

Abstract
White blood cells (WBCs) are a major component of immunity in response to pathogen invasion. Neutrophils are the most abundant WBCs in humans, playing a central role in acute inflammation induced by pathogens. Adhesion to vasculature and tissue infiltration of neutrophils are key processes in acute inflammation. Many inflammatory/autoimmune disorders and cancer therapies have been found to be involved in activation and tissue infiltration of neutrophils. A promising strategy to develop novel targeted drug delivery systems is the targeting and exploitation of activated neutrophils. Here, we review a new drug delivery platform based on neutrophils. There are two types of drug delivery systems: neutrophils as carriers and neutrophil membrane-derived nanovesicles. We will discuss how nanoparticles (NPs) hijack neutrophils in vivo to deliver therapeutics across blood vessel barriers and how neutrophil membrane-derived nanovesicles target inflamed vasculature. Finally, we present the potential applications of neutrophil-based drug delivery systems in treating inflammation and cancers.
Keywords: Targeted drug delivery, Neutrophils, Nanovesicles, Inflammation, Cancer
1. Introduction
Inflammation is a process in which the immune system is activated to prevent human bodies from foreign invaders, such as bacteria or viruses.[1] In this process, leukocytes (WBCs) are activated and move directly to the invading pathogens to subsequently sequester them. Neutrophils, a type of leukocyte, play a central role in innate immunity. Neutrophils are the most abundant (40 to 75%) type of WBCs in humans and represent the body’s first line of defense against invading pathogens.[2] Neutrophils circulate in the bloodstream, and their half-life is approximately 8 h.[3] Neutrophils are ready to respond to pathophysiological changes (infections or tissue damage) via the initiation of inflammation. For example, neutrophils rapidly activate in response to inflammatory stimuli and migrate from the circulation to infection sites by crossing the blood vessel endothelium (Figure 1).[4]
Figure 1.
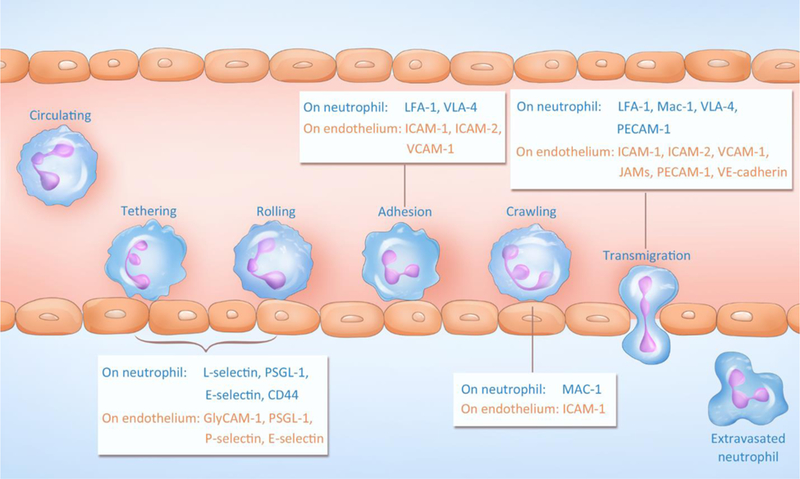
Adhesion and transmigration of neutrophils during inflammation. Adhesive molecules on both neutrophils and the endothelium are indicated. PSGL-1: P-selectin glycoprotein ligand-1; GlyCAM-1: Glycosylation-dependent cell adhesion molecule-1; LFA-1: Lymphocyte function-associated antigen-1; VLA-4: Integrin α4β1 (Very Late Antigen-4); ICAM-1: Intercellular Adhesion Molecule-1; ICAM-2: Intercellular Adhesion Molecule-2; VCAM-1: Vascular cell adhesion protein-1; Mac-1: Macrophage-1 antigen; PECAM-1: Platelet endothelial cell adhesion molecule-1; JAMs: Junctional adhesion molecules; VE-cadherin: vascular endothelial cadherin.
Neutrophil trafficking is specific and highly regulated by several membrane proteins and cytokine/chemokine gradients. Neutrophil recruitment is a multistep process that includes tethering, rolling, adhesion, crawling and transmigration.[4b] For instance, tumor necrosis factor-α (TNF-α) is a primary inflammatory stimulus that initiates the activation of both neutrophils and endothelia.[5] After activation, membrane adhesion proteins are upregulated on neutrophils and endothelial cells to facilitate the activation and arrest of neutrophils.[6] Endothelial cells increase their expression of adhesion molecules after they are activated by pattern-recognition receptors that detect pathogens. Pre-stored P-selectin in Weibel-Palade bodies is transported to the membrane of endothelial cells, and E-selectin is subsequently synthesized and upregulated. Both P-selectin and E-selectin bind to P-selectin glycoprotein ligand-1 (PSGL-1) on neutrophils; consequently, circulating neutrophils are tethered (captured) to the endothelium surface and roll along the vessel. The rolling of neutrophils facilitates further neutrophil activation via chemokines on the endothelium, and subsequently, integrins ((lymphocyte function-associated antigen-1 (LFA-1) and macrophage-1 antigen (MAC-1)) on neutrophils bind to intercellular adhesion molecule-1 or 2 (ICAM-1 or 2) expressed on the endothelium. After they are fully adherent, the neutrophils start to crawl toward a preferential site for transmigration under the guidance of a chemokine gradient. In the final step, transmigration occurs between endothelial junctions via the interaction of surface ligands, such as platelet endothelial cell adhesion molecule-1 (PECAM-1) and ICAM-1, or neutrophils penetrate through an individual endothelial cell. Once neutrophils leave the circulation, they move towards an infection site or injured tissue.[7]
Nanomedicine is the medical application of nanotechnology, especially in the area of targeted drug delivery, and it has revolutionized pharmacy and medicine. The concept of drug delivery with NPs involves incorporating drugs within a particle in the size range of a few hundred nanometers. The endothelium, a layer of cells encompassing the blood vessel, is a barrier to drug deposition in tissues. An enhanced permeability and retention (EPR) effect exists in tumor vasculature, and EPR is the basis for tumor NP targeting in mouse tumor models. [8] However, few NP-based tumor delivery platforms have been used in clinical settings.[9] While recent advances in nanotherapeutic engineering are promising for tumor targeting delivery, the tumor vasculature is still a barrier that limits the delivery of drugs into deep tumor tissues, particularly in solid tumors.[10] Novel concepts and technologies need to be developed to allow NPs to cross the blood vessel barrier for enhanced drug delivery into tumors.
Wang et al. recently observed that NPs made from denatured albumin protein (albumin NPs) efficiently delivered an anti-inflammation agent to inflamed neutrophils, therefore mitigating acute lung inflammation/injury.[11] The intravital images showed NP uptake by neutrophils (Figure 2A and 2B), and it is interesting to observe that the unconjugated dyes in albumin NPs diffuse in the cells, indicating that dye molecules can be released from albumin NPs. This result suggests that albumin NPs are loaded with a drug that can be delivered to neutrophils. In contrast, albumin proteins and NPs coated with natural albumin do not exist in neutrophils (Figure 2C and 2D). Quantitative analysis of NP uptake by neutrophils showed that 90–95% of neutrophils took up albumin NPs (Figure 2E), implying that neutrophil uptake of albumin NPs is specific and associated with the receptors expressed on neutrophils. Fcγ receptors are highly expressed on activated neutrophils and recycle denatured proteins in the body. When Fcγ RIII was knocked out in a mouse model, the uptake of albumin NPs by neutrophils dramatically decreased compared to their uptake in WT (wild type) mice. This result indicates that Fcγ RIII is required to mediate neutrophil uptake of albumin NPs.
Figure 2.
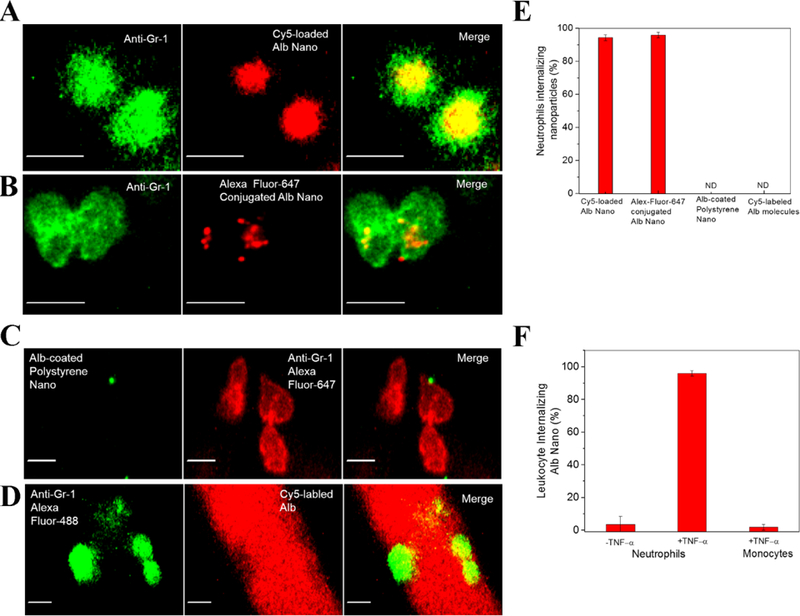
NP targeting of adherent neutrophils. (A) Cy5-loaded and (B) Alex Fluor 647-conjugated albumin NPs (red) were internalized by Gr-1-positive neutrophils (green). Scale bars, 10 µm. (C) Native albumin-conjugated polystyrene NPs (green) were bound to the neutrophil surface (red), and (D) Cy5-conjugated native albumin (red) was not internalized by Gr-1-positive neutrophils (green). Scale bars, 10 µm. (E) Quantitative analysis of percentage of Gr-1-positive neutrophils internalizing three types of NPs and Cy5-labeled albumin. (F) Quantitative analysis of uptake of Cy5-loaded and Cy5-conjugated albumin NPs was carried out by measuring fluorescence intensity per neutrophil. The results are shown as the mean ± s.e.m. (n = 13–20 vessels with three mice per group.) ND, not detected. Copyright 2014, Springer Nature.[11]
Activated neutrophils play a central role in inflammatory diseases; thus, targeting this population of neutrophils would be a novel strategy to treat inflammatory diseases. The authors examined whether albumin NPs specifically targeted activated neutrophils using intravital microscopy. The result showed that albumin NPs were not taken up by resting neutrophils or activated monocytes; however, 90% of activated neutrophils internalized albumin NPs (Figure 2F). When a mouse was administered albumin NPs loaded with spleen tyrosine kinase inhibitor, piceatannol, which blocks “outside-in” β2 integrin signaling in neutrophils, adherent neutrophils were detached, thus preventing neutrophil transmigration. Targeting inflammatory neutrophils using NPs offers a promising means to treat inflammatory diseases associated with neutrophil recruitment and vascular adhesion. This study demonstrates that NPs can specifically target activated neutrophils in situ and that NPs can possibly hijack neutrophils for drug delivery.
Transmigration of leukocytes (such as neutrophils) is a natural process from blood to tissue across the blood vessel barrier in response to pathogen invasion. The process is highly specific to disease sites; thus, neutrophils are natural carriers to transport drugs or nanotherapeutics and can improve current therapies if neutrophils can be hijacked in situ. As albumin NPs can specifically be internalized by activated neutrophils, it is possible that neutrophils can be employed as carriers to actively deliver nanotherapeutics across the blood vessel barrier, thus improving current therapies in inflammatory diseases and cancers.
Neutrophils are the first cell type to arrive at an inflammatory site. Monocytes/macrophages and T-lymphocytes usually appear in the late stage of acute inflammation (48 h later) or in chronic inflammation.[4b] Moreover, neutrophils are the most abundant cells during acute inflammation compared to monocytes/macrophages.[3] While other immune cells have been used as carriers for drug delivery,[12] this review is focused on recent progress in exploiting neutrophils for targeted drug delivery. In this review, we will discuss the status of neutrophil-based drug delivery systems and which disease models are used to test these drug delivery platforms (summarized in Figure 3 and Table 1). We will focus on three topics: (I) neutrophils as carriers to deliver nanotherapeutics across the blood vessel barrier; (II) neutrophil membrane-derived nanovesicles as a new drug delivery platform targeting inflamed vasculature; and (III) potential applications of neutrophil-based delivery systems in a wide range of diseases. We will also discuss the opportunities and challenges of neutrophil-based drug delivery systems.
Figure 3.
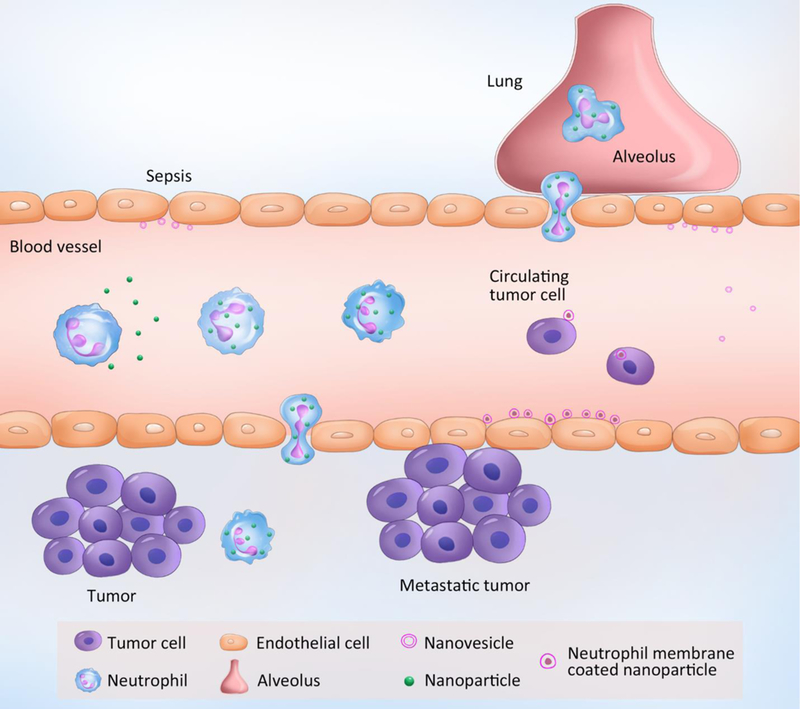
Neutrophil-mediated targeted delivery of nanotherapeutics and neutrophil membrane-derived vesicles for targeted drug delivery.
Table 1.
Neutrophils or neutrophil membrane-derived vesicles deliver therapeutics to different disease sites.
| NPs | Drug | Target | Disease model | Therapeutic methods |
Ref. |
|---|---|---|---|---|---|
| Anti-CD11b antibody- decorated polystyrene or gold NPs |
No | Activated neutrophils |
Lewis lung carcinoma with reactive oxygen species (ROS) induced-inflammation |
Photothermal Therapy (PTT) |
[13] |
| Denatured BSA NPs | Pyropheophor bide-a |
Activated neutrophils |
Melanoma with TA99 antibody induced- inflammation |
Photodynamic therapy (PDT) |
[14] |
| Denatured BSA NPs | TPCA-1 and cefoperazone acid |
Activated neutrophils |
LPS-induced lung inflammation and bacterial infection |
Anti-inflammation and antibacterial |
[15] |
| HL-60 cell membrane- formed nanovesicles |
TPCA-1 | Inflamed endothelium |
LPS-induced lung inflammation and bacterial infection |
Anti-inflammation and antibacterial |
[16] |
| Neutrophil membrane- derived nanovesicles |
Piceatannol | Inflamed endothelium |
LPS-induced lung inflammation and LPS-induced sepsis |
Anti-inflammation | [17] |
| Neutrophil membrane- coated NPs |
Carfilzomib | Circulating tumor cells (CTCs) and metastatic niche |
Circulating CTCs, early metastatic niche, and previously formed 4T1 lung metastasis |
Proteasome inhibitor (small molecule targeted therapy) |
[18] |
2. Neutrophils as Carriers in Targeted Drug Delivery
2.1. Targeting Tumor Microenvironments
Recently, Chu et al. proposed that the creation of acute inflammation in a tumor microenvironment promotes neutrophil infiltration to deliver nanotherapeutics. Photosensitizer pyropheophorbide-a (Ppa) is intravenously (i.v.) injected into a tumor-bearing mouse, and only the tumor tissue is exposed to light to generate acute inflammation because photosensitizers are excited by light and produce reactive oxygen species (ROS), leading to inflammation. Subsequently, neutrophils are activated to migrate into the tumor (Figure 4A).[13] CD11b antibodies are conjugated to the surface of NPs (size of 200 nm in diameter) for neutrophil targeting; neutrophils take up anti-CD11b-coated NPs because CD11b antigens are highly expressed on activated neutrophils.
Figure 4.
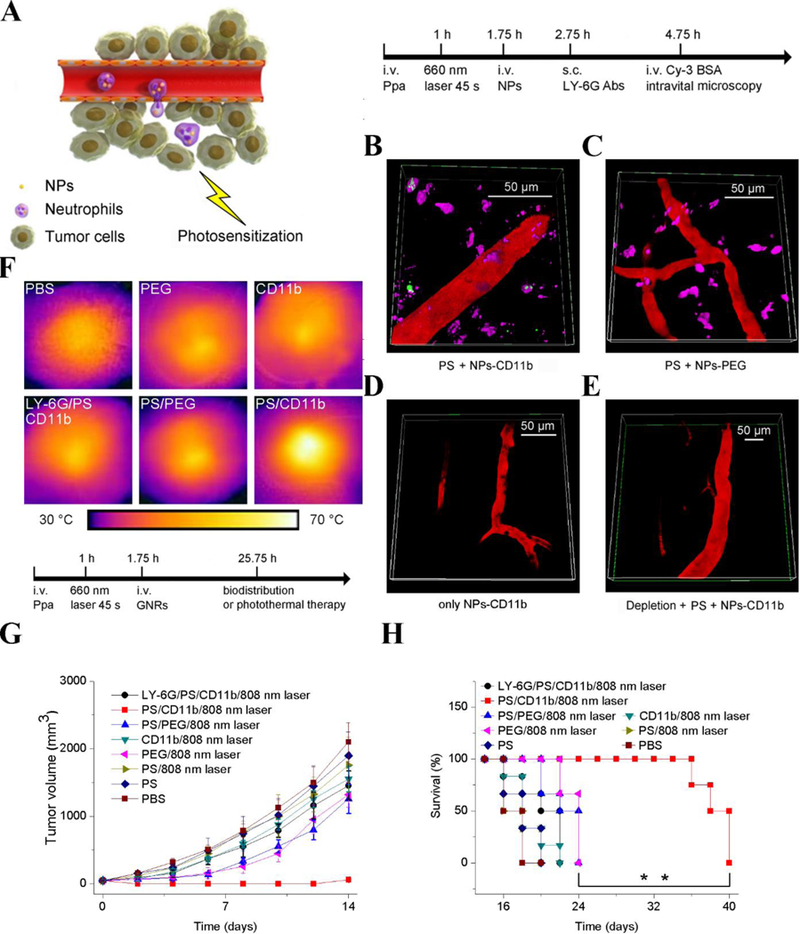
Neutrophil-mediated delivery of NPs to tumors across blood vessel walls. (A) The concept of neutrophil-mediated delivery of NPs to tumor tissues inflamed by photosensitization (PS). Intravital microscopic 3D images of mouse tumors treated with (B) both PS and NPs-CD11b (green), (C) PS and NPs-PEG (green), (D) NPs-CD11b only and (E) PS and NPs-CD11b after neutrophil depletion. The tumor was treated with PS (intravenous (i.v.) injection of pyropheophorbide-a (Ppa) and 660-nm laser). Approximately 0.75 h later, NPs were i.v. administered. LY-6G Abs (pink) was subcutaneously (s.c.) injected around the tumor to stain neutrophils. Cy3-BSA (red) was i.v. administered to visualize the blood vessel. Anti-LY-6G Abs were intraperitoneally (i.p.) injected 24 h before the administration of Ppa to deplete microphils. (F) Thermal graphic imaging of mouse tumors after the tumors were irradiated with a laser for 20 min. (G) Tumor size and (H) survival rate of the tumor-bearing mice. The mice were irradiated with an 808-nm laser after the injection of GNRs-PEG and GNRs-CD11b with or without PS (denoted as PS/PEG, PS/CD11b, PEG, and CD11b, respectively) or irradiated with an 808-nm laser after the treatment of both PS and GNRs-CD11b with neutrophil depletion (denoted as LY-6G/PS/CD11b). For PBS (without PS or laser irradiation at 808 nm), PS (with PS but without laser irradiation at 808 nm), and PS/808 nm laser (with both PS and laser irradiation at 808 nm), only PBS (pH 7.4) was injected. Data represent mean ± SD (n = 6 mice per group). Copyright 2017, Wiley.[13]
After tumor tissues were photosensitized (PS) at 660 nm, anti-CD11b-coated NPs (NPs-CD11b) were i.v. administered, and subsequently, Alexa fluor 647-labeled anti-LY-6G were subcutaneously injected. 3D images of a tumor microenvironment showed that neutrophils containing NPs-CD11b appeared outside of the blood vessel (Figure 4B). Several control experiments (with PS/PEG-coated NPs (NPs-PEG) and without PS/ NPs-CD11b) showed that PS and anti-CD11b are required for NP movement into tumor tissues (Figure 4C and 4D). Remarkably, NP transport was completely abolished when neutrophils were systemically depleted by an anti-LY6G antibody (Figure 4E). The amount of NPs was measured after the tumors were resected and homogenized to quantitatively analyze the tumor deposition of NPs. It was observed that there was 35 times more NPs-CD11b in tumors after PS than NPs-PEG (without targeting ligands). When neutrophils were systemically depleted, NPs-CD11b in tumors decreased to the level that occurred when NPs-PEG were used. Similarly, there was a very low level of NPs-CD11b in tumors without PS. The results show that neutrophil infiltration is the pathway to NPs-CD11b transport into tumor microenvironments.
PEG-, mouse IgG-, rat IgG- and rat anti-mouse CD11b Abs-decorated NPs were investigated to determine the specificity of NP neutrophil uptake.[13] Neutrophils were extracted from tumor tissues after the tumors were photosensitized, and they were analyzed by flow cytometry. The percentage of neutrophils internalizing NPs-CD11b was 10 times greater than that of neutrophils internalizing PEG-, rat IgG- and mouse IgG-coated NPs. Similarly, the mean fluorescence index (MFI) of NPs-CD11b was much higher than that of all other NPs, showing that anti-CD11b Abs are indispensable to mediating neutrophil uptake of NPs. Further studies show that NP uptake does not affect the activation and transmigration of neutrophils in vivo.[13]
Gold nanorods linked with anti-CD11b Abs were used as a model of therapeutics to translate this concept in cancer therapies. Gold nanorods can absorb infrared light to generate local heat due to surface plasmons produced by gold nanorods. Photothermal images showed that anti-CD11b-coated gold nanorods could produce very high local heat compared to the controls (Figure 4F). When NPs were administered to a tumor-bearing mouse, the combination of PS and photothermal therapies demonstrated that anti-CD11b-coated gold nanorods dramatically decreased tumor growth and increased survival in tumor-bearing mice (Figure 4G and 4H). This delivery system is promising for the treatment of solid tumors.
Manipulation of tumor microenvironments to activate neutrophil transmigration is a means to deliver nanotherapeutics via neutrophils. The previous study showed that administration of TA99 monoclonal antibodies could induce neutrophil infiltration via a mechanism of antibody dependent cellular cytotoxicity (ADCC).[19] Inspired by this finding, Chu et al. designed a strategy of combining TA99 and albumin NPs to treat melanoma in a mouse model.[14] The idea is that TA99 administration to a melanoma-bearing mouse activates the infiltration of neutrophils into tumors; subsequently, albumin NPs loaded with photodynamic therapy (PDT) agents were i.v. administered to target activated neutrophils for tumor delivery (Figure 5A). In the experiments, 48 h after administration of the TA99 antibody, the number of neutrophils in tumors significantly increased compared to the control group (Figure 5B and 5C). In addition, it was found that neutrophils internalized much more albumin NPs after the injection of TA99 than they did in the absence of TA99 (Figure 5D). Photosensitizers (Ppa) were loaded into albumin NPs to demonstrate the efficacy of drug-loaded albumin NPs for cancer therapy. PDT markedly inhibited tumor growth and increased survival compared to treatment with NPs or TA99 (Figure 5E and 5F). The study demonstrates a new avenue for the treatment of cancer with a combination of NPs targeting immune cells and ADCC. This strategy could be applied in combination with other immunotherapy approaches to induce neutrophil infiltration in tumors.
Figure 5.
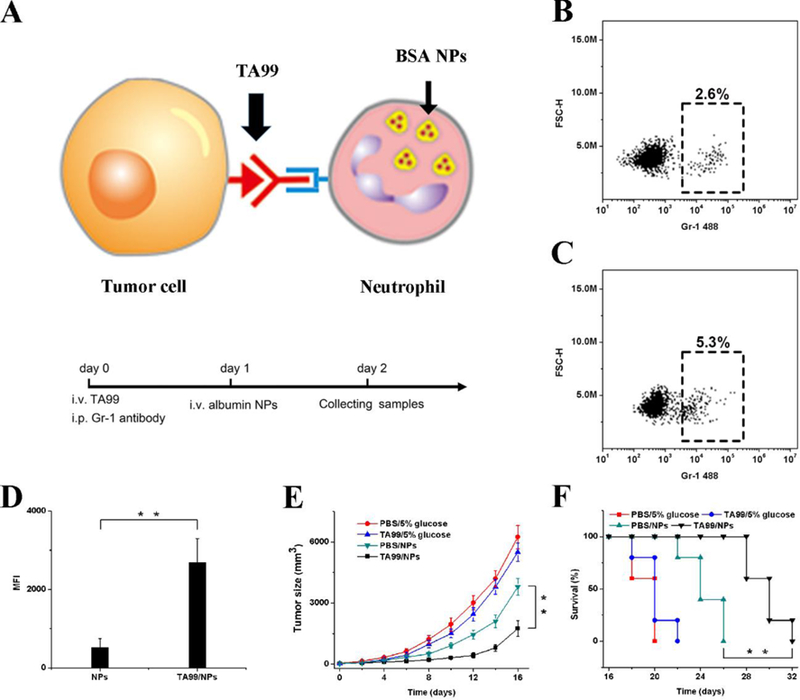
Neutrophils deliver NPs to tumors induced by antibody dependent cell-mediated cytotoxicity (ADCC). (A) Concept of NPs delivered by neutrophils to tumors mediated by ADCC. Percentage of neutrophils in tumor tissue with i.v. administration of (B) PBS and (C) TA99 at 48 h. The samples were single cell suspensions prepared from mouse tumors 48 h after treatment with PBS or TA99 in PBS. The cell suspension was analyzed by flow cytometry. (D) MFI of NPs in neutrophils in tumor by flow cytometry 48 h after the injection of Cy5-BSA NPs or both TA99 (i.v. injected at 24 h) and Cy5-BSA NPs (i.v. injected at 48 h) (n = 3). Neutrophils were stained with Alexa-488 anti-mouse Gr-1 antibody, and nucleus was stained by DAPI. (E) Tumor size and (F) survival rates of mice bearing melanoma illuminated with a 660-nm laser 48 h after i.v. injections of vehicles, TA99, Ppa-loaded NPs, or both TA99 (i.v. injected at 24 h) and Ppa-loaded NPs. Copyright 2016, Wiley.[14]
2.2. Targeting Lung Inflammation
Acute lung inflammation/injury (ALI) is a severe disease and is associated with dysregulated vascular inflammation. The cause of ALI is related to trauma and pathogen invasion, and ALI quickly develops acute respiratory distress syndrome (ARDS), which leads to injury of alveolar epithelium and vasculature. The estimated annual number of ALI/ARDS is approximately 200,000 adult patients in the United States with a mortality rate of 40%. ALI/ARDS is a big burden for the healthcare budget.[20] Because currently available drugs cannot efficiently target the lungs, a formally recommended pharmacological therapy is not available for ALI/ARDS.[20–21] In ALI, a large number of neutrophils migrate from the bloodstream into the lungs during the onset of ALI.[22] Therefore, exploiting neutrophils as carriers to deliver nanotherapeutics to the lungs is a means to improve the treatment of ALI/ARDS.
The lung has a unique feature of numerous tiny hollow cavities (called alveoli) surrounded by blood circulation.[23] The interface between blood and air is called the blood-air barrier (alveolar–capillary barrier), and it consists of the endothelium of capillaries, the epithelium of alveoli, and their shared basement membrane. When acute inflammation occurs in the airspace, neutrophils from the circulation accumulate in alveoli, where extravascular neutrophils can be analyzed after bronchoalveolar lavage fluid (BALF) is collected (Figure 6A). It has been shown that denatured bovine serum albumin (BSA) NPs specifically target activated neutrophils.[11] Albumin NPs were i.v. administered after a mouse was challenged with lipopolysaccharide (LPS) in the lungs, and lung neutrophils were examined by flow cytometry (Figure 6B and 6C) and confocal fluorescence microscopy (Figure 6D). The results showed that lung neutrophils took up albumin NPs and that the percentage increased with time. Further imaging of individual lung neutrophils confirmed the flow cytometry data. It is interesting to observe that albumin NPs in the lungs were not detected after neutrophils were depleted via anti-Gr-1 (Figure 6E), indicating that the transport of NPs is mediated by the transmigration of neutrophils. The uptake of albumin NPs did not affect neutrophil mobility or function in terms of the number of neutrophils and the concentrations of pro-inflammatory cytokines in BALF.[15]
Figure 6.
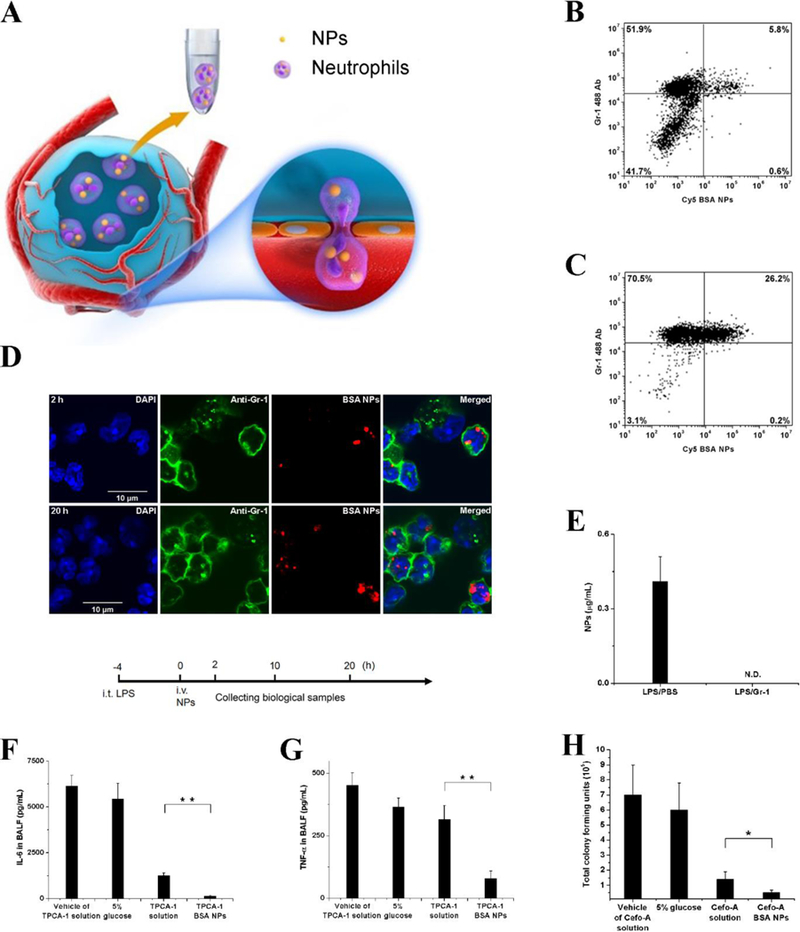
Neutrophil-mediated delivery of NPs to inflamed alveoli by crossing a blood-air barrier. (A) Schematic diagram of neutrophil-mediated delivery of NPs for the treatment of lung inflammation. Flow cytometry of bronchoalveolar lavage fluid (BALF) (B) 2 h and (C) 20 h after i.v. injection of Cy5-BSA NPs. (D) Fluorescence confocal microscopy of neutrophils from BALF 2 and 20 h after i.v. injection of Cy5-BSA NPs (red). Neutrophils were labeled with Gr-1 antibody (green). Nuclei were stained by DAPI (blue). (E) Cy5-BSA NPs in BALF with or without i.p. injection of Gr-1 antibody. Cy5-BSA NPs were i.v. injected 4 h after LPS challenge. Gr-1 antibodies were i.p. injected after LPS challenge. The samples were collected 20 h after the administration of NPs. NPs was administered in mice 4 h after LPS challenge (8 mg/kg). Concentrations of (F) IL-6 and (G) TNF-α in BALF 20 h after i.v. injection of vehicle of 2-[(Aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1) solution, 5% glucose, TPCA-1 solution or TPCA-1 BSA NPs in mice 4 h after LPS challenge (8 mg/kg). (H) Total colony forming units of P. aeruginosa in BALF after i.v. injection of 5% glucose, vehicle of cefoperazone acid (Cefo-A) solution, Cefo-A solution (25 mg/kg) and Cefo-A BSA NPs (25 mg/kg) in mice 12 h after P. aeruginosa infection. Samples were collected 12 h later. All data represent mean (SD) (3–4 mice per group). Copyright 2015, American Chemical Society.[15]
The authors established a mouse model of ALI induced by LPS or bacterial infection to demonstrate the usefulness of neutrophil-mediated delivery of albumin NPs. After i.v. injection of an anti-inflammation drug (2-[(Aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1)) loaded in albumin NPs to a mouse challenged by LPS in the lungs, lung inflammation was dramatically mitigated because of a decrease of cytokines (IL6 and TNF-a) (Figure 6F and 6G). The pathogenesis of ALI is highly associated with acute inflammation; therefore, decreased inflammation could alleviate ALI. P. aeruginosa is a major pathogen in lung infection; therefore, antibiotic-loaded albumin NPs were examined in a mouse model of P. aeruginosa infection. It was observed that albumin NPs dramatically prevent bacterial growth compared to free drugs (Figure 6H).[15] This research provided a new method to deliver therapeutics across air-blood barriers to treat lung inflammation and infection.
2.3. Design of NPs for Neutrophil Targeting
Neutrophils highly express several membrane proteins during acute inflammation as shown in Figure 1. Bio-functionalization of NPs is critical for developing novel drug delivery systems that specifically target activated neutrophils. For example, we have shown that conjugating anti-CD11b antibodies to polystyrene NPs or gold nanorods can facilitate uptake by activated neutrophils in situ and that neutrophils can mediate the transport of NPs in inflamed lungs or tumor microenvironments. Activated neutrophils can highly express Fcγ RIII, which functions to recycle denatured proteins (such as plasma proteins and antibodies) in the bloodstream. This mechanism is the natural process for macromolecule uptake. Recent publications inspired by this finding in nature discovered that albumin NPs made using organic solvents (such as ethanol) can specifically bind to Fcγ RIII receptors on activated neutrophils.[11] In addition, albumin NPs can be taken up by activated neutrophils in the bloodstream, and neutrophils mediate the delivery of nanotherapeutics in tumors and inflamed lungs.[14–15] These two examples demonstrate many opportunities for the rational design and engineering of NPs that target activated neutrophils in vivo for targeted drug delivery.
We have shown that NPs can deliver non-toxic agents (such as anti-inflammation agents, photosensitizers or gold NPs) via neutrophils into diseased sites. One should be cautious using neutrophils to deliver anti-cancer drugs to treat cancers. It is well known that paclitaxel and doxorubicin can significantly damage neutrophil functions, such as transmigration.[24] Even though these anti-cancer drugs are protected in NPs, it is difficult to prevent drug release after NPs are inside neutrophils. Therefore, rational engineering of NPs is very important for advancing novel neutrophil-based drug delivery systems to treat cancers and inflammatory disorders. The NPs release from neutrophils and uptake by targeted tissues also need to be considered when the delivery systems are designed, which have not been investigated yet by now.
3. Neutrophil Membrane-Derived Nanovesicles in Targeted Drug Delivery
3.1. Nitrogen Cavitation to Efficiently Generate Cell Membrane Nanovesicles
Biomimetic NPs are novel drug delivery platforms because they can avoid immune surveillance, resulting in targeting capabilities.[25] Cells in the body constantly liberate membrane vesicles to the extracellular space in the form of so-called extracellular vesicles (EVs).[26] EVs are a novel drug delivery platform, but it is difficult for current EV production techniques to meet clinical requirements, such as scalability, reproducibility, and safety.[27]
Novel approaches to efficiently produce EVs are needed. Studies [16–17, 28] have demonstrated that nitrogen cavitation is a novel means to generate EV-like nanovesicles. Nitrogen cavitation can generate mechanical forces in a chamber to rapidly disrupt cell membranes, and subsequently, the broken membrane spontaneously forms nanoscale vesicles.[29] A systemic approach has been established to generate cell membrane-derived nanovesicles (Figure 7A). The cryo-electron microscopy (cryo-TEM) image showed a vesicular structure with a wall thickness of 3–4 nm (Figure 7B).[17] The thickness of nanovesicles is similar to that of cell membrane, and an analysis of surface charges suggested that nanovesicles were directly formed from the cell membrane.[16] Further analysis of the proteomics of nanovesicles and their source cells indicated that the protein profiles of nanovesicles were similar to those of their parent cells, suggesting that nanovesicles possessed functions and features of cell membrane proteins (Figure 7C).[17]
Figure 7.
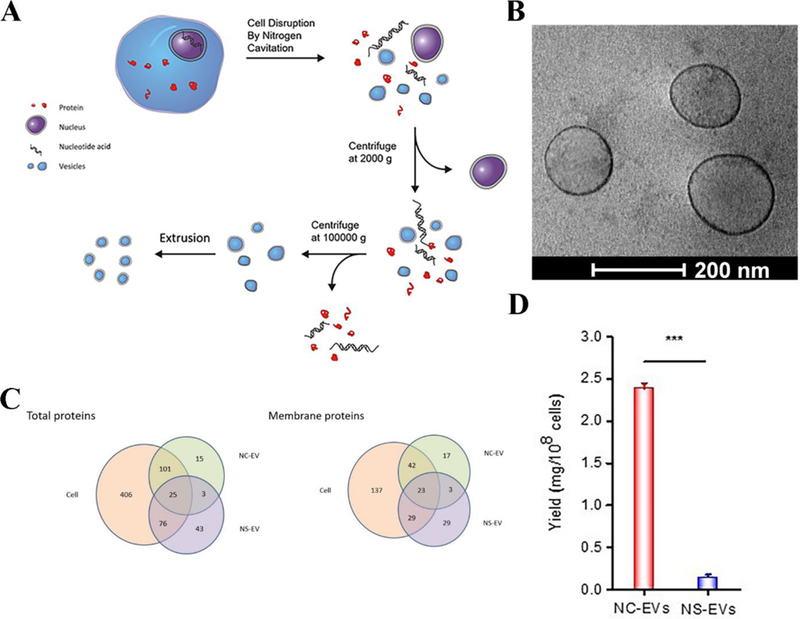
Preparation of cell membrane-derived nanovesicles by nitrogen cavitation. (A) The process to generate the nanovesicles via nitrogen cavitation, centrifugation and extrusion. Differentiated HL-60 cells were collected and disrupted by nitrogen cavitation under a pressure of approximately 350 psi, and membrane-formed vesicles, intracellular molecules, and the nucleus exist in the resulting solution. Afterwards, differential centrifugation was used to purify and obtain the needed vesicles. Copyright 2016, Elsevier.[16] (B) Cryo-TEM of cell membrane-formed vesicles, which are approximately 150 nm in diameter. (C) Proteomics of nanovesicles and their source cells. The total proteins (left) and membrane proteins (right) in nitrogen cavitation-generated EVs (NC-EVs), naturally secreted EVs (NS-EVs) and their parent cell analyzed by mass spectrometry. The shared proteins between EVs and their parent cells suggested that EVs were derived from their parent cells. (D) Quantification analysis of the yield of NC-EVs and NS-EVs indicated the reproductively of NC-EVs. Copyright 2017, Elsevier.[17]
The authors quantitatively measured the yield of nanovesicles to address the scalability of EV-like nanovesicles made using nitrogen cavitation.[17] The result showed that nitrogen cavitation can produce 16 times more nanovesicles higher than the conventional approach (secretion from cell culture medium) (Figure 7D). Nitrogen cavitation could reproducibly generate EV-like nanovesicles for clinical applications, unlike other physical and chemical approaches (homogenization, sonication, and cell lysis induced by reagents).[30] Nitrogen cavitation chambers can be scaled up to generate a great quantity of nanovesicles, but reproducibility is not lost with scale up because uniform forces are applied to cells and cell disruption occurs rapidly.[30] Nitrogen cavitation could be employed to produce nanovesicles from any cell type, except for neutrophils.
3.2. Neutrophil-Derived Nanovesicles Inhibit Acute Inflammation Induced by LPS
HL-60 cells are neutrophil-like cells that arise after differentiation induced by chemicals.[31] The binding of neutrophils to the endothelium occurs during ALI. Based on these intercellular interactions, it was proposed that nanovesicles made from neutrophil membranes could target inflamed lungs, thus increasing drug delivery. Nanovesicles were generated from HL-60 cells (HVs), and surface binding proteins (integrin β2) were analyzed on nanovesicles and their parent cells.[16] The results (Figure 8A) showed that nanovesicles highly expressed integrin β2, which is similar to their parent cells. Intravital microscopy of TNF-α-challenged cremaster venules showed that neutrophil-derived nanovesicles were adherent to the endothelium (Figure 8B and 8C). In the control, nanovesicles derived from red blood cells (without integrin β2) do not bind the activated endothelium.[16] An anti-inflammation agent, TPCA-1, was loaded into nanovesicles to examine whether nanovesicles could serve as drug carriers to deliver therapeutics to inflamed vasculature. In the LPS-induced acute lung inflammation model, neutrophil infiltration (Figure 8D) and the amount of cytokines TNF-α (Figure 8E) and IL-6 (Figure 8F) dramatically decreased after the administration of TPCA-1-nanovesicles (HV-TPCA-1) compared to free drugs and nanovesicles derived from red blood cells (non-targeted), indicating that neutrophil-derived nanovesicles can target inflamed lung vasculature, thus reversing acute lung inflammation. This work suggested that targeting blood vessels was a new method for treating inflamed tissue.
Figure 8.
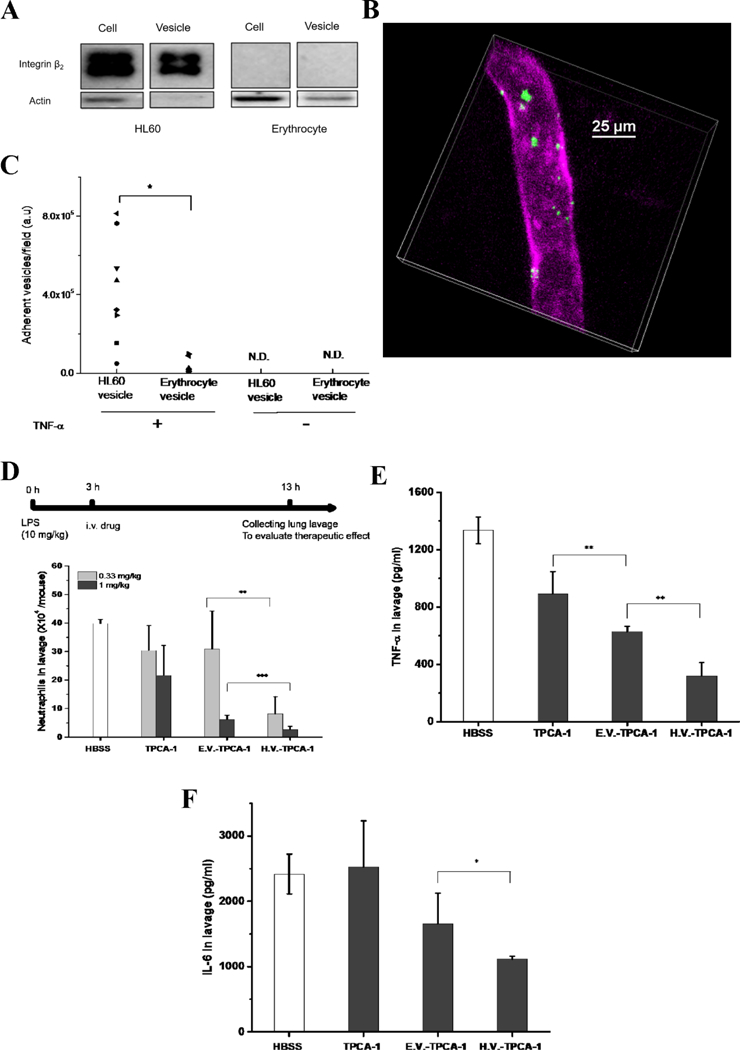
HL-60 cell membrane-formed nanovesicles target and treat inflammation. (A) Western blot of integrin β2 on HL-60 and erythrocyte cells and their nanovesicles. (B) Intravital images of the cremaster venule of a mouse treated with TNF-α and DiO-fluorescently labeled HVs (green). The endothelium was labeled by Alex Fluor-647-labeled anti-CD31 (pink) to indicate the blood vessel. Intravenously (i.v.) injected HV nanovesicles were found adherent to cremaster venules. (C) Quantification of the adherent HVs on cremaster venules based on intravital images. (n = 3). P<0.001. Numbers of neutrophils (D), TNF-α (E) and IL-6 (F) in BALF 10 h after injection of HBSS, erythrocyte vesicles-TPCA-1 (EV-TPCA-1) and HV-TPCA-1 in mice 3 h after LPS challenge. Compared with EV and free drug, HV showed a reduction of infiltrated neutrophils and amount of cytokines, which indicated the therapeutic efficacy of HV nanovesicles. *, **, and *** represent P<0.05, 0.01 and 0.001 in two-way t-test. Copyright 2016, Elsevier.[16]
3.3. Nanovesicles Target LPS-induced Sepsis
High concentrations of drugs must be loaded into preformed nitrogen cavitation-generated nanovesicles (NC-EVs) to increase therapeutic efficacy. In a recent study, a novel approach via a pH gradient was proposed to load drugs inside nanovesicles.[17] Nanovesicles derived from neutrophil-like cells (HL-60 cells) can be loaded with an anti-inflammation agent, piceatannol, via the pH gradient from pH = 7.4 outside of the nanovesicles to pH = 9.0 inside of the nanovesicles (Figure 9A).[17] Piceatannol loading increased 3-fold using this approach compared to the case without the pH gradient. Further studies showed that piceatannol-loaded nanovesicles (pic-NC-EVs) can inhibit the NF-κB pathway. When the nanovesicles were i.v. administered to a mouse challenged by LPS in the lung, neutrophil infiltration to the lungs was dramatically attenuated compared to administration of piceatannol alone (Figure 9B), implying that nanovesicles can enhance drug delivery in inflamed lungs. Furthermore, the nanovesicles were examined in a mouse model of sepsis. Sepsis is a severe disease associated with systemic inflammation induced by infections, such as bacterial infections. Systemic inflammation occurs, and vasculature highly expresses ICAM-1, which binds to integrin β2 on activated neutrophils.[32] In the LPS-induced mouse sepsis model by i.p. injection of LPS, pic-NC-EVs reduced systemic inflammation in the lung, liver and kidney because of the reduction of neutrophil infiltration (myeloperoxidase, MPO) (Figure 9C). This result might be associated with the increased survival (80%) observed after the administration of pic-NC-EVs (Figure 9D). The authors made mouse and human neutrophil-derived nanovesicles using nitrogen cavitation to address the translational possibility of this nanovesicle technology. Sepsis is a fatal condition, and neutrophil-derived nanovesicles represents a novel therapy for sepsis.
Figure 9.
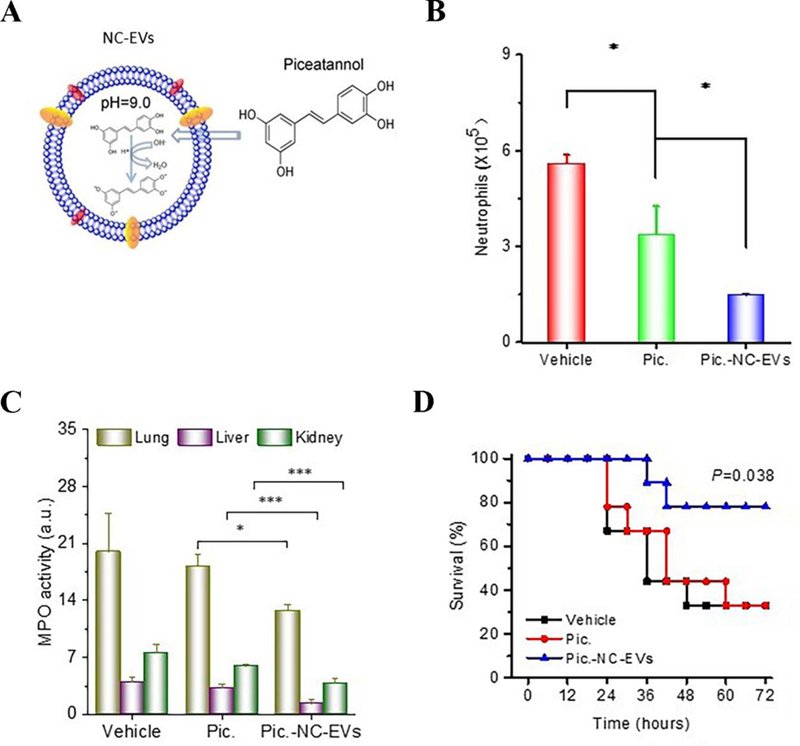
Drug-loaded neutrophil membrane vesicles for anti-inflammation therapy. (A) Schematic illustration of remote loading of piceatannol into cavitation-generated nanovesicles (NC-EVs) via pH gradient. (B) Neutrophil numbers in BALF 12 h after lung LPS challenge at a dose of 10 mg/kg. Vehicle, piceatannol (pic) (2 mg/kg) and pic-NC-EVs (pic at 2 mg/kg) were i.v. injected 2 h after LPS challenge. (C) MPO activity in mouse lung, liver and kidney 8 h after intraperitoneal (i.p.) LPS (22 mg/kg) challenge (LPS-induced sepsis model). Vehicle, piceatannol (3 mg/kg) and pic-NC-EVs (pic at 3 mg/kg) were i.v. injected 2 h after LPS challenge. The result showed that piceatannol-loaded NC-EVs decreased neutrophil tissue infiltration in all the three organs. (D) Survival rates after i.p. LPS (22 mg/kg) challenge (LPS-induced sepsis model). Vehicle, piceatannol (3 mg/kg) and pic-NC-EVs (pic at 3 mg/kg) were i.v. injected 2 h after i.p. LPS challenge. Piceatannol-loaded NC-EVs prevented death in 80% of mice. *, **, and *** represent P<0.05, 0.01 and 0.001 in two-way t-test. Copyright 2017, Elsevier.[17]
3.4. Nanovesicles Loaded with NPs Target Metastasis
When nanovesicles are translated into medical use, the challenge is how to effectively load therapeutic agents inside these nanovesicles. Polymeric NPs display effective encapsulation of drugs; therefore, inserting polymer NPs in nanovesicles would resolve the issue of low drug loading in nanovesicles.[33] A recent study reported that PLGA NPs are loaded in neutrophil membrane-derived nanovesicles, and this new formulation is capable of targeting circulation tumor cells (CTCs) and tumor vasculature (Figure 10).[18]
Figure 10.
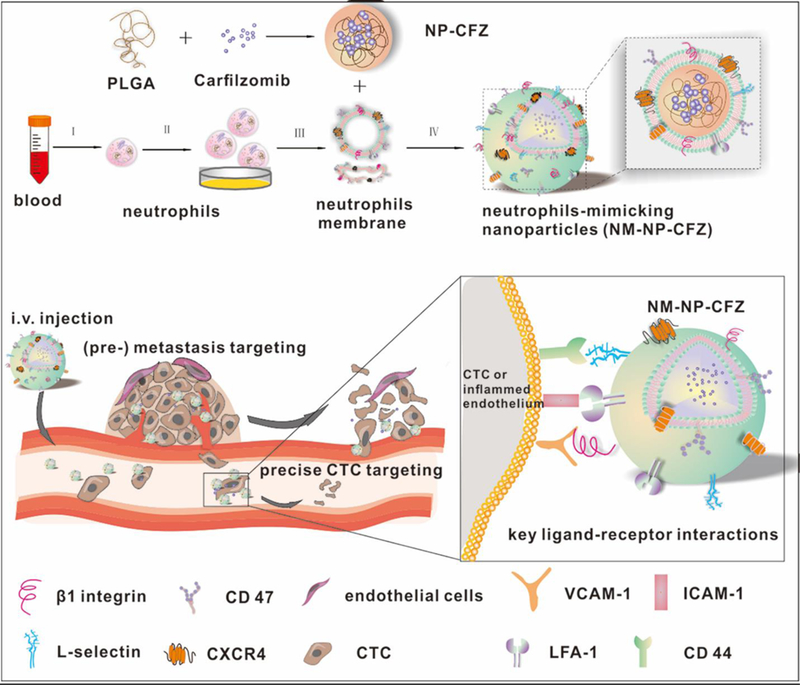
Scheme of Carfilzomib (CFZ), a second-generation proteasome inhibitor, loaded neutrophil-mimicking NPs (NM-NP-CFZ) that target CTCs and their site of colonization. Copyright 2017, American Chemical Society.[18]
In a tumor microenvironment, neutrophils interact with a metastatic niche and CTCs via intercellular adhesion molecules.[34]. Based on this finding, the authors made human neutrophil-derived nanovesicles and loaded polymeric NPs containing carfilzomib (CFZ), a second-generation proteasome inhibitor, inside nanovesicles (NM-NP-CFZ) to enhance the targeting of CTCs and tumor vasculature. The studies first demonstrated that NM-NP could bind with CTCs and home to the premetastatic endothelium in vitro, and NM-NP exhibited targeting ability and improved accumulation in the premetastatic niche in vivo. After nanovesicles were loaded with CFZ, their therapeutic effect was tested in three animal models. First, NM-NP-CFZ selectively eliminated CTCs in the circulation via the analysis of CTC apoptosis and leukocyte numbers (Figure 11A and 12B). In the second model, it was shown that NM-NP-CFZ prevented the formation of an early metastatic niche (Figure 11C). Finally, apoptosis was significantly improved (Figure 11D), and previously formed metastasis was inhibited by NM-NP-CFZ compared to other groups in the 4T1 lung metastasis model (Figure 11E). The neutrophil membrane-coated polymeric NPs represented a powerful approach as the delivery system using three different mechanisms to prevent tumor metastasis.
Figure 11.
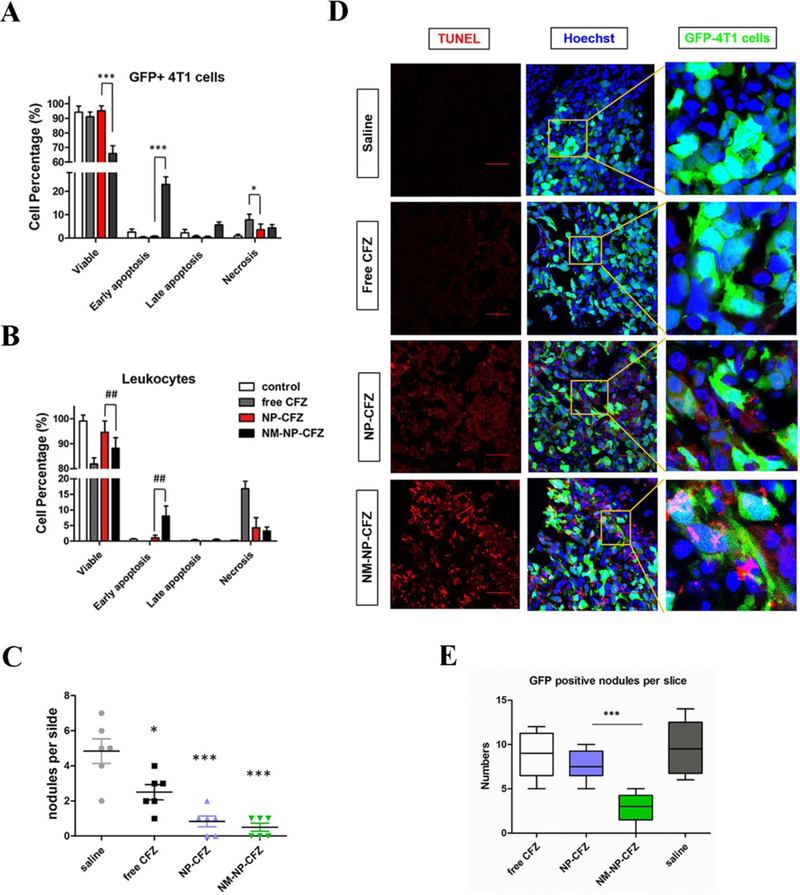
Neutrophil membrane-coated NPs treat cancer metastasis. Cell percentage of viable, early apoptotic, late apoptotic, and necrotic cells among GFP+ 4T1 cells (A) and leukocytes (B) in blood after treatment with free CFZ, NP-CFZ, and NM-NP-CFZ. (C) Quantification of metastasis nodules in lung tissue in the model of NM-NP-CFZ preventing the formation of early metastasis. Metastasis nodules were significantly reduced after treatment with NM-NP-CFZ compared to treatment with saline or free CFZ. (D) Imaging of in vivo apoptosis of successfully colonized GFP+ tumor cells in the lung by deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. 4T1 tumor metastasis models were established 16 days before TUNEL analysis. Apoptosis was significantly improved by NM-NP-CFZ compared to other groups in the previously formed 4T1 lung metastasis sites. Green indicated tumor cells, red indicated TUNEL labeling and nuclei were stained as blue. Scale bar is 100 μm. (E) Quantification of GFP+ nodules per slice in the model of NM-NP-CFZ inhibiting the development of previously formed lung metastasis. All data were represented as the mean ± SD. *** P<0.001, * P< 0.05 and ## P<0.01. Copyright 2017, American Chemical Society.[18]
4. Translational Potentials of Neutrophil-Based Drug Delivery Systems
4.1. Neutrophil-Mediated Pathogenesis and Disease
Neutrophils play a central role in preventing the spread of pathogens in the body through the release of cytokines/chemokines and ROS production.[35] On other hand, the inflammation induced by neutrophil infiltration also results in inflammatory diseases if it is unchecked and dysregulated. Due to an increased understanding of immunology in human immune systems, it is noted that many vascular diseases and inflammatory/autoimmune disorders are strongly linked to the functions and movement of neutrophils.[4] We will discuss several particular diseases in this section and show ways to treat them using neutrophil-based drug delivery systems.
Rheumatoid arthritis (RA).
Rheumatoid arthritis (RA), a chronic autoimmune disorder, primarily affects joints and results in swelling and pain in and around joints. Neutrophils play a role in the pathogenesis of joint destruction.[36] Activated neutrophils have been found in RA synovial fluid, synovial tissue and RA-associated skin disease. After their activation, neutrophils mediate damage to host tissues by degranulation, a process of releasing many signaling molecules, such as antimicrobial cytotoxic proteins in granules, ROS and proteases.[3, 37]
Ischemia-reperfusion injury (IRI).
Ischemia occurs due to blocked blood supply to organs, such as brain, heart, liver and kidney, resulting in injury and cell death.[38] Reperfusion of blood to ischemic tissues is a key surgical method in clinic treatments. However, reperfusion is always associated with the activation of inflammatory pathways, thus amplifying the secondary injury. This so-called ischemia-reperfusion injury (I/R) shares many characteristics with inflammatory responses. Activation and infiltration of neutrophils play central roles in acute inflammation during ischemia-reperfusion. I/R prompts the release of ROS, cytokines and other pro-inflammatory mediators that activate neutrophils. The recruitment of neutrophils is a process of transendothelial migration that may cause secondary injury during reperfusion, including endothelial dysfunction, microvascular collapse, blood flow defects, and cell apoptosis.[39]
Neurodegenerative disease.
The accumulation of neutrophils in the central nervous system (CNS) during the development of Alzheimer-associated diseases may cause chronic brain damage and inflammation, and neutrophil trafficking is regulated by LFA-1 integrin.[40] Neutrophil depletion or blockade of LFA-1 integrin in Alzheimer-associated mouse models reduced neuropathological features and recovered memory deficits, demonstrating a role of neutrophils in inducing cognitive dysfunction. Importantly, it was also found that the temporary inhibition of neutrophils during early stages of the disease had a beneficial long-term effect in older animals.[40] Neuroinflammation is also involved in Parkinson’s disease (PD), which is associated with decreased dopamine in the striatum and the loss of dopaminergic neurons in the substantia nigra.[41] The pathogenesis of PD is strongly related to the activation of neutrophils because blocking neutrophil activities reverses the development of PD.[42]
Atherosclerosis.
Neutrophils are observed in atherosclerotic lesions.[43] Studies have showed that the metabolism-dependent increase of neutrophils is associated with atherosclerosis development and closely correlated to the extension of early atherosclerosis formation under hyperlipidemic conditions.[44] Neutrophils participate in atherosclerosis via instructing other immune cells (such as monocytes) to enter atherosclerotic lesions.[45] Neutrophils release monocyte-attracting granule proteins, such as cathepsin G and cathelicidin, to recruit monocytes in atherosclerotic lesions.[46] Once monocytes have migrated into the arterial wall, they differentiate into macrophages and oxidize low density lipoproteins (LDL), thus damaging the arteries and accelerating the formation of atherosclerotic lesions.[47] ROS, which are released by either neutrophils or macrophages, can also oxidize LDL, thereby promoting foam cell formation.[45]
Pneumonia.
Pneumonia, an inflammatory condition in lung airways, is associated with the exuberant sequestration of peripheral neutrophils. The sequestration of neutrophils is regulated by cytokines produced by immune cells.[22] The major function of neutrophils is to kill microorganisms by producing ROS and antibacterial enzymes. After the death of neutrophils, neutrophils release their DNA and other antimicrobial granule proteins to form DNA-based neutrophil extracellular traps (NETs) that trap and kill microbial pathogens.[48] However, prolonged neutrophil activation and increased neutrophil survival may contribute to lung injury. Evidence shows that increased lung neutrophil apoptosis could resolve inflammation in pneumonia.[49] An effective pneumonia treatment requires the development of therapeutics that can attenuate excessive neutrophil accumulation while maintaining their critical role in antibacterial defense during bacterial pneumonia.[50]
Cancer.
In the 19th century, Rudolf Virchow first observed the presence of leukocytes within tumors. More studies indicate that the pathogenesis of cancer is strongly associated with inflammation.[51] It has been shown that inadequately resolved chronic inflammation may increase the risk of cancer.[52] Leukocytes, such as macrophages, neutrophils, T and B lymphocytes, dendritic cells, and natural killer cells, exist in tumor microenvironments. The infiltration of leukocytes is regulated by cytokines and chemokines produced by tumor cells.[53] In addition, inflammation might promote cancer metastasis.[34a]
There are two strategies to improve therapies for the diseases discussed above. Neutrophil infiltration is a natural process in the pathogenesis of these diseases; therefore, neutrophils can be hijacked to deliver various therapeutics to improve therapies. Second, most diseases are involved with inflammation that activates the vasculature, such as the lungs. Neutrophil membrane-derived nanovesicles may be a better choice to target the vasculature in these diseased tissues as neutrophils accelerate the progression of those kinds of diseases and the membrane vesicles do not have most of the biological functions of neutrophils.
4.2. Cancer Therapy-Mediated Acute Inflammation
When therapies are performed to treat cancers, the physical therapies and chemical/biological agents can cause activation of human immune systems, thus inducing acute inflammation. Acute inflammation is strongly correlated to neutrophil infiltration. We show some examples below.
Radiotherapy.
Radiotherapy is a major method to treat cancer patients and is also an interest of basic research.[54] There are two types of radiotherapies based on the method of radiation delivery. External radiotherapy refers to radiation delivered to tumors from outside of the body. Internal radiotherapy is associated with radioactive materials injected into the body, which accumulate in tumors to kill tumor cells. Radiotherapy is the process of high-energy radiation that kills tumor cells; however, radiation can damage healthy tissues adjacent to the tumors. Radiation therapies use gamma rays, X-rays and ultraviolet lights. The mechanism of radiotherapy for cancers is complex, but a simple mechanism is that radiation can damage double-stranded DNA molecules in the nucleus, thus preventing replication.[55] When DNA is damaged, the cells will initiate necrosis and apoptosis, which might activate immunity. Acute and chronic inflammation occur and are strongly linked to neutrophil infiltration.
Takeshima et al. observed a remarkable, but transient, infiltration of neutrophils into tumor microenvironments after focal radiation therapy in several tumor models, such as RN-9, 4T1 and EG7. Moreover, they discovered that the concurrent administration of granulocyte colony-stimulating factor (G-CSF) enhanced radiation therapy and mediated antitumor activity by further activating neutrophils.[56]
Photodynamic therapy (PDT).
Photodynamic therapy, abbreviated as PDT, is a type of phototreatment that involves a photosensitizer, light and tissue oxygen.[57] Photosensitizers are exposed to light to excite the electronic states of the photosensitizers. Subsequently, the triplet states of the photosensitizers react with oxygen existing in tissues, thus producing ROS that are highly toxic to cells.[58] Using this approach, tumors can be treated with light.[59] Although ROS can directly kill tumor cells using PDT, ROS also activates the immune system to recruit immune cells to tumor microenvironments.[57a, 60] For example, PDT-mediated neutrophil recruitment has been observed. [61] It is noted that the effect of PDT is also associated with neutrophil infiltration in tumors because the reduction of anti-cancer therapy by PDT was observed when neutrophils were systemically depleted after the administration of anti-Gr-1.[61c, 61d]
Antibody dependent cellular cytotoxicity (ADCC).
ADCC is an immune defense mechanism to kill tumor cells. After antibodies bind antigens expressed on the surface of tumor cells, immune effector cells can recognize the bound antibody via the Fc domain of antibodies. Subsequently, the immune effector cells bind tumor cells via antibodies.[62] The process of ADCC is dependent on the density of antibodies binding to tumor cells and the level of antigens expressed by tumor cells. After activation, immune effector cells release cytokines to attack tumors.[63] In a mouse melanoma model, the infiltration of neutrophils in tumors is strongly associated with the reduction of tumor burden after the injection of antitumor monoclonal antibody TA99. In contrast, neutropenic mice (lacking neutrophils) do not respond to TA99 therapy, indicating that neutrophils play a central role in ADCC.[19]
Cancer vaccines.
A novel strategy to treat cancers is harnessing the immune system via vaccination, and this approach has made a tremendous progress.[64] The ‘in situ vaccination’ immunotherapy strategy is a well-studied approach to treating cancer in which cancer vaccines are directly injected at a tumor site, generating a powerful immune response. Immune cells, such as neutrophils, dendritic cells and tissue-resident memory T cells, are involved, activating this immunomodulatory process.[65] Lizotte et al. have developed Cowpea Mosaic Virus (CPMV) as a vaccine for vaccination by direct injection of CPMV to tumors. They found that inhaled CPMV NPs were internalized by neutrophils and later activated neutrophils in the lung tumor microenvironment. Approximately 24 h after administration, they detected a marked increase of tumor-infiltrating neutrophils, particularly activated neutrophils.[65a] Cancer vaccination employing DCs has been widely investigated due to the abilities of DC cells to mediate innate and adaptive immunity. The major goal of DC vaccines is to activate tumor-specific CD8 T cells that can specifically shrink tumor size and can cause immunological memory to prevent tumor relapse. For subcutaneous vaccination, tumor-specific antigens were first provided to DC cells after the injection of an adjuvant that induces DC maturation and increases uptake, processing and presenting of antigens by DC cells and tumor-specific antigens.[66] When the growth of tumors was suppressed after vaccination, enhanced infiltration of neutrophils was also observed.[66e, 66f]
Combination of antibodies, checkpoint inhibitors, cancer vaccines and cytokines.
The elimination of aggressive tumors is a difficult challenge. A previous work showed that it is hard to elicit robust immunosuppressive tumor regression with single signaling checkpoints. Researchers at MIT recently found a promising method to overcome this problem by combining four biologic drugs: a tumor antigen specific antibody, a recombinant interleukin-2 (IL-2) with an extended half-life, anti-PD-1, and a powerful T cell vaccine. The therapeutic effect of the combination treatment was evaluated using different syngeneic tumor models: B16F10 melanoma, DD-Her2/neu (mouse breast cancer), TC-1 tumors (human HPV) and a genetically engineered mouse model of melanoma. Combinations of the three components and each component treatment alone showed modest therapeutic responses to large and advanced tumors. However, the four-component cancer immunotherapy led to robust and durable tumor regression in all tumor models. This four-component therapy can activate both the innate and adaptive immune system, thus effectively destroying tumors. Regarding the innate immune response, the number of neutrophils in tumors increased significantly, and neutrophil depletion caused significant reductions in overall survival rates. The authors noted that neutrophils can contribute to direct antibody dependent cytotoxicity against tumors and secrete inflammatory cytokines, which improved the activation and recruitment of additional immune cells.[67]
Based on the above discussions, we know that various cancer treatments can cause acute inflammation, involved in neutrophil infiltration in tumors and the activation of tumorous vasculature. Therefore, neutrophil-mediated NP delivery or neutrophil membrane-derived nanovesicles may deliver anti-cancer drugs into tumor microenvironments. As mentioned above, release of toxic drugs should be prevented after NPs are inside neutrophils. Other NPs could also be investigated after they are decorated with ligands targeting the inflamed vasculature in the tumor after the treatments discussed above, which can cause acute inflammation.
5. Conclusions and Challenges
In summary, we report the current status of targeted drug delivery based on the neutrophil infiltration pathway or via neutrophil membrane-derived nanovesicles and demonstrate that novel neutrophil-based technologies can improve current therapies for inflammatory disorders and cancers. Neutrophils are naturally involved in the pathogenesis of inflammatory/autoimmune diseases, and current nanotechnologies can design and generate NPs that specifically target activated neutrophils. Furthermore, neutrophils are the most abundant WBCs in humans, thus targeting activated neutrophils in situ to mediate the delivery of nanotherapeutics demonstrates a new paradigm in a wide range of therapies in inflammatory/autoimmune disorders, such as acute lung injury, RA, stroke, atherosclerosis, Alzheimer’s and PD. Most importantly, current cancer therapies (such as radiotherapies and PDT) spontaneously cause immune responses that activate infiltration of neutrophil into tumors. This effect provides many opportunities to develop strategies to target infiltrated neutrophils using nanotechnology. Indeed, in this review, we have shown several examples in which nanotherapeutics are delivered to tumor tissues via neutrophil infiltration, such as PDT-mediated delivery of gold nanorods for photothermal therapy and TA99-mediated delivery of albumin NPs. Based on the intercellular binding between neutrophils and the vasculature in inflammation, neutrophil membrane-formed nanovesicles can act as drug carriers with vasculature targeting to improve therapies for ALI, sepsis and cancer metastasis. Neutrophil membrane-formed nanovesicles may also be employed to treat various diseases as mentioned above.
While neutrophil-based drug delivery systems have shown potential in nanomedicines, many interesting questions and challenges still need to be addressed. When will NPs be delivered for better targeting of infiltrated neutrophils? The time course of neutrophil infiltration is strongly dependent on the pathogenesis of the disease and its stages. Studies on the dynamics of neutrophil activation and trafficking during acute inflammation are needed for better delivery of nanotherapeutics in disease sites. We have observed that most NPs accumulate in the liver, so how do we shift NP trafficking to targeted sites (tumors or inflamed tissues)? Rational design of NPs and increased targeting specificity to neutrophils might be interesting areas to explore, for example, conjugating antibodies to the NP surface to increase NP uptake in activated neutrophils. Chemotaxis and tissue transmigration of neutrophils are critical for delivering drugs after nanotherapeutics are internalized by neutrophils. Choosing drugs and carriers that would not affect the functions of neutrophils is the most important task to accomplish delivery to diseased sites via neutrophils. If the drugs reduce neutrophil transmigration, smart delivery systems might be considered in which drugs loaded into NPs do not release in neutrophils before they arrive at disease sites. For example, a design of responsive drug release NPs might control drugs loaded in NPs so that they are not released in neutrophils because neutrophil intracellular environments protect the disassembly of NPs. This design is challenging, but advances in engineering pH/enzyme responsive NPs might offer hope in delivering toxic agents via neutrophils. Since neutrophils contribute to the progression of some types of disease, the benefit/damage balance should be considered when neutrophils are used as carriers to deliver drugs.
In the case of neutrophil membrane-derived nanovesicles for drug delivery, drug loading efficiency and drug release profiles must be considered because it is challenging to load large amounts of drugs into nanovesicles after they are produced. It was also found that most neutrophil membrane-derived nanovesicles accumulated in the liver after systemic injection. Neutrophil membrane-derived vesicles could be made of gene-engineered neutrophils expressing much higher antigens than activated neutrophils alone to improve their targeting abilities. Furthermore, reproducibility and scalability are required in nanovesicle generation for clinical translation.
Therefore, new nanomaterials must be developed and novel strategies and tools must be created to address the challenges of neutrophil-based drug delivery systems. In summary, the neutrophil-based drug delivery platform would have a great impact on nanomedicine.
Acknowledgments
This work was supported by National Institute of Health grant RO1GM116823 to Z.W.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
Contributor Information
Dafeng Chu, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Spokane, Washington 99210, United States.
Xinyue Dong, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Spokane, Washington 99210, United States.
Xutong Shi, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Spokane, Washington 99210, United States.
Canyang Zhang, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Spokane, Washington 99210, United States.
Zhenjia Wang, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Spokane, Washington 99210, United States.
References
- [1].Fullerton JN, Gilroy DW, Nat Rev Drug Discov 2016, 15, 551. [DOI] [PubMed] [Google Scholar]
- [2].Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER, Trends Immunol 2010, 31, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wright HL, Moots RJ, Bucknall RC, Edwards SW, Rheumatology (Oxford) 2010, 49, 1618. [DOI] [PubMed] [Google Scholar]
- [4].Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, Benarafa C, Roos D, Skokowa J, Hartl D, PLoS Pathog 2015, 11, e1004651; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P, Nat Rev Immunol 2013, 13, 159; [DOI] [PubMed] [Google Scholar]
- Pham CT, Nat Rev Immunol 2006, 6, 541. [DOI] [PubMed] [Google Scholar]
- [5].Williams MR, Azcutia V, Newton G, Alcaide P, Luscinskas FW, Trends Immunol 2011, 32, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zarbock A, Ley K, McEver RP, Hidalgo A, Blood 2011, 118, 6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Phillipson M, Kubes P, Nat Med 2011, 17, 1381; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S, Nat Rev Immunol 2007, 7, 678. [DOI] [PubMed] [Google Scholar]
- [8].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Nature Reviews Materials 2016, 1.
- [9].Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST, Adv Drug Deliv Rev 2017, 108, 25. [DOI] [PubMed] [Google Scholar]
- [10].Bae YH, Park K, Journal of Controlled Release 2011, 153, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang Z, Li J, Cho J, Malik AB, Nat Nanotechnol 2014, 9, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dong X, Chu D, Wang Z, Theranostics 2017, 7, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chu D, Dong X, Zhao Q, Gu J, Wang Z, Adv Mater 2017, 29. [DOI] [PMC free article] [PubMed]
- [14].Chu D, Zhao Q, Yu J, Zhang F, Zhang H, Wang Z, Adv Healthc Mater 2016, 5, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chu D, Gao J, Wang Z, ACS Nano 2015, 9, 11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gao J, Chu D, Wang Z, J Control Release 2016, 224, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gao J, Wang S, Wang Z, Biomaterials 2017, 135, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kang T, Zhu Q, Wei D, Feng J, Yao J, Jiang T, Song Q, Wei X, Chen H, Gao X, Chen J, ACS Nano 2017, 11, 1397. [DOI] [PubMed] [Google Scholar]
- [19].Albanesi M, Mancardi DA, Jonsson F, Iannascoli B, Fiette L, Di Santo JP, Lowell CA, Bruhns P, Blood 2013, 122, 3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Matthay MA, Zemans RL, Annu Rev Pathol 2011, 6, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Matthay MA, Ware LB, Zimmerman GA, J Clin Invest 2012, 122, 2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Craig A, Mai J, Cai S, Jeyaseelan S, Infect Immun 2009, 77, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, McFadden G, Kubes P, Cell Host Microbe 2013, 13, 169. [DOI] [PubMed] [Google Scholar]
- [24].Mendonca MA, Cunha FQ, Murta EF, Tavares-Murta BM, Cancer Chemother Pharmacol 2006, 57, 663; [DOI] [PubMed] [Google Scholar]
- b) MacFadden DK, Saito S, Pruzanski W, J Clin Oncol 1985, 3, 415. [DOI] [PubMed] [Google Scholar]
- [25].Tan S, Wu T, Zhang D, Zhang Z, Theranostics 2015, 5, 863; [DOI] [PMC free article] [PubMed] [Google Scholar]
- b) Sawdon A, Peng CA, Ther Deliv 2013, 4, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Budnik V, Ruiz-Canada C, Wendler F, Nat Rev Neurosci 2016, 17, 160; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minciacchi VR, Freeman MR, Di Vizio D, Semin Cell Dev Biol 2015, 40, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gimona M, Pachler K, Laner-Plamberger S, Schallmoser K, Rohde E, Int J Mol Sci 2017, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gottlieb RA, Adachi S, Methods Enzymol 2000, 322, 213. [DOI] [PubMed] [Google Scholar]
- [29].Simpson RJ, Cold Spring Harb Protoc 2010, 2010, pdb.prot5513. [DOI] [PubMed]
- [30].Del Buono BJ, Luscinskas FW, Simons ER, J Cell Physiol 1989, 141, 636. [DOI] [PubMed] [Google Scholar]
- [31].Martin SJ, Bradley JG, Cotter TG, Clin Exp Immunol 1990, 79, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG, Annu Rev Pathol 2011, 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L, Proc Natl Acad Sci U S A 2011, 108, 10980; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Sun W, Qian C, Wang C, Bomba HN, Gu Z, Adv Mater 2015, 27, 7043; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L, Nature 2015, 526, 118; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang RH, Hu CM, Luk BT, Gao W, Copp JA, Tai Y, O’Connor DE, Zhang L, Nano Lett 2014, 14, 2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wculek SK, Malanchi I, Nature 2015, 528, 413; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJM, Ciampricotti M, Hawinkels L, Jonkers J, de Visser KE, Nature 2015, 522, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mayadas TN, Cullere X, Lowell CA, Annu Rev Pathol 2014, 9, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kaplan MJ, Arthritis Res Ther 2013, 15, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wright HL, Moots RJ, Edwards SW, Nat Rev Rheumatol 2014, 10, 593. [DOI] [PubMed] [Google Scholar]
- [38].Schofield ZV, Woodruff TM, Halai R, Wu MC, Cooper MA, Shock 2013, 40, 463. [DOI] [PubMed] [Google Scholar]
- [39].Jordan JE, Zhao ZQ, Vinten-Johansen J, Cardiovasc Res 1999, 43, 860. [DOI] [PubMed] [Google Scholar]
- [40].Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S, Montresor A, Carlucci T, Nani S, Tosadori G, Calciano L, Catalucci D, Berton G, Bonetti B, Constantin G, Nat Med 2015, 21, 880. [DOI] [PubMed] [Google Scholar]
- [41].Qian L, Flood PM, Hong JS, J Neural Transm (Vienna) 2010, 117, 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang F, Qian L, Flood PM, Shi JS, Hong JS, Gao HM, J Pharmacol Exp Ther 2010, 333, 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Soehnlein O, Circ Res 2012, 110, 875. [DOI] [PubMed] [Google Scholar]
- [44].Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O, Circulation 2010, 122, 1837. [DOI] [PubMed] [Google Scholar]
- [45].Hartwig H, Silvestre Roig C, Daemen M, Lutgens E, Soehnlein O, Hamostaseologie 2015, 35, 121. [DOI] [PubMed] [Google Scholar]
- [46].Soehnlein O, Lindbom L, Weber C, Blood 2009, 114, 4613. [DOI] [PubMed] [Google Scholar]
- [47].Bondjers G, Olofsson SO, Fager A, Wiklund O, Acta Med Scand Suppl 1986, 712, 10. [DOI] [PubMed] [Google Scholar]
- [48].Bordon J, Aliberti S, Fernandez-Botran R, Uriarte SM, Rane MJ, Duvvuri P, Peyrani P, Morlacchi LC, Blasi F, Ramirez JA, Int J Infect Dis 2013, 17, e76. [DOI] [PubMed] [Google Scholar]
- [49].Moret I, Lorenzo MJ, Sarria B, Cases E, Morcillo E, Perpina M, Molina JM, Menendez R, Eur Respir J 2011, 38, 1158. [DOI] [PubMed] [Google Scholar]
- [50].Balamayooran G, Batra S, Fessler MB, Happel KI, Jeyaseelan S, Am J Respir Cell Mol Biol 2010, 43, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Grivennikov SI, Greten FR, Karin M, Cell 2010, 140, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA, J Immunol Res 2014, 2014, 149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Balkwill FR, Mantovani A, Semin Cancer Biol 2012, 22, 33. [DOI] [PubMed] [Google Scholar]
- [54].Gregoire V, Langendijk JA, Nuyts S, J Clin Oncol 2015, 33, 3277; [DOI] [PubMed] [Google Scholar]
- Bartelink H, Roelofsen F, Eschwege F, Rougier P, Bosset JF, Gonzalez DG, Peiffert D, van Glabbeke M, Pierart M, J Clin Oncol 1997, 15, 2040. [DOI] [PubMed] [Google Scholar]
- [55].Formenti SC, Demaria S, J Natl Cancer Inst 2013, 105, 256; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichselbaum RR, Liang H, Deng L, Fu YX, Nat Rev Clin Oncol 2017, 14, 365; [DOI] [PubMed] [Google Scholar]
- De Meerleer G, Khoo V, Escudier B, Joniau S, Bossi A, Ost P, Briganti A, Fonteyne V, Van Vulpen M, Lumen N, Spahn M, Mareel M, Lancet Oncol 2014, 15, e170; [DOI] [PubMed] [Google Scholar]
- Schaue D, McBride WH, Nat Rev Clin Oncol 2015, 12, 527; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Chen Y, Liang C, Yi X, Liu J, Sun X, Shen S, Yang K, Liu Z, Adv Mater 2016, 28, 7143; [DOI] [PubMed] [Google Scholar]
- Zhang XD, Luo Z, Chen J, Shen X, Song S, Sun Y, Fan S, Fan F, Leong DT, Xie J, Adv Mater 2014, 26, 4565; [DOI] [PubMed] [Google Scholar]
- Santivasi WL, Xia F, Antioxid Redox Signal 2014, 21, 251. [DOI] [PubMed] [Google Scholar]
- [56].Takeshima T, Pop LM, Laine A, Iyengar P, Vitetta ES, Hannan R, Proc Natl Acad Sci U S A 2016, 113, 11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Castano AP, Mroz P, Hamblin MR, Nat Rev Cancer 2006, 6, 535; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Song J, Nie L, Chen X, Chem Soc Rev 2016, 45, 6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Fan W, Huang P, Chen X, Chem Soc Rev 2016, 45, 6488. [DOI] [PubMed] [Google Scholar]
- [59].Dolmans DE, Fukumura D, Jain RK, Nat Rev Cancer 2003, 3, 380. [DOI] [PubMed] [Google Scholar]
- [60].Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q, J Natl Cancer Inst 1998, 90, 889; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechet D, Mordon SR, Guillemin F, Barberi-Heyob MA, Cancer Treat Rev 2014, 40, 229. [DOI] [PubMed] [Google Scholar]
- [61].Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J, CA Cancer J Clin 2011, 61, 250; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbelik M, Cecic I, Cancer Lett 1999, 137, 91; [DOI] [PubMed] [Google Scholar]
- de Vree WJ, Essers MC, Koster JF, Sluiter W, Cancer Res 1997, 57, 2555; [PubMed] [Google Scholar]
- de Vree WJ, Essers MC, de Bruijn HS, Star WM, Koster JF, Sluiter W, Cancer Res 1996, 56, 2908; [PubMed] [Google Scholar]
- Gollnick SO, Evans SS, Baumann H, Owczarczak B, Maier P, Vaughan L, Wang WC, Unger E, Henderson BW, Br J Cancer 2003, 88, 1772; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Mironov O, Foster TH, Theranostics 2012, 2, 840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rouse BT, Grewal AS, Babiuk LA, Nature 1977, 266, 456. [DOI] [PubMed] [Google Scholar]
- [63].Greenberg AH, Hudson L, Shen L, Roitt IM, Nat New Biol 1973, 242, 111; [DOI] [PubMed] [Google Scholar]
- Koren HS, Anderson SJ, Larrick JW, Nature 1979, 279, 328; [DOI] [PubMed] [Google Scholar]
- Bar RS, Kahn CR, Koren HS, Nature 1977, 265, 632; [DOI] [PubMed] [Google Scholar]
- Herberman RR, Ortaldo JR, Bonnard GD, Nature 1979, 277, 221; [DOI] [PubMed] [Google Scholar]
- Weiner GJ, Nat Rev Cancer 2015, 15, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Umar A, Dunn BK, Greenwald P, Nat Rev Cancer 2012, 12, 835. [DOI] [PubMed] [Google Scholar]
- [65].Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S, Nat Nanotechnol 2016, 11, 295; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, Marcheteau E, Fabre E, Mandavit M, Terme M, Blanc C, Escudie JB, Gibault L, Barthes FLP, Granier C, Ferreira LCS, Badoual C, Johannes L, Tartour E, Nat Commun 2017, 8, 15221; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencherif SA, Warren Sands R, Ali OA, Li WA, Lewin SA, Braschler TM, Shih TY, Verbeke CS, Bhatta D, Dranoff G, Mooney DJ, Nat Commun 2015, 6, 7556; [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ, Nat Rev Cancer 2016, 16, 219. [DOI] [PubMed] [Google Scholar]
- [66].Palucka K, Banchereau J, Nat Rev Cancer 2012, 12, 265; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana F, Shahbazi MA, Liu D, Zhang H, Makila E, Salonen J, Hirvonen JT, Santos HA, Adv Mater 2017, 29; [DOI] [PubMed] [Google Scholar]
- Luo Z, Wu Q, Yang C, Wang H, He T, Wang Y, Wang Z, Chen H, Li X, Gong C, Yang Z, Adv Mater 2017, 29; [DOI] [PubMed] [Google Scholar]
- Wang C, Ye Y, Hu Q, A. Bellotti, Gu Z, Adv Mater 2017, 29; [DOI] [PubMed] [Google Scholar]
- Huo M, Zhao Y, Satterlee AB, Wang Y, Xu Y, Huang L, J Control Release 2017, 245, 81; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Huo M, Xu Z, Wang Y, Huang L, Biomaterials 2015, 68, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, Kumari S, Kelly RL, Kwan BH, Abraham W, Hu K, Mehta NK, Kauke MJ, Suh H, Cochran JR, Lauffenburger DA, Wittrup KD, Irvine DJ, Nat Med 2016, 22, 1402. [DOI] [PMC free article] [PubMed] [Google Scholar]