstream (<50 bp) to these available TF binding sites and none of them share the same TF with their cognate gene (Table S8).
Our dRNA-seq data was obtained sampling different time points in standard H. salinarum NCR-1 growth curve assays to increase sampling power, but it can be break down into time-series for qualitative differential expression since not all libraries were sequenced at similar depths. Post-transcriptional and expression regulation controls turn direct inferences of protein-level differential expression based on transcript-level differential expression not straightforward [49] but is it still a reasonable and accepted general proxy for it. Fold-change between stationary phase (37h) and exponential phase (17h) and between gas vesicle release phase (86h) and exponential phase (17h) were analyzed considering TEX+ data of intraRNAs for which intraORFs were established. A comparison with their respective cognate gene TEX+ signal shows few cases of opposite patterns: cognate up-regulation and intraRNA down-regulation and vice-versa (Table S9).
From the set of cognate gene/intraRNAs with opposite expression patterns, we highlight the gene htr15 (TSS_7082_3), which encodes for a transducer protein. The detected intraRNA expression is 4-fold up-regulated in stationary phase relative to exponential phase and 2-fold up-regulated in gas vesicles release phase relative to exponential phase, meanwhile htr15 is down-regulated >2-fold at the same time-points. The existence of ChIP-seq binding sites for transcription factors TfbB and TfbG [48] within 10 bp upstream of the iTSS and tiling microarray data showing temporal modulation consistent with our findings validates the intraRNA expression pattern (Fig. S10). The htr15 gene is located in the archaellum operon, upstream of the flaD gene. Literature data on the whole archaellum flaDEFGHIJK gene cluster indicates that the transcript starting from htr15 locus covers the downstream gene flaD but not from flaE on [50]. The algorithmic TIS selection always chooses the first ATG or GTG but individual inspection for this case shows a putative ribosome binding site downstream of the automatically chosen TIS, located 17 nt upstream of the second potential start codon: GTGC(…)GAGGAGATCGCGACCTCCGTGGAC(…). The established open reading frame could be interpreted equivalently as a htr15 intraORF or as a flaD uORF (upstream open reading frame).
Finally, we noticed that although the replicated libraries taken at the gas vesicles release phase (86h) yielded the least total amount of sequenced TEX+ reads by a factor of half, there is a single intraRNA (TSS_13451_3) almost exclusively identified from this phase: 24 pairs of reads vs just 1 in each of the other phases. This putative intraRNA is related to an important H. salinarum gene, gvpC1, which encodes for one of the gas vesicle structural proteins. Although extensively studied [51], there are no reports of transcripts equivalent to the intraRNA found here, probably because growing H. salinarum for such relatively long time was not usual until the gas vesicle release phenomenon was proposed [52]. Secondary structure prediction with different methods did not show evidence of structures that would generate dRNA-seq false positives (data not shown). Also, it is possible to identify: (i) the general BRE-TATAbox-PPE-TSS layout, as established for TFB binding in H. salinarum [48], (ii) a pyrimidine at position −1 from the iTSS, and (iii) the presence of a SD-like sequence located 20 nt upstream of the first available ATG start codon (Fig. S11). This example did not make it into our stringent intraORF set because there are no paired-end reads with at least 173 nt (quantile 95% cutoff). However, there are several reads above a more moderate ad hoc cutoff of 45 nt, the TEX+ dataset median read length. Almost all reads surpass the putative TIS since the predicted 5ʹ intraUTR is 32 nt. Interestingly, a western blot experiment published twenty-five years ago [53] shows two protein bands at the predicted apparent MW for both GvpC1 and the putative isoform (Fig. S11). The gvpC1 gene encodes for a 42.4 kDa protein and, if translated, the intraRNA encodes for a 29.4 kDa isoform. Acidic correction places the GvpC1 band at 62.2 kDa and the isoform at 43.0 kDa, which is consistent with experimental data.
Involvement of asRNAs in translation initiation site selection
We noticed that there are instances of intraRNAs that are accompanied by antisense signals (asRNA) for much of their extension. We evaluated experimentally if such coincidence would interfere with the translation of a potentially coding intraRNA. For this purpose we selected the orc4 gene, which encodes for a cell division protein.
We detected a moderate statistically significant iTSS inside orc4 locus (TSS_13093_3, p-value = 4.4 · 10−6), a TSS signature at approximately the same location could be also detected in tiling microarray data [4]. This iTSS presented no evidence of being a false positive due to downstream secondary structure formation (Fig. S12). At the same time, there is a clear aTSS signal (TSS_11645_3, p-value <10−15) 194 nt downstream of the iTSS position. Visual inspection of sequenced reads from this region shows that the asRNA/intraRNA intersection covers all the intraRNA fraction (Fig. S13). In the aTSS vicinity (forward strand, antisense to orc4) there is a set of reads in the TEX- dataset that vanishes in the TEX+ dataset (Fig. S14). Among the longest reads for the intraRNA, there is a relatively abrupt termination pattern. These evidences lead us to speculate that there is some sort of asRNA mediated transcript processing of the intraRNA. Since this putative processing site is located near a GTG codon (Fig. 1C, position 291,623 reverse strand) in-frame with orc4, we hypothesize that the asRNA guide the TIS selection skipping all other in-frame initiation codons that would define longer intraORFs. To test this hypothesis we constructed a recombinant H. salinarum NRC-1 strain carrying the FLAG tag at the 3ʹ end of orc4 gene and performed western blot assays on tricine-SDS-PAGE gels to improve discriminatory resolution on low MW proteins.
Orc4 is a 45.6 kDa protein which migrates as having apparent MW of 53.7 kDa after addition of FLAG-tag residues (DYKDDDDK) and acidity correction. The protein isoform expected by the aforementioned hypothesis is 9.1 kDa and is predicted to migrate as 13.0 kDa after corrections. The experimental data is consistent with the prediction (Fig. 2C). Predictions using the next upstream (GTG) or downstream (ATG) neighbor frequent start codons would result in bands at 16.0 kDa and 10.9 kDa apparent MW, respectively, less consistent with the western blot result. Additional experiments to confirm and eventually elucidate the mechanistic details of the proposed asRNA involvement in isoform translation are the next logical step of this work.
Isoform translation from intraRNAs is an ancient phenomenon
In order to gain some insight on how ancient is the translation of intraRNAs, we focus on fusA, which encodes for the translational elongation factor 2, a universal protein probably present in the Last Universal Common Ancestor (LUCA) [54,55]. This elongation factor has homologues in all three domains of life: EF-G in bacteria, eEF-2 in eukaryotes and aEF-2 in archaea and is composed of five domains, a GTPase domain and domains II to V [45].
Contrary to eukaryotes, it is not commonly expected from bacteria or archaea to generate protein diversity by translating alternative isoforms from RNA which share the same genomic locus. Mass spectrometry data-mining revealed a semi-tryptic peptide mapping to H. salinarum aEF-2 consistent with a moderate statistically significant intraRNA TEX+ > TEX- signal (TSS_10415_3, p-value = 4.4 · 10−5). The dRNA-seq analysis method estimated an uncertainty of 3 bp on the TSS position which turns it compatible with an ATG start codon and explains the moderate statistical support. This zero-length 5ʹ UTR putative intraRNA would translate a protein for which both tryptic and semi-tryptic peptides are available in PeptideAtlas: PAp02311109 and PAp00628707, respectively (Table S11). This intraRNA would code for a 19.0 kDa protein that contains the aEF-2’s domain V (PF00679) and a part of domain IV (PF03764). When it was first characterized thirty years ago, this protein presented at least two putative isoforms in SDS-PAGE purification gels [56] but the absence of MW markers hindered isoform identification. IntraRNAs upstream of aEF-2’s domain IV and V were detected by our re-analysis of publicly available dRNA-seq data for several archaea: Haloferax volcanii (TSS_004167, p-value <10−15), Methanocaldococcus jannaschii (TSS_007660, p-value = 8.2 · 10−4), Thermococcus kodakarensis (TSS_005251, p-value = 8.6 · 10−5) and Thermococcus onnurineus (TSS_005310, p-value = 6.5 · 10−6) (Table S10). We speculate that these other organisms may also have a similar isoform translated.
It is no novelty that eukaryotes produce protein diversity by translating alternatively spliced mRNA isoforms. For meaningful comparisons we chose Saccharomyces cerevisiae since it has no introns in its eEF-2 coding gene. We analyzed yeast’s public RNA-seq dataset that presents the highest coverage available (Jan/2018) in SRA database [57] (PRJNA408327). The most frequent 5ʹ end from reads aligned inside EFT2 locus (which encodes for eEF-2) identifies an intraRNA and corresponding intraORF that contains the elongation factor IV and V domains (Fig. S15). Other longer intraRNAs corresponding to the next two RNA-seq peaks could also be translated, predicting isoforms of 24.9 kDa, 53.7 kDa and 67.1 kDa, respectively. Efforts to crystallize eEF-2 (predicted MW of 93.2 kDa) fifteen years ago showed isoforms of ~25 kDa, ~55 kDa, and ~70 kDa, regarded then as undesirable byproducts from the main ~94 kDa protein [58] (Fig. S15).
We found similar intraRNA translation evidence for aEF-2’s bacterial homolog EF-G (fusA gene, locus b3340) in E. coli K-12. For that, we took advantage of the well established RegulonDB [59] since this database includes TSS and promoters datasets. The database registers a putative intraRNA (TSS_3889 cluster) that would translate a 30.1 kDa product starting at an ATG codon 6 nt downstream of the iTSS, with predicted pI of 5.79 and acidity-correct apparent MW of 32.7 kDa. We searched the two-dimensional gel electrophoresis image database SWISS-2DPAGE [61] for duplicated EF-G spots placed in accordance with the isoform prediction and retrieved the spot ID 2D-001WR8 (32.1 kDa, pI 5.78). Efforts to purify E. coli EF-G forty-five years ago revealed two isoforms [62] that were washed away in successive purification rounds. This isoform contains the elongation factor domains IV and V (Fig. S16). IntraRNAs upstream of EF-G’s domain IV and V could be detected by our re-analysis of publicly available long-reads RNA-seq data or TAP-treated RNA-seq data in two additional bacteria: Mycoplasma hyopneumoniae and Listeria monocytogenes (Fig. S17).
Taken together, these results allow us to speculate that intraRNA translation is an ancient phenomenon, perhaps present since the LUCA.
Isoform translation from intraRNAs is present in horizontally acquired genes
We note that the extensively studied gene dnaK, which encodes for molecular chaperone DnaK (aka Hsp70), also presents a putative intraRNA that would translate observed isoforms in archaea and bacteria. The presence of dnaK in archaeal genomes is not always verified, in remarkable contrast with all bacteria and eukaryotes [63,64]. It was proposed that dnaK and its companion co-chaperones, dnaJ and grpE, were acquired by Halobacteriales from bacteria, lost and reacquired [65,66].
Our H. salinarum dRNA-seq data shows an intraRNA (TSS_6356_3) inside dnaK with borderline characteristics that did not grant it membership in our stringent intraORF set but for which translation evidence is available (p-value = 2.2 · 10−10, maximum read length 156 nt). Searches on PeptideAtlas MS database retrieve the semi-tryptic peptide PAp02314977 and its correspondent tryptic cognate peptide PAp00368901 (Table S11). This tryptic peptide is indistinguishable from a putative semi-tryptic that did not cleave the N-terminal methionine since arginine is the previous amino acid residue. A DnaK 311 amino acid long protein isoform (318 N-terminal amino acids left out) would have pI 3.57 and 32.9 kDa MW, and is predicted to migrate as a 45.2 kDa band in gels due to its acidic characteristics. DnaK itself has MW of 67.4 kDa, from which its alias Hsp70 was historically coined, but is predicted to migrate as 88.9 kDa in gels. A western blot performed seventeen years ago to study a nucleoside diphosphate kinase [67] also shows a clear ~45 kDa band with the same expression patterns of the DnaK band [68] (Fig. S18). The absence of secondary structure upstream of the iTSS accessed with several prediction methods (data not shown) and the presence of a strong SD-like ribosome binding site 21 nt upstream of the first available ATG start codon (Fig. S18) indicate that this intraRNA translates a DnaK isoform.
In bacteria, from which archaea probably acquired dnaK, we were able to see a similar phenomenon searching on RegulonDB [59] for iTSS and SWISS-2DPAGE [61] for 2Dgel/MS data. In E. coli there is an iTSS (TSS_31 cluster) inside dnaK CDS (locus b0014) which defines an intraORF that has no 5ʹ UTR, starts from an ATG codon and would translate a 37.0 kDa MW isoform with pI of 4.84. Searches on 2D-gel images for duplicated DnaK spots with the acidity-corrected apparent MWs of 78.1 kDa and 42.6 kDa, corresponding respectively to the full-length protein and its putative isoform, retrieved a gel (ECOLI4-5, spots 2D-001HF6 and 2D-001HIJ) consistent with the predictions (Fig. S19B).
Such kind of conservation allow us to predict that intraRNA translation in a given organism may be present by extrapolation of observations made in other organisms. As an example we highlight the cydA gene, which encodes for the cytochrome bd ubiquinol oxidase subunit I, and was probably acquired from bacteria along with other aerobic respiration genes [69]. We found that in E. coli this gene presents an iTSS (TSS_873, locus b0733) for a putative intraRNA that, if translated using the first available ATG start codon 29 nt downstream, would produce a 43.9 kDa isoform of the 58.2 kDa CydA cognate protein. Mass spectrometry identification of spots in 2D gels of cytoplasmic membranes performed ten years ago [70] showed CydA (among others) spots with observed MW incompatible with full-length protein but consistent with isoform. Contrary to previous examples, this protein is not highly acidic and do not meet the criteria for MW correction. It was reported a 44.0 kDa CydA spot, consistent with isoform predicted MW. A similar iTSS pattern is found in H. salinarum in which there is a clear TEX+ >TEX- signal in both cydA1 and cydA2 (TSS_10696_3 and TSS_13533_3, respectively). Interestingly, high-throughput sequencing of asRNAs that are in a double-stranded form with their cognate mRNAs (dsRNA-seq) in E. coli [71] reveals a moderate interaction signal in the region upstream of the iTSS (Fig. S20). Although there are no dsRNA-seq data available for H. salinarum, our dRNA-seq data shows a clear aTSS signal in an equivalent region which is differentially expressed along the growth curve (Fig. S21). These conserved features seem to have withstood the horizontal transference between both domains of life. In light of a strong RBS signature 16 nt upstream of an ATG start codon, we predict that the intraRNA translation in H. salinarum would also be conserved. We speculate that the intraRNA could be somehow bypassing the asRNA mediated putative regulation of cydA by translating an isoform containing 86% of CydA’s PF01654 protein domain.
Taken together, these results allow us to infer that intraRNA translation is present in genes transferred horizontally among bacteria and archaea, in spite of differences in transcription and translation molecular machinery.
Discussion
In this work, we propose that internal RNAs overlapping coding sequences can drive the production of protein isoforms in archaea. In vivo detection of the full length and smaller protein isoform reinforces the ‘hidden’ protein repertoire derived from intraRNAs. In summary, we verified the intraRNA translation phenomenon, with varying evidence strengths, for 17 genes in organisms from all three domains of life (Table S11).
IntraRNAs translation as an alternative hypothesis to proteolysis
Post-translational processing is the most common hypothesis for prokaryote isoform biogenesis but we argue that intraRNA translation should also be considered. It is becoming unusual practice nowadays to publish whole protein gels, where unspecific, proteolysis derived, unexpected MW or inexplicable bands commonly appear. It is interesting to note that high-throughput genome wide datasets can now help explain some of these features that could be valuable experimental resources to be reused.
For instance, several ‘outliers’ 2D-gel spots increased their intensities when H. volcanii was cultivated with a proteasome-specific inhibitor [72] emphasizing the role of regulated proteolysis on central functions of the cell. Some of the spots with unexpected MW and pI pairs are consistent with intraRNA translation such as: hmgB, which encodes for hydroxymethylglutaryl-CoA synthase (locus HVO_2419); and tef1a1, which encodes for elongation factor 1-alpha/Tu (locus HVO_0359). Our re-analysis of H. volcanii dRNA-Seq data shows an iTSS inside hmgB gene locus (TSS_026134) that would translate a predicted 11.0 kDa isoform (14.7 kDa corrected MW) consistent with the 15 kDa observed isoform. Analogously, tef1a1 (TSS_004261) would translate a predicted 17.6 kDa isoform (19.9 kDa corrected MW) consistent with the 20 kDa observed isoform spot (Table S10).
The H. salinarum R1 2D-gel database HaloLex [73] has several ‘outliers’ spots, with MW and pI inconsistent with fingerprint peptides extracted from them. Also, sometimes the low number of fingerprint peptide matches put into question the identification. Both features would be explained by isoforms or proteolysis. We observed that intraRNA translation is consistent with some of such discrepant spots. For example, the gel G839 showed at least 3 outliers, citB, cxp, and rpoB2; that would be consistent with the overall experiment under the intraRNA translation biogenesis hypothesis (Fig. S22). Interestingly, rpoB2, which encodes for the RNA polymerase subunit B’’, is currently misannotated as a pseudogene due to an early stop codon which is actually the separation between subunits B’’ and H, the upstream CDS (protein P0CX06) [74]. The true rpoB2 TSS was detected by our dRNA-Seq analysis as an iTSS (TSS_10473_3) inside the longer misannotated CDS and the ‘isoform’ has pI and MW values consistent with the true RpoB2 (Fig. S22).
We speculate that plenty of overlooked 2D-gel spots accumulated in the literature over the years could be explained by intraRNA translation instead of proteolysis.
Potential roles of intraRNA derived protein isoform
There is still much to be debated concerning the relevance or functionality of intraRNAs and their translated product. Also, there is a controversy of whether protein isoforms themselves are indeed functionally relevant [22,23]. However, as it has been found for sORFs or ncRNAs, intraRNAs encoded isoforms might present fine tuning regulatory roles that are yet to be discovered. It is also possible that the isoform itself does not play a role as protein but, similarly to what is known for uORF products [20,75], its translation process could regulate a downstream gene translation via ribosome interference if the intraRNA is part of a polycistronic transcriptional unit. In this scenario, it would be conceptually appropriate to rethink an intraORF as an uORF of the following gene. Sequencing technologies that can read >1kb nt would be able to sort out RNA isoforms and address these issues.
Evidently a protein which has one out of two domains excluded would function differently. Even small differences in crucial N-terminal parts such as export signals could make qualitative differences due to localization. The gas vesicle structural protein GvpC1, mentioned as one validation example, showed evidence of an isoform in whole lysate cells but not in purified gas vesicles (Fig. S11).
From the protein point of view, given an isoform, it seems unlikely that the biogenesis route (intraRNA, proteolysis or 2nd ribosome binding site) matters. Conversely, from a regulatory perspective, it would be probably important to discriminate between the cases. Our data on differential expression considered only four conditions, which is still limited compared to the compendium accumulated over the years using microarrays [25] or tiling arrays [4], none of which have the spatial resolution necessary to pinpoint intraRNA regulation deconvoluted from the cognate gene. Even though, some differential modulation between both overlapping transcripts was inferred in this work. Sequencing technologies that can read uninterrupted molecules could, as some sort of high-throughput version of northern blots, discriminate RNA isoform by length and follow unconverted differential expression on different contexts.
IntraRNA translation in conserved proteins
The indicative that intraRNAs can produce alternative proteins and it is probably an ancient phenomenon reinforces the modular nature of protein domains [76–78]. We demonstrated the plausibility of such idea by showing examples of 3 genes conserved in many organisms, fusA, dnaK and cydA; that seem to produce isoforms from intraRNAs.
The existence of multiple promoter regions inside coding sequences, together with the modular evolution of proteins could contribute to the existence of protein domains being transcribed and translated independently, even after fusion processes to compose multidomain proteins [76,78]. The fact that many intraORFs identified in H. salinarum are also found as independent stand-alone putative proteins in other organisms is taken as evidence of such modularity. Detailed research would be necessary to assert if the intraRNA is prior to the dismantling of a cognate gene in modular blocks or if blocks were assembled taking the original promoter regions to the new fused coding sequence and thus creating an intraRNA.
In conclusion, recapitulating the phenomenon of intraRNA translation in archaea, bacteria and eukaryotes, we close the gap and establish its ubiquitousness in all three domains of life.
Caveats and limitations
This work makes use of reasonable implicit and explicit assumptions that need to be acknowledged. Moreover, there are technical limitations that still preclude indisputable evidence and should be diligently addressed in future research. Ideally, mutagenesis perturbations that stop transcription leading to disappearing protein traces would better establish intraRNA translation. Aware that ‘correlation does not imply causation’ (cum hoc ergo propter hoc fallacy), we explicitly assume causality if a protein is found with the properties (molecular weight or semi-tryptic peptide) predicted by an RNA molecule (lex parsimoniae principle).
Connected to the fundamental explicit assumption, there are three important implicit assumptions derived from technical limitations: (i) sequenced reads length; (ii) acidity correction and (iii) methionine cleavage. Almost all validation cases we presented based on semi-tryptic peptide finding were matches without the first (methionine) amino acid. Therefore it is assumed implicitly that these proteins had their N-terminal methionine cleaved and the mature form was recorded in the database, not being a degradative proteolysis event. All validation cases based on protein gels were acidity-corrected [37] implicitly assuming that the equation is universally applicable and grounded on the fact that the known full-length protein MWs were properly adjusted. Finally, the current generation sequencing platforms allows, even with the paired-end improvement, a limited coverage of RNA lengths. It is rare to be able to observe a full-length transcript so we implicitly assume that if a given intraRNA candidate presents reads that extend near the maximum capacity provided by the technology, then it would go full-length if not censored. Moreover, it is implicitly assumed that the intraRNA full-length contains a stop codon. This could be a source of false positives on our intraORFs list regarding coding potential. The lack of trial-and-error rounds on our western blot validation efforts (3 out of 3 chosen) suggests that the aforementioned assumption is reasonable. The small total number of intraORFs found relative to the whole genome CDS number can be seen as an indication that most intraRNAs are likely non-coding instead of indication of our selection stringency.
We cannot completely rule out that the smaller proteins can be the result of protein cleavage/degradation. In this scenario, we are not able to explain such coincident coupling between transcriptional and post-translational processes in general since we understand that such correlations could hardly be obtained by chance alone.
The most competitive scenario against the intraRNA translation hypothesis is the alternative/secondary ribosome binding site (RBS) usage. In the absence of point-wise mutagenesis experiments, the observables from intraRNA translation or internal RBS usage would be the same if the iTSS is a false positive created by a secondary structure on TEX+ libraries. This could be a source of false positives on our intraORFs list in spite of our stringent cutoffs on iTSS definition. Future work on computational/experimental RNA structuromics [79] can better evaluate putative iTSS to discriminate both cases since it is not expected that large-scale synonymous mutations efforts to shoot down intraRNAs can be practically undertaken. We implicitly assume that ‘in all other things being equal’ intraRNA translation is favorable to RBS usage on grounds of parsimony principle.
In order to mitigate unavoidable false positive cases, we applied a stringent set of inclusion rules on our intraRNA and intraORF lists. Moreover, to mitigate false negative cases we made available all data, computer code and visualization tools at http://labpib.fmrp.usp.br/~rvencio/intrarna/ to allow alternative re-analysis with less or more stringent criteria.
Materials and methods
Strains, media and growth conditions
Halobacterium salinarum NRC-1 were grown in complex media (CM) (250 g/L NaCl, 20 g/L MgSO4, 2 g/L KCl, 3 g/L Sodium citrate, 10 g/L bacteriological peptone (Oxoid)) at 37°C, under light and constant agitation of 125 rpm. Samples were taken at 3 different time points in a growth curve: middle of exponential phase (17h, OD600 ~ 0.3, aka early-log phase [4]), end of stationary phase (37h, OD600 ~ 0.5, aka mid-log phase) and gas vesicles release phase (86h, OD600 ~ 1.1, aka late-log phase) (Fig. S1). Reference samples were cultured under standard growth conditions (37ºC, 225 rpm, constant light) and sampled at mid-log phase (OD600 ~ 0.5) [80].
dRNA-seq library preparation and sequencing
Total RNA extraction was performed using mirVana miRNA isolation kit (Ambion) from biological duplicates. DNA contamination was removed with treatment with Turbo DNAse (Ambion) and verified by PCR. dRNA-seq was performed as described in a previous study [9]. Briefly, TEX+ sample was treated with TEX (Epicentre TER51020) and TEX- was incubated only with buffer at 30°C for 60 min and purified with RNeasy MinElute Cleanup (Qiagen). Samples were treated with TAP (Epicentre), purified and quantified by Quant-iT RiboGreen RNA Assay (Invitrogen). For strand specific dRNA-seq library preparation, TruSeq Small RNA Sample Preparation (Illumina) was used as described in [81]. Sequencing was performed using MiSeq Reagent v2 300 cycles. The raw sequencing data is available at the NCBI SRA database under the accession ID: SRP137801.
Sequencing data analysis of genome-wide datasets
We developed a semi-automatic protocol to process RNA-seq data, available at https://github.com/alanlorenzetti/frtc/. Libraries were downloaded from NCBI Sequence Read Archive (SRA) [82] and converted to FASTQ format using either SRAdb v1.40.0 [83] or fastq-dump v2.8.2 [82]. We preprocessed paired-end and single-end libraries using Trimmomatic v0.36 [84] in order to trim known adapters and/or low quality ends. Reads were trimmed to the end if the mean Phred of a four nucleotide sliding window was less than 30 and only reads satisfying the minimum length of 20 nucleotides were allowed to survive. Reads surviving as a pair were aligned to reference genomes in a paired-end fashion using HISAT2 v2.1.0 [85], suppressing alignments resulting in fragments longer than 1000 nucleotides. Orphan R1 and R2 sequences from paired-end libraries and those coming from single-end runs were aligned using the single-end mode. We allowed the program to report multi-mappers aligning up to 1000 times and required it to suppress spliced, soft-clipped, gapped, discordant and mixed alignments. The output SAM files were converted to BAM using SAMtools v1.3.1 [86] and input in MMR [87] to find the most likely position for each multi-mapper. Briefly, the software computes the genome-wide coverage considering only uniquely aligned reads, and then assign a unique position to each multi-mapper based on its potential of reducing the local variance of coverage. Paired-end alignments adjusted by MMR may lack conformity if the fragments are too small and/or the reads align entirely to direct repeats, so we removed these particular cases to avoid uncertainty. Genome-wide coverage was computed for every library by deepTools v2.5.3 [88], taking into consideration the extension of entire fragments for paired-end alignments and the fact that orphan R2 reads align to the opposite strand of the real RNA fragment. Furthermore, we used bedtools v2.2.26 [89] to compute 5ʹ and 3ʹ profiles for each library, employing the aligned R1 and R2 reads, respectively, once more taking into consideration the orientation of reads in relation to the original RNA fragment in the sample. Data visualization was performed using IGV v2.4.6 [90] and Gaggle Genome Browser [26]. Additionally to Halobacterium salinarum, this genome-wide dataset analysis was performed for: Haloferax volcanii DS2 (PRJNA324298) [8], Methanocaldococcus jannaschii DSM 2661 (PRJNA342613) [91], Thermococcus kodakarensis KOD1 (PRJNA242777) [10] and Thermococcus onnurineus NA1 (PRJNA339284) [92] (Table S10).
Sequencing data analysis of partial datasets (gene-centric)
Analyses of specific gene transcription using regular RNA-seq were performed using straightforward BLAST searches at SRA databases (aka SRA BLAST) using NCBI’s web interface. Gene full-length sequences were used as query sequence and transcriptome sequencing datasets as search database. Discontiguous megablast algorithm with default parameters was used except the number of target sequences retrieved, which was set to the maximum allowed 20,000, to retrieve as much aligned reads as possible. Quantitative information regarding the number of reads aligning at each position, for coverage or simple histogram (counts) was extracted from BLAST outputs with simple R parsers. This gene-centric analysis was performed for: fusA gene in Mycoplasma hyopneumoniae 7448 (PRJNA255516), Listeria monocytogenes EGD-e (PRJNA151809), Escherichia coli K-12 (PRJNA348358); EFT2 gene in Saccharomyces cerevisiae (PRJNA408327); and dnaK gene in Escherichia coli K-12 (DRP003075).
Transcription start site detection
TSS were identified from dRNA-Seq experiments using TSSAR java client version 1457945232 [93]. For iTSS, gTSS detection and UTR length estimation p-value < 10−15 was used as statistical significance cutoff, with a minimum of 4 reads per position and the grouping of TSS with a distance of at least 5 nt. Other less stringent p-values were considered in a case-by-case basis for few relevant genes depending on auxiliary additional evidence but never > 10−3. Multiple test adjustments were not used.
Filtering iTSS in Halobacterium salinarum NRC-1
H. salinarum NRC-1 annotation from RefSeq database, updated in 2017 according to the Prokaryotic Genome Annotation Pipeline [94] was used as reference (Table S1). Additionally, curated H. salinarum R1 annotation from HaloLex database [95] was considered when RefSeq CDS were missing. iTSS located within 90 bp or 10% of CDS’ annotated edges were disregarded as intraRNA’s TSS and filtered out in next steps to avoid simple downstream gene promoters. Potentially structured regions were filtered out by calculating folding minimum free energy (MFE) along the whole genome sequence. A sliding window tiled the genome on several sub-sequences of size 25 + 1 + 25 = 51 with an offset of 10 nt (thus 41 nt superposition between adjacent sub-sequences). All sub-sequences were subjected to secondary structure prediction using RNAfold v2.0.5 [96] with default parameters. The distribution of MFE obtained for the tiled genome was compared with the distribution obtained for only sub-sequences immediately downstream of iTSS. The 33.3% quantile in the whole-genome MFE distribution was arbitrarily chosen as cutoff for potentially forming structures and thus putative false positive iTSS.
Chromosomal tagging and western blot
FLAG epitope was inserted at the 3ʹ end of genes kef1 (VNG_RS07995/VNG2068C), acs3 (VNG_RS05220/VNG1339C), and orc4 (VNG_RS11810/VNG6363G) as described in [97] using the modular vector pHSal-S [98]. Oligonucleotides used for cloning:
VNG2068-F1: CGCGGAATTCACCGTTCACAGCGGCGAC,
VNG2068-R1: CTACTTGTCGTCATCGTCTTTGTAGTCCCCGTCGTCGGGGTG,
VNG2068-F2: GACGATGACGACAAGTAGTCGCGCCCGCTCACT,
VNG2068-R2: AGCTAAGCTTTGGCGGCGCGTGGCAGCG,
VNG1339-F1: AGCTGAATTCGGATCGACGCCCGCATCG,
VNG1339-R1: CTACTTGTCGTCATCGTCTTTGTAGTCGTCCTCGTCGGGGAC,
VNG1339-F2: GACGATGACGACAAGTGACCCGGCGCGACCCGC,
VNG1339-R2: CGCGAAGCTTGTGGCGAGTCACCTCCATCTC,
VNG6363-F1: AGCTGAATTCGGCGTCCTTTCCGAGGAC,
VNG6363-R1: TTACTTGTCGTCATCGTCTTTGTAGTCCTCGCGAGTCCAGTC,
VNG6363-F2: GACGATGACGACAAGTAATGACCCCTACTATTG,
VNG6363-R2: AGCTAAGCTTGACGAACTGCAGGCGGGC.
Recombinant strains were verified by sequencing and grown in CM supplemented with uracil. Cell pellets were lysed in triple lysis buffer (TrisHCl 10mM pH 8.0, NaCl 50mM, Triton X-100 1%, SDS 0.1%, sodium deoxilate 6nM) and protease inhibitor cocktail (Sigma Aldrich), incubated on ice for 20 min, and sonicated. After centrifugation at 13,500 rpm at 4°C, the supernatant was collected and quantified by Bradford assay (Bio-Rad, Hercules, CA, USA). Samples were denatured at 95°C for 10 min, run in tricine-SDS-PAGE 15% and transferred to 0.2 µm nitrocellulose membrane (GE Healthcare) (Tris 24.7 mM, glycine 192 mM, ethanol 18.46%) at 80 V for 1h. Membranes were blocked in non-fat milk 5% in TBS-Tween 0.1% and incubated with monoclonal antibody anti-FLAG conjugated to HRP (Sigma A8592) diluted 1:1000 in TBS-T for 1h. Membranes were washed 5 times with TBS-T and detection was performed by ECL (GE Healthcare Bio-Sciences). Images were acquired in Bio-Rad ChemiDoc Imaging System (Bio-Rad Laboratories, Hercules, CA). Parent H. salinarum NRC-1 strain without the FLAG-tag was included in an auxiliary blot to demonstrate that the antibody binds specifically to the FLAG-tag (data not shown).
Protein molecular weight and semi-tryptic analysis
Protein validation was based on MW accurate estimation or semi-tryptic peptide identification. Apparent MW due to differential migration of acidic protein was calculated by the equations Mc = M + ∆M(M, s) and ∆M(M, s) = n(s) · (276.5· k(s)/n(s) – 31.33), where: M is the predicted protein MW; Mc is the apparent MW after correction; n(s) is the number of amino acid residues and k(s) is the number of glutamic acid (E) or aspartic acid (D) amino acid residues in the protein sequence s [37]. However, ∆M(M, s) = 0 if k(s)/n(s) < 0.114 or if k(s)/n(s) > 0.511, i.e., there is no correction. When necessary, the appropriate flag mass and amino acid sequence were included. In the case of FLAG-tag constructs created for this work, the sequence is DYKDDDDK. Semi-tryptic peptides, i.e. sequences with arginine (R) or lysine (K) as their last C-terminal amino acid residue but not at N-terminal, were searched in PeptideAtlas curated database (www.peptideatlas.org) against the latest public H. salinarum NRC-1 build (2014–11) [38] and against H. salinarum R1 SWATH data (PRIDE ID PXD003667) [39]. Two semi-tryptic peptides variations were considered, full peptides and methionine cleaved peptides (thus missing first M residue). Putative intraRNA translation products that were indistinguishable from tryptic peptide versions (M preceded by R or K) were not considered valid since they could be generated from cognate protein by technical trypsin cuts.
Funding Statement
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo [2015/21038-1]; [2011/07487-7]; [2017/03052-2]; [2011/14455-4]; [2015/12012-9]; Fundação de Apoio ao Ensino, Pesquisa e Assistência do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo [415/2018]; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) [Finance Code 001]; Conselho Nacional de Desenvolvimento Científico e Tecnológico [166166/2014].
Acknowledgments
We thank Silvia Epifânio for technical support. We thank João Paulo Pereira de Almeida for testing the sequencing analysis pipeline. We thank the anonymous reviewers for extremely helpful ideas and criticism which led to better validation and caveats clarification.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article can be accessed here.
References
- [1].Babski J, Maier L-K, Heyer R, et al. Small regulatory RNAs in Archaea. RNA Biol. 2014;11:484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hör J, Gorski SA, Vogel J.. Bacterial RNA biology on a genome scale. Mol Cell. 2018;70:785–799. [DOI] [PubMed] [Google Scholar]
- [3].Güell M, Van Noort V, Yus E, et al. Transcriptome complexity in a genome-reduced bacterium. Science. 2009;326:1268–1271. [DOI] [PubMed] [Google Scholar]
- [4].Koide T, Reiss DJ, Bare JC, et al. Prevalence of transcription promoters within archaeal operons and coding sequences. Mol Syst Biol. 2009;5:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wade JT, Grainger DC. Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat Rev Microbiol. 2014;12:647–653. [DOI] [PubMed] [Google Scholar]
- [6].Dornenburg JE, Devita AM, Palumbo MJ, et al. Widespread antisense transcription in Escherichia coli. mBio. 2010;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thomason MK, Bischler T, Eisenbart SK, et al. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol. 2015;197:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Babski J, Ponsawat J, Tongsima S, et al. Genome-wide identification of transcriptional start sites in the haloarchaeon Haloferax volcanii based on differential RNA-Seq (dRNA-Seq). BMC Genomics. 2016;17:629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sharma CM, Snider GS, Kuekes PJ, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. [DOI] [PubMed] [Google Scholar]
- [10].Jäger D, Förstner KU, Sharma CM, et al. Primary transcriptome map of the hyperthermophilic archaeon Thermococcus kodakarensis. BMC Genomics. 2014;15:684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bilusic I, Popitsch N, Rescheneder P, et al. Revisiting the coding potential of the E. coli genome through Hfq co-immunoprecipitation. RNA Biol. 2014;11:641–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Popitsch N, Bilusic I, Rescheneder P, et al. Temperature-dependent sRNA transcriptome of the Lyme disease spirochete. BMC Genomics. 2017;18:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shao W, Price MN, Deutschbauer AM, et al. Conservation of transcription start sites within genes across a bacterial genus. mBio. 2014;5:e01398–01314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cohen O, Doron S, Wurtzel O, et al. Comparative transcriptomics across the prokaryotic tree of life. Nucleic Acids Res. 2016;44:W46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Koubova JM, Hu Y-C, Bhattacharyya T, et al The coding and noncoding architecture of the Caulobacter crescentus genome. PLoS Genet. 2014;10:e1004463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Čuklina J, Ponsawat J, Tongsima S, et al. Genome-wide transcription start site mapping of Bradyrhizobium japonicum grown free-living or in symbiosis - a rich resource to identify new transcripts, proteins and to study gene regulation. BMC Genomics. 2016;17:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Di Martino ML, Romilly C, Wagner EGH, et al. One gene and two proteins: a leaderless mRNA supports the translation of a shorter form of the shigella VirF regulator. mBio. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shell SS, Wang J, Lapierre P, et al. Leaderless transcripts and small proteins are common features of the mycobacterial translational landscape. PLoS Genet. 2015;11:e1005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nakagawa S, Niimura Y, Gojobori T. Comparative genomic analysis of translation initiation mechanisms for genes lacking the Shine-Dalgarno sequence in prokaryotes. Nucleic Acids Res. 2017;45:3922–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cabrera-Quio LE, Herberg S, Pauli A. Decoding sORF translation - from small proteins to gene regulation. RNA Biol. 2016;13:1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Prasse D, Thomsen J, De Santis R, et al. First description of small proteins encoded by spRNAs in methanosarcina mazei strain Gö1. Biochimie. 2015;117:138–148. [DOI] [PubMed] [Google Scholar]
- [22].Tress ML, Abascal F, Valencia A. Most alternative isoforms are not functionally important. Trends Biochem Sci. 2017;42:408–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Blencowe BJ. The relationship between alternative splicing and proteomic complexity. Trends Biochem Sci. 2017;42:407–408. [DOI] [PubMed] [Google Scholar]
- [24].Ng WV, Kennedy SP, Mahairas GG, et al. Genome sequence of halobacterium species NRC-1. Proc Natl Acad Sci. 2000;97:12176–12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Brooks AN, Reiss DJ, Allard A, et al. A system-level model for the microbial regulatory genome. Mol Syst Biol. 2014;10:740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bare JC, Koide T, Reiss DJ, et al. Integration and visualization of systems biology data in context of the genome. BMC Bioinformatics. 2010;11:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Price A, Garhyan J, Gibas C. The impact of RNA secondary structure on read start locations on the Illumina sequencing platform. PloS One. 2017;12:e0173023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Soppa J. Normalized nucleotide frequencies allow the definition of archaeal promoter elements for different archaeal groups and reveal base-specific TFB contacts upstream of the TATA box. Mol Microbiol. 1999;31:1589–1592. [DOI] [PubMed] [Google Scholar]
- [29].Wurtzel O, Sapra R, Chen F, et al. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010;20:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Slupska MM, King AG, Fitz-Gibbon S, et al. Leaderless transcripts of the crenarchaeal hyperthermophile pyrobaculum aerophilum. J Mol Biol. 2001;309:347–360. [DOI] [PubMed] [Google Scholar]
- [31].Lloréns-Rico V, Waters MR, Perrotti A, et al. Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci Adv. 2016;2:e1501363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Giess A, Jonckheere V, Ndah E, et al. Ribosome signatures aid bacterial translation initiation site identification. BMC Biol. 2017;15:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nakahigashi K., Takai Y, Kimura M, et al. Comprehensive identification of translation start sites by tetracycline-inhibited ribosome profiling. DNA Res Int J Rapid Publ Rep Genes Genomes. 2016;23:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sartorius-Neef S, Pfeifer F. In vivo studies on putative Shine-Dalgarno sequences of the halophilic archaeon halobacterium salinarum. Mol Microbiol. 2004;51:579–588. [DOI] [PubMed] [Google Scholar]
- [35].Hering O, Brenneis M, Beer J, et al. A novel mechanism for translation initiation operates in haloarchaea. Mol Microbiol. 2009;71:1451–1463. [DOI] [PubMed] [Google Scholar]
- [36].Oren A. Microbial life at high salt concentrations: phylogenetic and metabolic diversity. Saline Syst. 2008;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guan Y, Zhu Q, Huang D, et al. An equation to estimate the difference between theoretically predicted and SDS PAGE-displayed molecular weights for an acidic peptide. Sci Rep. 2015;5:13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Van PT, Schmid AK, King NL, et al. Halobacterium salinarum NRC-1 PeptideAtlas: toward strategies for targeted proteomics and improved proteome coverage. J Proteome Res. 2008;7:3755–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Losensky G, Jung K, Urlaub H, et al. Shedding light on biofilm formation of Halobacterium salinarum R1 by SWATH-LC/MS/MS analysis of planktonic and sessile cells. Proteomics. 2017;17:1600111. [DOI] [PubMed] [Google Scholar]
- [40].Falb M, Aivaliotis M, Garcia-Rizo C, et al. Archaeal N-terminal protein maturation commonly involves N-terminal acetylation: a large-scale proteomics survey. J Mol Biol. 2006;362:915–924. [DOI] [PubMed] [Google Scholar]
- [41].Nakagawa S, Niimura Y, Miura K, et al. Dynamic evolution of translation initiation mechanisms in prokaryotes. Proc Natl Acad Sci U S A. 2010;107:6382–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Omotajo D, Tate T, Cho H, et al. Distribution and diversity of ribosome binding sites in prokaryotic genomes. BMC Genomics. 2015;16:604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chang B, Halgamuge S, Tang S-L. Analysis of SD sequences in completed microbial genomes: non-SD-led genes are as common as SD-led genes. Gene. 2006;373:90–99. [DOI] [PubMed] [Google Scholar]
- [44].Kramer P, Gäbel K, Pfeiffer F, et al. Haloferax volcanii, a prokaryotic species that does not use the Shine Dalgarno mechanism for translation initiation at 5ʹ-UTRs. PloS One. 2014;9:e94979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Finn RD, Coggill P, Eberhardt RY, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bolhuis A. Protein transport in the halophilic archaeon halobacterium sp. NRC-1: a major role for the twin-arginine translocation pathway? Microbiology. 2002;148:3335–3346. [DOI] [PubMed] [Google Scholar]
- [47].Martinez-Pastor M, Tonner PD, Darnell CL, et al. transcriptional regulation in archaea: from individual genes to global regulatory networks. Annu Rev Genet. 2017;51:143–170. [DOI] [PubMed] [Google Scholar]
- [48].Seitzer P, Wilbanks EG, Larsen DJ, et al. Carlo-based framework enhances the discovery and interpretation of regulatory sequence motifs. BMC Bioinformatics. 2012;13:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Schmid A. K., Reiss DJ, Kaur A, et al. The anatomy of microbial cell state transitions in response to oxygen. Genome Res. 2007;17:1399–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Patenge N, Berendes A, Engelhardt H, et al. The fla gene cluster is involved in the biogenesis of flagella in halobacterium salinarum. Mol Microbiol. 2001;41:653–663. [DOI] [PubMed] [Google Scholar]
- [51].Pfeifer F. Haloarchaea and the formation of gas vesicles. Life Basel Switz. 2015;5:385–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yao AI, Facciotti MT. Regulatory multidimensionality of gas vesicle biogenesis in Halobacterium salinarum NRC-1. Archaea Vanc BC. 2011;716456:2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Halladay JT, Jones JG, Lin F, et al. The rightward gas vesicle operon in Halobacterium plasmid pNRC100: identification of the gvpA and gvpC gene products by use of antibody probes and genetic analysis of the region downstream of gvpC. J Bacteriol. 1993;175:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Weiss MC, Sousa FL, Mrnjavac N, et al. The physiology and habitat of the last universal common ancestor. Nat Microbiol. 2016;1:16116. [DOI] [PubMed] [Google Scholar]
- [55].Atkinson GC, Baldauf SL. Evolution of elongation factor G and the origins of mitochondrial and chloroplast forms. Mol Biol Evol. 2011;28:1281–1292. [DOI] [PubMed] [Google Scholar]
- [56].Saruyama H, Sasaki S. Purification and characterization of peptide-elongation factor 2 (aEF-2) from an extremely halophilic archaebacterium halobacterium halobium. Eur J Biochem. 1988;170:499–505. [DOI] [PubMed] [Google Scholar]
- [57].Garalde DR, Snell EA, Jachimowicz D, et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods. 2018;15:201–206. [DOI] [PubMed] [Google Scholar]
- [58].Jørgensen R, Carr-Schmid A, Ortiz PA, et al. Purification and crystallization of the yeast elongation factor eEF2. Acta Crystallogr D Biol Crystallogr. 2002;58:712–715. [DOI] [PubMed] [Google Scholar]
- [59].Gama-Castro S, Salgado H, Santos-Zavaleta A, et al. RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 2016;44:D133–D143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Appel RD, Sanchez JC, Bairoch A, et al. SWISS-2DPAGE: a database of two-dimensional gel electrophoresis images. Electrophoresis. 1993;14:1232–1238. [DOI] [PubMed] [Google Scholar]
- [61].Rohrbach MS, Dempsey ME, Bodley JW. Preparation of homogeneous elongation factor G and examination of the mechanism of guanosine triphosphate hydrolysis. J Biol Chem. 1974;249:5094–5101. [PubMed] [Google Scholar]
- [62].Zmijewski MA, Macario AJL, Lipińska B. Functional similarities and differences of an archaeal Hsp70(DnaK) stress protein compared with its homologue from the bacterium Escherichia coli. J Mol Biol. 2004;336:539–549. [DOI] [PubMed] [Google Scholar]
- [63].Kabani M, Martineau CN. Multiple hsp70 isoforms in the eukaryotic cytosol: mere redundancy or functional specificity? Curr Genomics. 2008;9:248–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Macario AJL, Brocchieri L, Shenoy AR, et al. Evolution of a protein-folding machine: genomic and evolutionary analyses reveal three lineages of the archaeal hsp70(dnaK) gene. J Mol Evol. 2006;63:74–86. [DOI] [PubMed] [Google Scholar]
- [65].Petitjean C, Moreira D, López-García P, et al. Horizontal gene transfer of a chloroplast DnaJ-Fer protein to thaumarchaeota and the evolutionary history of the DnaK chaperone system in archaea. BMC Evol Biol. 2012;12:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ishibashi M, Tokunaga H, Hiratsuka K, et al. NaCl-activated nucleoside diphosphate kinase from extremely halophilic archaeon, halobacterium salinarum, maintains native conformation without salt. FEBS Lett. 2001;493:134–138. [DOI] [PubMed] [Google Scholar]
- [67].Tokunaga H, Hara S, Arakawa T, et al. Identification and partial purification of DnaK homologue from extremely halophilic archaebacteria, halobacterium cutirubrum. J Protein Chem. 1999;18:837–844. [DOI] [PubMed] [Google Scholar]
- [68].Kennedy SP, Ng WV, Salzberg SL, et al. Understanding the adaptation of Halobacterium species NRC-1 to its extreme environment through computational analysis of its genome sequence. Genome Res. 2001;11:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wagner S, Baars L, Ytterberg AJ, et al. Consequences of membrane protein overexpression in escherichia coli. Mol Cell Proteomics MCP. 2007;6:1527–1550. [DOI] [PubMed] [Google Scholar]
- [70].Lybecker M, Zimmermann B, Bilusic I, et al. The double-stranded transcriptome of escherichia coli. Proc Natl Acad Sci. 2014;111:3134–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kirkland PA, Reuter CJ, Maupin-Furlow JA. Effect of proteasome inhibitor clasto-lactacystin-beta-lactone on the proteome of the haloarchaeon haloferax volcanii. Microbiol Read Engl. 2007;153:2271–2280. [DOI] [PubMed] [Google Scholar]
- [72].Pfeiffer F, Denger K, Weinitschke S, et al. Genome information management and integrated data analysis with HaloLex. Arch Microbiol. 2008;190:281–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jun S-H, Reichlen MJ, Tajiri M, et al. Archaeal RNA polymerase and transcription regulation. Crit Rev Biochem Mol Biol. 2011;46:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dar D, Sorek R. Regulation of antibiotic-resistance by non-coding RNAs in bacteria. Curr Opin Microbiol. 2017;36:111–117. [DOI] [PubMed] [Google Scholar]
- [75].Björklund AK, Ekman D, Light S, et al. Domain rearrangements in protein evolution. J Mol Biol. 2005;353:911–923. [DOI] [PubMed] [Google Scholar]
- [76].Karamichali I, Koumandou VL, Karagouni AD, et al. Frequent gene fissions associated with human pathogenic bacteria. Genomics. 2014;103:65–75. [DOI] [PubMed] [Google Scholar]
- [77].Pasek S, Risler J-L, Brézellec P. Gene fusion/fission is a major contributor to evolution of multi-domain bacterial proteins. Bioinforma Oxf Engl. 2006;22:1418–1423. [DOI] [PubMed] [Google Scholar]
- [78].Vinogradova SV, Sutormin RA, Mironov AA, et al. Probing-directed identification of novel structured RNAs. RNA Biol. 2016;13:232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Baliga NS, DasSarma S. Saturation mutagenesis of the TATA box and upstream activator sequence in the haloarchaeal bop gene promoter. J Bacteriol. 1999;181:2513–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zaramela LS, Vêncio RZN, ten-Caten F, et al. Transcription start site associated RNAs (TSSaRNAs) are ubiquitous in all domains of life. PLoS ONE. 2014;9:e107680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Leinonen R, Sugawara H, Shumway M. International nucleotide sequence database collaboration. The sequence read archive. Nucleic Acids Res. 2011;39:D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zhu Y, Stephens RM, Meltzer PS, et al. SRAdb: query and use public next-generation sequencing data from within R. BMC Bioinformatics. 2013;14:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kahles A, Behr J, Rätsch G. MMR: a tool for read multi-mapper resolution. Bioinforma Oxf Engl. 2016;32:770–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ramírez F, Ryan DP, Grüning B, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinforma Oxf Engl. 2010;26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Robinson JT, Razick S, Turner B, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Smollett K, Blombach F, Reichelt R, et al. A global analysis of transcription reveals two modes of Spt4/5 recruitment to archaeal RNA polymerase. Nat Microbiol. 2017;2:17021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Cho S, Kim M-S, Jeong Y, et al. Genome-wide primary transcriptome analysis of H2-producing archaeon Thermococcus onnurineus NA1. Sci Rep. 2017;7:43044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Amman F, Wolfinger MT, Lorenz R, et al. TSSAR: TSS annotation regime for dRNA-seq data. BMC Bioinformatics. 2014;15:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Tatusova T, DiCuccio M, Badretdin A, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Pfeiffer F, Oesterhelt D. A manual curation strategy to improve genome annotation: application to a set of haloarchael genomes. Life Basel Switz. 2015;5:1427–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lorenz R, Bernhart SH, Höner Zu Siederdissen C, et al. ViennaRNA package 2.0. Algorithms Mol Biol AMB. 2011;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wilbanks EG, Larsen DJ, Neches RY, et al. A workflow for genome-wide mapping of archaeal transcription factors with ChIP-seq. Nucleic Acids Res. 2012;40:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Silva-Rocha R, Pontelli MC, Furtado GP, et al. Development of new modular genetic tools for engineering the halophilic archaeon halobacterium salinarum. PLoS One. 2015;10:e0129215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.