

Abstract
Reconstructions of vascular plant mitochondrial genomes (mt-genomes) are notoriously complicated by rampant recombination that has resulted in comparatively few plant mt-genomes being available. The dearth of plant mitochondrial resources has limited our understanding of mt-genome structural diversity, complex patterns of RNA editing, and the origins of novel mt-genome elements. Here, we use an efficient long read (PacBio) iterative assembly pipeline to generate mt-genome assemblies for Leucaena trichandra (Leguminosae: Caesalpinioideae: mimosoid clade), providing the first assessment of non-papilionoid legume mt-genome content and structure to date. The efficiency of the assembly approach facilitated the exploration of alternative structures that are common place among plant mitochondrial genomes. A compact version (729 kbp) of the recovered assemblies was used to investigate sources of mt-genome size variation among legumes and mt-genome sequence similarity to the legume associated root holoparasite Lophophytum. The genome and an associated suite of transcriptome data from select species of Leucaena permitted an in-depth exploration of RNA editing in a diverse clade of closely related species that includes hybrid lineages. RNA editing in the allotetraploid, Leucaena leucocephala, is consistent with co-option of nearly equal maternal and paternal C-to-U edit components, generating novel combinations of RNA edited sites. A preliminary investigation of L. leucocephala C-to-U edit frequencies identified the potential for a hybrid to generate unique pools of alleles from parental variation through edit frequencies shared with one parental lineage, those intermediate between parents, and transgressive patterns.
Keywords: Caesalpinoideae, mimosoid clade, iterative assembler, PacBio, transgressive, hybrid
Introduction
Mitochondrial genomes (mt-genomes) are known to be highly reduced in terms of genic content when compared with their alphaproteobacterial ancestors. The transfer of mitochondrial genes to the nucleus is well-known across eukaryotes, where gene content can vary slightly even among closely related taxa (Kubo and Newton 2008). In addition, the sizes of mt-genomes have been shown to vary over 100-fold across eukaryotes (Wu, Cuthbert, et al. 2015), wherein the land plants display some of the largest and most variable assemblies (Gualberto and Newton 2017). Major differences in mt-genome size are attributed to noncoding sequences, as variation in the total number of genes and their combined lengths account for a limited amount of the observed inter genomic variation. Acquisition of noncoding sequences is due in part to horizontal DNA transfer between other organelles and exogenous sources (Plitmann 1993; Bergthorsson et al. 2003; Mower et al. 2010; Warren et al. 2016), but a large portion of noncoding sequences are not conserved between closely related species. Thus, their origins often remain unclear.
Variation in plant mt-genome structure is also widespread, even among members of the same species. This is mostly due to the presence of dispersed repeats that contribute to extensive homologous recombination (Stern and Palmer 1984; Alverson et al. 2010; Gualberto and Newton 2017). During the early days of mt-genome analysis, plants were believed to have circular mt-genomes like their metazoan counterparts. Continued work involving microscopy and related techniques in plant mt-DNAs recovered mostly linear molecules of varying size, with most all much larger than the standard ∼16 kbp metazoan mitochondrial genomes that had been characterized at the time (Ward et al. 1981). Restriction endonuclease analyses then gave way to circular fragment maps, which showed that the mt-genomes of plants and fungi were highly variable in size, but circular mapping (Sparks and Dale 1980; Palmer and Shields 1984; Palmer and Herbo 1987; Oda et al. 1992). The linear fragments observed previously were deemed artifacts of extraction and thus ignored whereas the circular model of mt-genome structure became, and has remained, predominant in the literature. Interestingly, later studies utilizing pulsed-field gel electrophoresis showed that linear rather than circular molecules were the dominant structure in mt-genomes of plants and fungi, and that these molecules can vary greatly in length within a single cell, existing as mt-DNA “chromosomes” (Bendich 1993; Jacobs et al. 1996; Sloan 2013). The size and number of these chromosomes can vary across species within a single genus (Sloan, Alverson, et al. 2012), within individuals (Sloan et al. 2012), and is further complicated by the presence of autonomous DNA elements of unknown function (Wu, Cuthbert, et al. 2015; Warren et al. 2016).
Mitochondrial genomes and their genetics are further complicated by extensive C-to-U RNA editing that forms, at least in part, a corrective mechanism for overcoming potentially deleterious mutations (e.g., Maier et al. 2008; Chateigner-Boutin and Small 2010; Ichinose and Sugita 2017). The gene families and mechanisms behind different classes of RNA edits vary between major lineages of eukaryotes and even between genomic compartments within lineages (e.g., Grice and Degnan 2015; Moreira et al. 2016; Yang et al. 2017). The nuclear encoded pentatrichopeptide repeat (PPR) gene family that is primarily responsible for these RNA-edits in land plant mitochondrial genetics has experienced massive expansion in the vascular plants, where it represents the largest gene family found in several angiosperm genomes to date (e.g., Lurin 2004; Fujii and Small 2011). Additional investigations of mitochondrial RNA editing among closely related species of angiosperms are needed to better understand the degree of variation in RNA editing between recently diverged lineages as well as the impact of interspecific hybridization on patterns of editing. Such variation has been posited to provide potentially important variation contributing to adaptation and population-level divergence (Gommans et al. 2009).
The combination of land plant mt-genome structural variation and their RNA-editing features illustrate several important features associated with the complex nature of these genomes and their transcriptomes. However, these dynamic genomes remain among the least well surveyed eukaryotic organellar genomic systems (e.g., Richardson et al. 2013). Within the economically and ecologically important legume plant family there are currently seven mt-genomes available for comparative studies (table 1), but these are all representatives of subfamily Papilionoideae. Even within this phylogenetically restricted set of representatives, there is considerable mt-genome variation, including a lack of recombinationally active dispersed repeats in Vigna (Alverson et al. 2011), which contrasts with what is known in Glycine (Chang et al. 2013) and the majority of flowering plants. Given limited representation of legume mt-genomes, it is likely that considerably more variation is present across the family. Additional genomes are needed to assess important elements of mitochondrial genetics, like RNA editing.
Table 1.
Mitochondrial Genome Characteristics for Exemplar Taxa
| Lineage | Species | Accessions | Genome Size (bp) | Unique Protein Coding Genes | Unique tRNA Genes | Unique rRNA Genes | Percent GC |
|---|---|---|---|---|---|---|---|
| Mimosoid member of Caesalpinoideae | Leucaena trichandra | MH717173/MH717174 | 729,504 | 36 | 16 | 3 | 44.90 |
| Papilionoid legumes | Glycine max | NC_020455.1 | 402,558 | 36 | 19 | 3 | 45.03 |
| Lotus japonicus | NC_016743.2 | 380,861 | 31 | 20 | 3 | 45.40 | |
| Medicago truncatula | NC_029641.1 | 271,618 | 31 | 16 | 3 | 45.39 | |
| Millettia pinnata | NC_016742.1 | 425,718 | 33 | 22 | 3 | 45.00 | |
| Vicia faba | KC189947 | 588,000 | 32 | 17 | 3 | 45.03 | |
| Vigna angularis | NC_021092.1 | 404,466 | 32 | 16 | 3 | 45.19 | |
| Vigna radiata | NC_015121.1 | 401,262 | 31 | 16 | 3 | 45.11 | |
| Other angiosperm representatives | Arabidopsis thaliana | NC_001284.2 | 366,924 | 32 | 22 | 3 | 44.77 |
| Beta vulgaris | NC_002511.2 | 368,799 | 29 | 25 | 3 | 43.86 | |
| Curcurbita pepo | NC_014050.1 | 982,833 | 38 | 13 | 3 | 42.80 | |
| Geranium maderense | NC_027000.1 | 737,091 | 31 | 27 | 3 | 42.32 | |
| Gossypium hirsutum | NC_027406.1 | 668,584 | 35 | 29 | 3 | 44.98 | |
| Malus × domestica | NC_018554.1 | 396,947 | 32 | 17 | 3 | 45.4 | |
| Oryza sativa | NC_011033.1 | 490,520 | 35 | 17 | 3 | 43.85 | |
| Vitis vinifera | NC_012119.1 | 773,279 | 37 | 20 | 3 | 44.14 |
Perhaps the most significant impediment to the development of mitochondrial genomic resources in land plants are dispersed repeat systems (e.g., Alverson et al. 2010) that lead to frequent intragenomic recombination and structural rearrangements within and among mt-genomes. This variation often foiled traditional PCR-based investigations of mt-genomes as well as modern short-read high-throughput sequencing studies for genome assembly (Ogihara et al. 2005; Cahill et al. 2010; Iorizzo et al. 2012). Such issues create major headaches for mt-genome assembly, resulting in many vascular plant nuclear genomes being published without their mitochondrial counterparts, further slowing progress toward a comprehensive understanding of vascular plant mitochondrial genetics (Richardson et al. 2013).
Here, we use a single molecule long-read (amenable to Pac-Bio or Nanopore) iterative assembly pipeline to assess genome content and structure for a member of the species-rich mimosoid lineage of subfamily Caesalpinioideae (LPWG 2017), Leucaena trichandra. The genus Leucaena comprises 25 species of trees primarily distributed in seasonally dry tropical forests in Mexico, Central America, and northern South America (Hughes 1998b). Leucaena includes species used as minor food plants in south-central Mexico since pre-Colombian times (Hughes et al. 2007), as well as one species, Leucaenaleucocephala (an allotetraploid), which is pantropically cultivated as a forage and multipurpose tree that has escaped cultivation to become a devastating tropical woody invasive (Hughes 1998a; Lowe et al. 2000).
We use a compact version of the mt-genome assemblies generated herein to further characterize legume mt-genome content and structure as well as sequence shared with the legume associated root holoparasite Lophophytum. We then use the genome and a suite of complementary data to characterize patterns of RNA editing across the genus, including patterns associated with the allopolyploid L. leucocephala.
Materials and Methods
Sampling, DNA Extraction, and Sequencing
High molecular weight DNA (>20 kbp) was extracted using a modified Aquagenomic DNA extraction protocol (MultiTarget Pharmaceuticals, LLC). For each extraction 10 mg of fresh young leaf material was obtained from a L. trichandra sapling (seed accession 53/88/09) (Hughes 1998a) that had been kept in the dark for 24 h to reduce polysaccharide concentration. Tissues were homogenized in 250 µl of Aquagenomic buffer plus 4 µl of 100 mg/ml RNAse A, incubated at 65 °C for 10 min, and then centrifuged for 5 min at 21,000 × g. 200 µl of supernatant was recovered and one volume (200 µl) of 2 M NaCl was added and gently mixed by inversion. Two volumes (800 µl) of 70% ethanol (rather than the recommended isopropanol) were added and gently mixed by inversion followed by a 5 min incubation on ice and 3 min spin at 21,000 × g. The DNA pellet was washed three times with 500 µl 70% ethanol, then air dried for 10 min and re-suspended in 50 µl of 10 mM Tris, pH 8.5. 19 µg of DNA with an average fragment size of 21 kbp was submitted for sequencing. Libraries (PacBio P6-C4 chemistry) and sequencing reactions were carried out by the University of Delaware DNA Sequencing & Genotyping Center.
Genome Assembly
Mitochondrial genome assembly followed an iterative approach that begins with the assembly of highly conserved regions and extends from that starting point. This protocol involves: (1) mapping reads to a reference genome(s) to identify a subset of raw reads that likely belong to the novel mitochondrial genome of interest, (2) assembling those reads de novo, and (3) repeating the process using the new draft genome derived from the prior round as the subsequent reference to recruit additional reads that belong to the novel genome, resulting in extension of the genome (fig. 1). Each round of assembly ends with the de novo reconstruction of the genome from recruited reads so that the reference structure does not influence the assembly. The pipeline involved using BLASR (Chaisson and Tesler 2012) to map raw reads against the reference, filtering hits by a minimum aligned length (500 bp), recovering the qualifying reads to a new fastq file using seqtk (version 1.0-r31 from Github), and assembling reads with CANU (version1.3 + 101 commits, Koren et al. 2016). In this way, novel reads that overlap with the ends of the prior de novo assembly are added, extending the assembly in each round. The process was repeated until the number of contigs recovered and the total size of the genome stabilized for at least three cycles. For this study, the starting reference genome was a multi-fasta file containing soybean and cotton (table 1) mitochondrial genomes (see “Results and Discussion” section for the final parameters used). A copy of the code applied for these assemblies is available on github (https://github.com/cdb3ny/Mitochondrial-Genome-Scripts; last accessed August 28, 2018). Blobtools represents another option for filtering mapped reads that one could apply (https://github.com/DRL/blobtools; last accessed August 28, 2018). The project primarily employed an AMD7252 32 core server with 256GB of RAM.
Fig. 1.
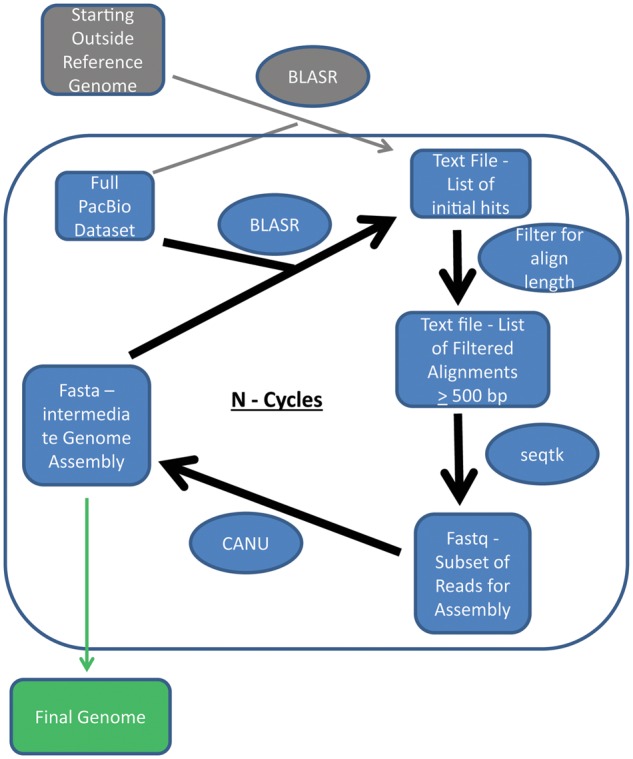
—An overview of the assembly pipeline. The upper gray symbols and lines indicate the starting pass alignment that uses an outside reference to identify conserved mitochondrial sequence from the complete PacBio data set. The central cyclical portion of the figure (inside the blue box) continues until the size of the genome and contig number in the assembly stabilizes through multiple cycles resulting in the recovered of the draft genome.
Circularization, Genome Polishing, and Annotation
CANU did not indicate that any of the assemblies were likely to be circular in nature. To check this, we next applied manual approaches to try to circularize contigs, but failed to identify appropriate regions of overlap or reads spanning the ends. Finally, we used Circlator (Hunt et al. 2015) to investigate the potential to (1) trim and circularize, (2) extend and circularize, or (3) merge and then circularize mitochondrial contigs recovered in each assembly (Hunt et al. 2015). The final assembly was polished with Pilon (version 1.20) (Walker et al. 2014) by mapping 71,065,617 (100 bp Illumina) reads (available in SRA305491) to the mt-genome assembly with bowtie2 (version 2.2.6) (Langmead and Salzberg 2012) using default parameters. Annotated regions were initially identified using the program Mitofy (Alverson et al. 2010). To detect genes that may not be recovered by Mitofy, we also transferred annotations from the Glycine (NC_020455.1), Vigna (NC_021092.1), and Carica (NC_012116) mt-genomes using a 70% minimum match and the “Find Annotations” and associated default options in Geneious version 6.1.6 (Kearse et al. 2012). Open reading frames (ORFs) for CDS regions and tRNA boundaries were verified/identified using Geneious (Kearse et al. 2012) (standard genetic code) and tRNA-scan (Lowe and Eddy 1997), respectively. The genome was further scanned for regions with unusually high sequence coverage. These regions were subject to default blastn queries against the nucleotide database in GenBank and the L. trichandra plastome (KT428297, Dugas et al. 2015) to identify other novel regions worthy of note or annotation.
Repeat Analysis
The characterization of mononucleotide, tandem, and dispersed repeats followed prior work (Dugas et al. 2015). In short, we applied the Tandem Repeat Finder online interface (Benson 1999), an in-house script for mononucleotide strings ≥8 bp (https://github.com/cdb3ny/Mitochondrial-Genome-Scripts; last accessed August 28, 2018), and a self-blastn-on-blastn approach for dispersed repeats.
Characterization of Conserved Regions and Expressed Regions Outside of Annotated Genes
Intergenic regions of interest were characterized by identifying conservation to other angiosperm mitochondrial genomes as well as expression of intergenic sequences. First, intergenic sequences were extracted by generating a multi-fasta file of all regions in the genome not including CDS, tRNA, and rRNA. These sequences were queried with blastn against the angiosperm database in NCBI GenBank (taxid: 3398) with default options and optimized for highly similar sequences (megablast). This returned a hit table containing “conserved regions.” Intergenic sequences were also probed for ORFs >300 bp in length. These ORFs were then analyzed for sequence conservation by using blastn (version 2.2.31) against the same angiosperm database used on the intergenic sequence multi-fasta file. Expression of these ORFs was quantified using the TopHat2 (version 2.1.0) (Kim et al. 2013)/cufflinks version v2.2.1. (Trapnell et al. 2010) pipeline. ORFs were determined to be of interest if >=90% of their length aligned with another angiosperm sequence and was expressed at RPKM > 1.
Shared mitochondrial–nuclear regions within L. trichandra were inferred using a blastn search with our draft nuclear genome contigs (Bailey C. Donovan et al. unpublished data) as queries and the mt-genome as the subject (options perc_identity = 70, max_target_seqs 2–word_size 100). Blast hit-table output was then used to determine the lengths of mitochondrial–nuclear shared regions in the nucleus as well as in the mt-genome.
RNA Editing
RNA editing was assessed empirically through a combination of additional RNA-seq and DNA-seq data resources. RNA-seq data generated from three biological replicates per sample (three whole seedlings harvested at the third-leaf stage of development) that were combined, extracted, sequenced, and mapped to the L. trichandra mt-genome. Each of these samples was subjected to two-independent library preps and sequencing runs (technical replicates). These RNA-seq data (SRP103307) were developed from divergent Leucaena species to represent at least one member of each well-supported clade (genome group) within the genus (Govindarajulu, Hughes, et al. 2011), including L. cruziana (Oxford Forestry Institute seedlot 43/85/16), L. cuspidata (83/94), L. esculenta (47/87/01), L. pulverulenta (84/87/05), and L. trichandra (4/91/15), as well as the allotetraploid hybrid of L. cruziana and L. pulverulenta, L. leucocephala (80/92/02) (Govindarajulu, Hughes, Alexander, et al. 2011). RNA was extracted using the Norgen Plant RNA/DNA Purification Kit (Norgen Corp., Toronto, CA) and treated with plant rRNA depletion reagents (courtesy of New England Biolabs) prior to library construction using the NEBNext Ultra II RNA First Strand Synthesis Module (#E7771), Ultra II Directional RNA Second Strand Synthesis Module (#E7550), Ultra II DNA Library Prep Kit for Illumina (#E7645), and Multiplex Oligos for Illumina (Index Primers Sets 1 & 2) (#E7335 and #E7500). Pooled libraries were sequenced on an Illumina NextSeq 500 (2 × 150 paired-end), and the untrimmed reads are available via the NCBI SRA project PRJNA379675. Raw reads were trimmed using Trimmomatic (Bolger et al. 2014) version 0.32 (with options—ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10 LEADING:25 TRAILING:25 SLIDINGWINDOW:5:25 MINLEN:65).
At least 20 million 65–150 bp total reads (paired and unpaired) per sample passed the trimming step and were mapped to the reference L. trichandra mt-genome with Tophat2 version 2.1.0. In deciding on the final parameters to map the transcriptomes to the genome, an important issue arose. We initially used default Bowtie2 mapping, which has been applied in some RNA editing work. Given that the cumulative percentage of RNA edits across plant mitochondria genomes is low (e.g., <0.2%), using default Bowtie, which maps reads with at most two subsitutions/100 bp (≤2% divergence) by default, seemed sufficient. However, careful inspection of our initial results revealed that the number of reads with multiple edits per 100 bp was underestimated. This is a consequence of the clustered distribution of RNA edits in CDS regions. Some regions in the L. trichandra mt-genome contain as many as 6 edits per 100 bp (6% divergence), resulting in partially edited transcripts (e.g., only those with ≤2 substitutions per 100 bp read) being mapped whereas fully edited reads (>2 substitutions per 100 bp read) were not mapped. We ultimately selected default settings plus “-N 10—read-edit-dist 10—library-type fr-firststrand” to solve this issue. Strand specific edits were recovered using a combination of “samtools–view” options (to generate the appropriate BAM files) and the “Find Variants/SNPs” function in Geneious to retrieve the characteristics of potential edits (both the type and frequency). Edits were retained if they occurred in both replicates/sample with a minimum of 50× coverage and variant frequency ≥20%.
To minimize the potential influence of underlying genotypic variation, we mapped 2 × 100 bp Illumina reads derived from gDNA libraries representing one species from mitochondrial representatives of each genome group (Govindarajulu, Hughes, et al. 2011) to the L. trichandra mt-genome to identify genomic SNPs [genome groups 1, 2, and 3 were represented by L. trichandra {SRA305491}, L pueblana {PRJNA379675}, and L. leucocephala {PRJNA379675}, respectively]. These resulted in a minimum mean sequence coverage of 90× and inferred genomic SNP positions were eliminated from consideration as potential RNA edits.
Frequency of Individual RNA Edits
For the allotetraploid L. leucocephala and its diploid parents L. cruziana and L. pulverulenta, we also investigated variation in the frequency of the edited nucleotide (in the RNA-seq data), versus the genomic nucleotide (based on the genome sequence), among transcripts on a site-by-site basis to identify whether the frequency of editing per site in the hybrid polyploid followed a maternal, paternal, intermediate, or transgressive pattern. The proportion of genomic and edited nucleotides at each site was compared via contingency table analysis of likelihood ratios. This resulted in 291 comparisons in the forward strand and 339 in the reverse strand. To adjust for multiple comparisons, we set the alpha value for significance by dividing 0.05 by the number of comparisons (Bonferroni correction). For example, for the forward strand alpha equaled 0.05 divided by 291, or 0.0002, and only comparisons which yielded P < 0.0002 were deemed significant. Nonetheless, because of the high level of sequence coverage, a large number of comparisons were significant, but likely trivial in nature. For each position with a significant outcome we then calculated the percentage deviation from expected {[(observed=-expected)/expected] * 100} for each value; only those that deviated at least 10% from one another (e.g., the observed variant frequency for L. leucocephala was at least 10% different from that of either parent) were considered further. Each of these positions was categorized into one of five groups that assigned the allotetraploid variant frequency to one of the following patterns: (1) maternal, (2) paternal, (3) intermediate, (4) transgressive increase, or (5) transgressive decrease.
Results and Discussion
Genome Assembly and Genome Content
The L. trichandra PacBio reads included >60× nuclear genome coverage with an N50 of 14.5 kbp (Bailey, C. Donovan et al., unpublished data), providing sufficient long read data to also assemble the mitochondrial genome. Nonetheless, when we identified likely mt-genome contigs recovered from assemblies derived from all the available reads (which includes mitochondrial, nuclear, and plastid data in large computationally intensive analyses), the mitochondrial portion was moderately fragmented (>7 contigs). This led us to the idea of an assembler that aims to isolate mitochondrial reads (or any target) from read data derived from multiple genomes (e.g., a mixture of nuclear, plastid, and mitochondrial reads or even many genomes in a metagenomic study) and iteratively assemble them from an initial starting point (see “Materials and Methods” section) (Ortega-Rodriguez and Bailey 2016). We reasoned such an approach could be useful when the sequence data includes multiple genomes, for example organellar and/or metagenomics assemblies.
Preliminary testing of this strategy to develop a L. trichandra mt-genome involved a wide range of parameters for sampled reads [including data sets ranging from all raw reads down to smaller randomly selected sets and multiple genome size estimates {400–2,000 kbp}]. From the resulting assemblies we identified mitochondrial fragments in each by shared sequence coverage and annotated mitochondrial genes found in the recovered set of contigs. Through this we concluded that unique mitochondrial elements comprised about 700–850 kbp of DNA, and that the 500 bp minimum aligned length (from the BLASR step) provided consistent results. Subsequently, we conducted a series of assemblies that included different samples of reads (i.e., random samples), different numbers of reads, and number of raw reads and genome size estimates focused around 800 kbp (table 2). The assembly generated using 2 M raw reads and an 800 kbp estimated genome size provided the most compact representation of the genome (721 kbp) and is the only assembly configuration to resolve to a single mitochondrial contig. Remapping the PacBio reads to this assembly resulted in 48× continuous coverage, and 90–100× coverage across most regions. The portions of the 721 kbp assembly recovered in the alternative assemblies are presented in table 2 and supplementary figure S1, Supplementary Material online.
Table 2.
Assembly Pipeline Parameters and Results Applied to the Final Assemblies
| Raw Reads | Genome Size Parameter | Mt-Genome Contigs Recovered | Inferred mt-Genome Size (bp) | Other Contigs Assembled | Cycles to Stabilize |
|---|---|---|---|---|---|
| 2M | 800K | 1 | 721,986 | 2 cpDNA | 3 |
| All—4,623,744 | 800K | 3 | 776,020 | 3 cpDNA | 4 |
| 2M | 2,000K | 4 | 826,820 | 4 cpDNA | 3 |
| 1M | 1,000k | 2 | 741,430 | 1 cpDNA | 4 |
| 1M | 800K | 1 | 741,421 | 1 cpDNA | 4 |
| 500K | 800K | 4 | 751,403 | 2 cpDNA | 5 |
| 250K | 800K | 4 | 681,477 | 1 cpDNA | 5 |
Note.—Raw reads refer to the number of randomly selected PacBio reads used in the assembly.
M, million; K, thousand.
Circlator merged two mitochondrial contigs into larger mitochondrial scaffolds in two of the assemblies, the one that included all PacBio reads and the one that included 250 K reads (table 2), but it failed to circularize those merged molecules, or any of the single mitochondrial contigs, in any assembly. Only plastid genomes, generated as a byproduct of the assembly approach, were circularized. These findings, from multiple different configurations, suggest that this mt-genome is not frequently in a circular form in the tissues and conditions sampled.
The 721 kbp assembly includes the unique elements found in all other assemblies except for one 7.5 kbp fragment found in the alternative assemblies. This fragment contains no recognized annotated mitochondrial regions and matches nothing of relevance via a GenBank blastn query. For simplicity we selected the single 721 kbp contig plus this anonymous 7.5 kbp segment to represent the core L. trichandra mt-genome (hereafter “Assembly 1”) as a representative assembly of numerous possible genomic configurations (discussed below).
By varying the set of sampled raw reads and genome size parameter in the assembly process, we recovered alternative assemblies that, at first glance, provided grounds for concern due to the large number alternative configurations (table 2). However, despite clearly differing in absolute structure and length, the alternative constructs all share large collinear segments with Assembly 1, whereas the latter includes all the unique elements found in the alternative assemblies (supplementary fig. S1, Supplementary Material online) and all of the same unique annotated features. The observed structural variation between assemblies was primarily associated with dispersed repeats greater than 400 bp (fig. 2, table 3, and supplementary fig. S1, Supplementary Material online). Recombination between these types of repeats is the primary mechanism underlying the frequent structural rearrangements found in many other plant mitochondrial genomes, including the extremes noted in another legume, Glycine (Chang et al. 2013). By tweaking the assembly parameters (genome size and input reads) we appear to be able to be generate alternative configurations. Any or all of these alternatives may be present among the population of mt-genomes sampled from multiple cells in a single individual.
Fig. 2.

—A circularized representation of the unique L. trichandra mitochondrial genome components. The black outer circle represents the genome within which light blue, green, and orange blocks identify dispersed repeats (>400 bp), plastome derived regions, and the concatenated anonymous 7.5 kbp fragment, respectively. Forward and reverse strand genes names are denoted by black and red fonts, respectively. Within the circle, light blue ribbons connect copies of the same dispersed repeat type and the interior dark blue arc circumscribes the trans-spliced nad5 genic boundary.
Table 3.
Dispersed Repeats >400 bp in the L. trichandra Mt-Genome
| Repeat Name | P Similarity | Length | Query Start | Query End | Subject Start | Subject End | Relative Direction |
|---|---|---|---|---|---|---|---|
| 1a | 99.97 | 22,210 | 699,782 | 721,986 | 513,531 | 491,323 | Forward |
| 1b | 99.97 | 22,210 | 491,323 | 513,531 | 721,986 | 699,782 | Reverse |
| 2a | 99.99 | 17,429 | 198,567 | 215,995 | 17,428 | 1 | Forward |
| 2b | 99.99 | 17,429 | 1 | 17,428 | 215,995 | 198,567 | Reverse |
| 3a | 99.89 | 14,132 | 455,579 | 469,709 | 49,820 | 63,937 | Forward |
| 3b | 99.89 | 14,132 | 49,820 | 63,937 | 455,579 | 469,709 | Forward |
| 4a | 99.9 | 7,713 | 230,519 | 238,231 | 127,773 | 135,485 | Forward |
| 4b | 99.9 | 7,713 | 127,773 | 135,485 | 230,519 | 238,231 | Forward |
| 5a | 97.12 | 903 | 718,455 | 719,348 | 428,106 | 427,209 | Forward |
| 5b | 97.12 | 903 | 493,964 | 494,857 | 427,209 | 428,106 | Reverse |
| 5c | 97.12 | 903 | 427,209 | 428,106 | 493,964 | 494,857 | Reverse |
| 6a | 95.54 | 583 | 642,081 | 642,663 | 290,900 | 290,327 | Forward |
| 6b | 95.54 | 583 | 290,327 | 290,900 | 642,663 | 642,081 | Reverse |
| 7a | 95.77 | 449 | 247,544 | 247,992 | 124,242 | 123,807 | Forward |
| 7b | 95.77 | 449 | 123,807 | 124,242 | 247,992 | 247,544 | Forward |
Assembly 1 was polished using Illumina genomic DNA reads and Pilon. 3.3% of the L. trichandra Illumina reads mapped to the mitochondrial genome resulting in minimum coverage of 46× and average coverage of 459× (supplementary fig. S2, Supplementary Material online). Pilon polishing resulted in the confirmation of 99.9% of base calls and suggested corrections for 39 sites [4 SNPs, 29 single base insertions {mostly in G/C mononucleotide repeats}, and six 1 bp deletions]. Just two of these corrections were located in annotated genic regions (cox1 position 249,787 and matR position 399,927). Because the mt-genome includes several horizontally transferred plastid regions (discussed below), Pilon identified short clusters of suggested changes in those regions that were not accepted as valid edits because these appeared to be plastid short reads mapping to the mitochondrial genome. The final corrected assembly was 729,504 bp (contig 1 plus the 7.5 kbp fragment—fig. 2).
It is important to acknowledge that the assembly pipeline developed here relies on linkage between elements. Separate chromosomes in the target genome must each have at least one recovered match to the reference genome (i.e., one 500 bp aligned region would be enough to initiate the assembly of a chromosome of any size). Without a match, there is no initial assembly point for that target segment. We raise this point because the approach taken here could miss unusual potentially autonomous elements with little sequence conservation or known function that are known from a handful of mitochondrial systems (Sloan et al. 2012; Warren et al. 2016; Sanchez-Puerta et al. 2017). The anonymous 7.5 kbp fragment recovered in this study, which was only assembled under some assembly parameters, suggests that it may be linked to known mitochondrial elements in some mt-genome configurations but not others, which raises further questions about these interesting elements. Future work on these regions will hopefully shed light on their frequency, function, and genomic configurations in general.
Because angiosperm mt-genome assemblies are complicated by a dizzying array of possible configurations and/or number of copies of sub elements found within a cell, between cells, and between tissues, estimating genome size variation between species is a complex issue. For L. trichandra we have explored the genome configurations estimated by long read data and have intentionally presented the most compact form recovered that included all unique annotated elements in the alternative assemblies. Some elements are duplicated in alternate assemblies, as indicated in supplementary figures S1 and S2, Supplementary Material online. Based on the general consistency of shared gene sets in most cases, we have accepted that other published mitochondrial assemblies (table 1) are similarly compact versions for the comparisons conducted below.
The annotated genome contains 55 unique genes [36 protein coding, 3 rRNA genes, and 16 tRNA {table 1}], four of which have duplicate copies (atp1, nad3, rrL, and trnFm). One copy of atp1 does not maintain an ORF and is a likely pseudogene. Seven genes contain multiple exons: ccmFc (2 exons), nad2 (2 exons), nad4 (4 exons), nad5 (5 exons), nad7 (5 exons), rps3 (2 exons), and rps10 (2 exons).
Default blastn comparisons recovered five plastid derived regions with ≥80% similarity and a minimum length of 300 bp. These ranged from 588 to 3,049 bp (fig. 2), representing 1% of the mt-genome. Two of these segments included potentially functional genes, a psaA cpDNA-like gene (with a full 2,106 bp ORF) and the trnP, trnW, and petG cassette in the other (fig. 2). The latter cpDNA cassette is known from other plant mt-genomes (Kubo et al. 1995; Kanno et al. 1997), including sugar beet, wheat, rice, and others, suggesting an ancient origin with subsequent losses or numerous parallel origins. The cpDNA derived trnW appears to represent the only functional copy whereas the plastid trnP gene exists in addition to the mitochondrial form.
We also extracted ORFs >300 bp in non-coding regions of the genome and used expression data and blastn results to isolate potentially expressed CDS regions (expressed >1 RPKM and matching 90% with another angiosperm sequence). Using this approach, we found 41 ORF regions of interest. Of these, 13 occurred on the forward strand and 28 on the reverse strand. Eleven were assigned GO-terms when searched against a database in TRAPID (Van Bel et al. 2013). All 41 ORFs and their corresponding GO-terms (if applicable) are located in supplementary table S1, Supplementary Material online. In total, these ORFs make up 18,847 bp, or 2.82% of intergenic space. Figure 3 shows the location of high-confidence ORFs in the genome in addition to regions of similarity with 15 other angiosperm mt-genomes.
Fig. 3.

—Circos plot illustrating sequence conservation relative to the L. trichandra mt-genome. The outer circle is the L. trichandra circularized assembly (fig. 2). Inner track 1 identifies conserved ORFs with black and red lines denoting forward and reverse strand ORFs, respectively. Tracks 2–17 denote regions of sequence conservation between specific taxa and L. trichandra. Track: 2—Vicia faba, 3—Lotus japonicus, 4—Medicago truncatula, 5—Millettia pinnata, 6—Glycine max, 7—Vigna angularis, 8—Vigna radiata, 9—Cucurbita, 10—Malus, 11—Arabidopsis, 12—Geranium, 13—Gossypium, 14—Vitis, 15—Beta, 16—Oryza, and 17—the holoparasite Lophophytum.
Genome Size Variation and Sequence Conservation
Available angiosperm mt-genome assemblies range from 66 kbp in the parasite Viscum scurruloideum (Skippington et al. 2015) to 11+ mbp in Silene (Sloan et al. 2012), and even closely related plant species can display massive variation in absolute length and configuration. For example, despite having diverged relatively recently, Silene noctiflora and Sileneconica have been found to be >40-fold and >25-fold different in size compared with other Silene species, with more than 90% of that variation attributable to intergenic sequences (Sloan et al. 2012). Genic sequences accounted for just 1% of genome content in those two species, and >85% of intergenic sequence lacked detectible similarity with other plant genomes (nuclear, plastid, or mitochondrial) (Sloan et al. 2012). The dramatic size variation in Silene mt-genomes derives from increases in rates of insertion and deletion events that correlate with increased rates of coding sequence substitution (Sloan et al. 2012).
Within this context of angiosperm mitochondrial genomes, the 729 kbp L. trichandra assembly is particularly neither small nor large, but it is nearly twice the size of other published legume mitochondrial assemblies (table 1). Genic regions (excluding the trans-spliced nad5 gene) and CDS regions (including nad5) account for just 8.3% and 4.3% of L. trichandra mt-genome, respectively. There is little variation in gene or CDS content among the available legume mitochondrial genomes (table 1). Because intergenic regions are known to be highly variable among plant mitochondrial genomes, we explored intergenic DNA sequence conservation among mt-genomes. Based on blastn comparisons, just 0.01% of intergenic sequence in the L. trichandra mt-genome is conserved with any of the eight outgroup eudicot representatives (fig. 3). Regions showing conservation with any of the seven published legume mitochondrial genomes comprised just 19.4% of the mt-genome and 36.9% of those overlap with annotated genic regions (fig. 3). This leaves the identity and origin of 80% (588,000 bp) of the L. trichandra mt-genome without detectable similarity to other legume/rosid genomes, consistent with prior findings for intergenic regions in plant mt-genomes (Mower et al. 2012; Wu, Stone, et al. 2015). Below we investigate a number of potential contributors to intergenic content among these genomes.
Repeats
The most common contributors to organellar genome size variation include various classes of repeats. We investigated mononucleotide, tandem, and dispersed repeat (two classes, 100–1,000 bp and >1,000 bp) content to characterize their contributions to these genomes. Variation in dispersed repeat content (fig. 4A) is a major contributor to mt-genome size in these legumes and representative angiosperms. Among the legume mt-genomes, dispersed repeats are clearly important sources of mt-genome size variation, but comparatively little of this variation is attributable to the 100–1,000 bp repeat size category. Three of the four legumes with the smallest mt-genomes (the two Vigna and one Medicago species) lack dispersed repeats >1,000 bp and display less than 2.3% in the 100–1,000 bp category. In contrast, at least 12% of the second smallest legume mitochondrial genome (Lotus) and all the other legume genomes are attributable to larger dispersed repeats. With 68% dispersed repeat content, Vicia is particularly extreme and similar to Oryza (57%) (fig. 4A). In addition to contributing important recombinationally active regions, the 1,000+ bp dispersed repeats contribute considerably to mt-genome length variation in legumes and represent over 17% (>128 kbp) of the L. trichandra mt-genome. Our comparisons identify little sequence conservation between the dispersed repeats of L. trichandra and the other legume mt-genomes (fig. 3), underscoring their dynamic nature and complex history (e.g., Havird et al. 2017).
Fig. 4.

—The percentage of representative legume and rosid mt-genomes occupied by repeats. (A) Dispersed repeats 100–1,000 bp and >1,000 bp. (B) Tandem repeats (>2 repeats). (C) Mononucleotide repeats >8 bp.
In contrast to the importance of dispersed repeats to legume mt-genome length variation, tandem repeats (0.2–3.15%, fig. 4B) and mononucleotide repeats (0.30–0.47%, fig. 4C) contribute little to size variation among these legume mt-genomes.
Horizontal Genome Transfer
Species of the holoparasitic plant genus Lophophytum (Balanophoraceae) are known to grow only from the roots of Anadenanthera, Parapiptadenia, Piptadenia, and Apuleia species (Hansen 1980), which are all members of the recently re-circumscribed legume subfamily Caesalpinioideae (LPWG 2017), the first three being members of the mimosoid clade nested within Caesalpinioideae, and the fourth in the Cassieae clade. Recent sequencing and investigation of the Lophophytum mirabile mt-genome identified massive horizontal transfer (HGT) of genic regions from its mimosoid legume hosts into the Lophophytum sequence (Sanchez-Puerta et al. 2017). In fact, 80% of 44 genes investigated in the parasite genome were supported to be of legume origin (Sanchez-Puerta et al. 2017), providing another example of extreme protein coding HGT in plant mitochondria (Rice et al. 2013; Xi et al. 2013). However, without complete mitochondrial genomes from both host and parasite (these studies lacked mt-genomes of the host plants), it has not been possible to determine if non-coding DNA may have also been transferred from host to parasite.
As we explored the potential origin(s) of non-conserved regions in the L. trichandra mt-genome using default NCBI blastn searches on L. trichandra intergenic regions to the NCBI nucleotide database, it became apparent that alignments with the L.mirabile mt-genome (KU992322-KU992380) dominated the results. A subsequent standalone blastn comparison between the Lophophytum and L. trichandra mt-genomes recovered 281 kbp of shared sequence (fig. 3, supplementary table S2, Supplementary Material online). This is twice the level of sequence conservation observed between L. trichandra and any of the papilionoid legume mt-genomes (ca. 141 kbp) (fig. 3), reinforcing the conclusions of Sanchez-Puerta et al. (2017) about the degree of horizontal transfer that has occurred. With the exception of one Lophophytum chromosome (KU992332), all of the other Lophophytum chromosomes (53 of 54) shared sequence with L. trichandra mtDNA.
Whereas there is evidence for both RNA- and DNA-mediated HGT in the Lophophytum mt-genome (Sanchez-Puerta et al. 2017), the inclusion of large spans of mimosoid legume non-coding sequence in the Lophophytum mt-genome suggest that DNA-mediated mechanisms have played the predominant role. This is consistent with previous suggestions for both Rafflesia (Xi et al. 2013) and Lophophytum (Sanchez-Puerta et al. 2017). However, this single example of a mimosoid mitochondrial genome for Leucaena (which is relatively closely related to Anadenanthera, Parapiptadenia, and Piptadenia, but which is not the Lophophytum host plant) does not include a structure comprised many subgenomic circular elements (unlike Glycine), which have been hypothesized to play a role in the evolution of the Lophophytum sequence (Sanchez-Puerta et al. 2017). Furthermore, the large dispersed repeats that influence recombination in the Leucaena and other mitochondrial genomes (fig. 2) are not particularly conserved with Lophophytum (fig. 3), suggesting these may not be involved in homologous recombination between host and parasite. The sequencing of the mt-genome of the primary host of L. mirabile, Anadenanthera colubrina (Sanchez-Puerta et al. 2017), and additional strategically selected caesalpinioid legumes (LPWG 2017) will be needed to provide further insight into the mechanism(s) driving HGT in the mt-genome of Lophophytum, as well as the frequency of these HGT events; however, the dominance of mimosoid-derived genes (Sanchez-Puerta et al. 2017) and the presence of large amounts of mimosoid-derived non-coding sequence in the Lophophtyum mt-genome are consistent with the capture of an entire mimosoid mitochondrial genome(s) during the evolutionary history of the Lophophytum parasite.
Patterns of RNA Editing
The system developed as part of this study offered the opportunity to empirically investigate and document patterns of mitochondrial RNA editing among a closely related group of species that includes a well characterized hybrid tetraploid (allotetraploid) (Hughes et al. 2007; Govindarajulu, Hughes, et al. 2011; Govindarajulu, Hughes, Alexander, et al. 2011). We were particularly interested in the allotetraploid because of its potential to identify whether divergent nuclear encoded PPRs have combined in a hybrid lineage to generate novel RNA-editing patterns on the maternally inherited haploid genome or if maternal editing predominates.
Leucaena mitochondrial RNA edits were investigated from three perspectives. We first sought to demonstrate the utility of the data generated by investigating RNA-editing in L. trichandra and comparing the findings to those from prior studies of individual species. Next, we identified a conservative estimate of shared coding and non-coding editing across all three major genome groups (clades) of Leucaena to both identify these patterns and further demonstrate the utility of the system. Having illustrated the consistency and utility of the C-to-U RNA edit data, we studied the impact of hybridization on mitochondrial RNA editing in the allotetraploid. These assessments involved mapping stranded ribo-depleted RNA-seq reads from each accession to the L. trichandra mitome sequence and the extraction of strand-specific edits. Positions associated with genomic SNP variants in divergent lineages were removed to minimize misinterpretation as RNA-edits (see “Materials and Methods” section).
Leucaena trichandra Editing
For the L. trichandra-only investigation, 996 potential RNA edits (supplementary table S3, Supplementary Material online) were inferred across the mt-genome (fig. 5). These include 740 C-to-U edits, a value that is at the high end of what has been identified for other angiosperm mitochondria (e.g, Mower et al. 2012), but similar to estimates for ancestral angiosperm mitochondrial RNA editing (Richardson et al. 2013). When considering annotated CDS regions, only the plastome-derived petG gene showed no signs of RNA editing. Excluding the plastome-derived psaA gene, which likely has plastid RNA reads mapping to it (Wu, Cuthbert, et al. 2015), 513 of 521 (98.5%) inferred CDS edits were the expected C-to-U type and 89% of these code for an alternative amino acid.
Fig. 5.

—The percentage of RNA variants sites in L. trichandra plotted for each of three genomic regions (CDS, intron, intergenic). Plots for each region include one for all variant types (20 classes) combined versus the C-to-U class expected of plant mitochondrial RNA editing.
Alternative classes of RNA editing (non C-to-U) are not characteristic of plant mitochondrial RNA editing, leading us to carefully investigate the remaining CDS associated sites (8 sites). Several different sources of evidence clarified their status as other variants versus RNA substitution edits. First, the cox2 variant (one G-to-C) came from a poorly aligned region spanning an indel in the transcripts relative to the genome, eliminating confidence as a substitution edit. Second, we used blasr alignments to search the draft nuclear genome for potential nuclear copies of the remaining CDS with non C-to-U variants. The three sdh4 (G-to-A), one rps19 (C-to-A), and one ccmFn (one U-to-C) variants were each associated with a nuclear gene copy of each respective gene and the sdh4 nuclear copy is consistent with prior evidence for legumes (Adams et al. 2001). The two remaining variants [atp4 {A-to-G} and rp15 {C-to-A}] have clean alignments and we found no evidence for alternative nuclear copies of these genes. We suspect these result from minor variation in mitochondrial genomes between individuals rather than RNA-edits. We also investigated whether the properties of known CDS associated edits differ from the general properties of those inferred outside characterized CDS. Comparisons of mean edit frequency in the CDS (84.3% ± 3.9) versus non-CDS (68.1% ± 7.8) variants were significantly different (P < 0.001), further supporting that the CDS region edits are distinctive. Overall, the broad pattern of RNA editing in L. trichandra CDS regions is consistent with the vast majority (>99.6%, 517 of 519) of changes representing genuine C-to-U edits.
Extensive patterns of potential editing outside of annotated CDS regions in the L. trichandra mt-genome are also interesting, but more complicated (fig. 5). When we compared C-to-U editing to all other classes inferred outside of CDS regions, the C-to-U type clearly dominates (fig. 6). Forty-eight percent (229) of 475 edits were the C-to-U type, while the remaining changes were distributed across 19 alternative variant classes with an average of 13 variants/class (ranges 1–39) (supplementary table S3, Supplementary Material online and fig. 6). These genomic regions evolve more quickly at the DNA level than CDS regions and our analyses could miss genomic variants between individuals (i.e., differences between individuals used for the DNA-seq and RNA-seq), resulting in genomic SNPs from these other classes. The relatively large proportion of the C-to-U class variants, versus any other single class of variant across the non-coding regions of the mt-genome suggests that considerable C-to-U editing is going on in understudied portions of these mitochondria. However, the mean frequency of each C-to-U edit in these unannotated regions (69.1% ± 8.2) versus characterized CDS (84.3% ± 3.9) are notably different (P < 0.001), identifying differences in these sites between the CDS and unannotated regions. Furthermore, the mean frequency of C-to-U change (69.1% ± 8.2) is not significantly different from the other classes of change (66.3% ± 7.3) when comparing unannotated regions. Further work is needed to identify if the C-to-U edits outside of annotated regions are important or perhaps represent misfiring of edit machinery, underlying genomic SNPs, or some combination thereof. The number of C-to-U conversions outside of known CDS (219) is higher than, but in line with, that reported for a smattering of other angiosperms, including Silene (97 positions) (Wu, Cuthbert, et al. 2015), Nicotiana (73 positions) (Grimes et al. 2014), or Brassica (37 positions) (Grewe et al. 2014).
Fig. 6.

—Classes of non-CDS variant in L. trichandra. The number of edits from each of 20 classes of nucleotide variant is plotted for non-coding regions of the mt-genome.
Genus-Wide Evaluation
The estimate of edited sites shared by all three genome groups in Leucaena identified 607 conserved positions (supplementary table S4, Supplementary Material online). Those within known CDS regions (463) were all C-to-U and 96% altered the encoded amino acid. Outside of CDS regions, 90% (129 of 144) were C-to-U, further demonstrating the likely occurrence of additional RNA editing in these understudied portions of the genome (Wu, Stone, et al. 2015). The remaining 10% (15 variant positions), conserved across the genus, included 10 positions found in cpDNA associated regions and two positions found in ribosomal genes. These likely reflect incorrectly mapped reads from alternative genomic compartments. The three remaining non C-to-U edits all occur on the intron side of intron-exon boundaries in nad7 (one each in introns 1, 3, 4). These are associated with RNA-seq reads that are poorly aligned length variants across several bases in these regions, suggesting they come from an alternative genomic compartment or form of RNA-processing. Thus all conserved high confidence RNA-edits within and outside of annotated CDS regions, derived from a genus-wide comparison, were of the C-to-U type expected for plant mitochondria. These findings further diminish idea that any non C-to-U changes are real substitution RNA edits in L. trichandra (see above) but supports a large proportion of C-to-U edits outside of coding regions as real edits. The majority of shared conserved C-to-U editing outside of annotated CDS are concentrated in regions that retain an ORF greater than 100 bp. Furthermore, some of these ORFs share high sequence similarity (blastn searches) with diverse angiosperms, supporting the idea that they may have conserved but unknown mitochondrial function.
Editing in an Allotetraploid
When considering high confidence CDS associated C-to-U edits in Leucaena, considerable variation was observed between the robust estimate for a single species (L. trichandra with 517 edits) and those shared across all species in the genus (463 edits). This variation in RNA editing, among closely related species, presents a useful system to explore such differences. Next, we compared patterns of C-to-U RNA editing (CDS and non-CDS) in the allotetraploid L. leucocephala relative to its diploid maternal and paternal progenitors, L. pulverulenta and L. cruziana, respectively (Hughes et al. 2002; Govindarajulu, Hughes, Alexander, et al. 2011). Leucaena cruziana, shared 21 (14 CDS/7 non-coding) exclusive edits with L. leucocephala whereas L. pulverulenta shared 26 (12 CDS/14 non-coding) exclusive edits with the allotetraploid (supplementary table S5, Supplementary Material online). The two divergent diploids shared 10 (4 CDS/6 non-coding) edits that were not found in L. leucocephala (supplementary table S5, Supplementary Material online). A maternal origin of the mt-genome and plastid genomes is supported by transcriptome derived plastome and mt-genome phylogenies (Kovar, Lynsey et al., unpublished data) and previous work (Govindarajulu, Hughes, Alexander, et al. 2011). Thus, we expected to observe a predominance of maternal editing in the allotetraploid, but this was clearly not the case. Within annotated regions of the L. leucocephala mt-genome, unique elements of RNA editing are retained with each of its parents. This included 8 maternal plus tetraploid and 7 paternal plus tetraploid (hereafter “10/8”) genes with edits, 5/9 total amino acid substitutions, and 7/5 synonymous substitutions.
These findings are consistent with what is known about plant mitochondrial genetics. The complex interaction of a nuclear encoded gene family of RNA editing factors with the “simple” haploid genome can create functional alleles that differ from their haploid template. These interactions result in the population of different of alleles, which can vary across time and developmental phase (Meier et al. 2016) from the unedited primary transcript to the fully edited functional form, ultimately presenting a complex assemblage of possible alleles. The evidence from the RNA editing in the tetraploid hybrid, L. leucocephala, supports the idea that divergent elements of editing can combine to generate novel transcripts and combinations of transcripts from different genes on which selection may act, further supporting such potential for RNA editing (Gott 2003; Gommans et al. 2009).
Site-by-Site Evaluation of Nucleotide Frequencies at Edited Positions
When discussing RNA editing of a position in a genome, it is easy to overlook the continuous nature of these edits among all transcripts per allele. An edited position is rarely converted in 100% of transcripts at a given point in time (Huntley et al. 2016), meaning that the relative frequency of the genomic nucleotide, versus the edited nucleotide, ranges from 0% to 100% in the observed transcripts. If the allele has more than one edited site, each will be edited from 0% to 100% and those site-by-site percentages may or may not be correlated by gene or allele (i.e., one site might have 20% editing versus another with 80% for the same allele). Such variation can add to the diverse population of alleles on which selection may act. Indeed, recent studies have uncovered variation in the frequency of an RNA edit versus the genomic nucleotide at individual positions between tissues and/or conditions that may prove functionally significant (Meng et al. 2010; Picardi et al. 2010; Tseng et al. 2013), supporting the idea that plant mitochondrial RNA editing may multiply the number of possible alleles rather than just correct underlying deleterious mutations (Gott 2003; Gommans et al. 2009).
Through the comparative analysis of the expected versus observed RNA-edit frequencies between L. cruziana and L. pulverulenta, we identified 141 high confidence (C-to-U) edits with significant variation in editing frequency between taxa. When comparing the sites between taxa, each is “edited” to the same non-genomic nucleotide (C-to-U), but at significantly different frequencies. Therefore, these parents of the allotetraploid L.leucoceophala appear to retain differentially heritable variation in editing frequency at these positions. Though the study lacks distinctive RNA-seq biological replicates (replicates were combined), the combination of samples and data used permit a preliminary investigation into the behavior of RNA edit frequency in an allopolyploid lineage.
Next, we explored variation in the frequency of each edited nucleotide versus its genomic counterpart between all three taxa (diploid parents and the allotetraploid). To limit comparisons with the highest confidence RNA edit sites, we only considered C-to-U edits found in annotated CDS regions (which represent 75% of all sites recovered in this analysis). We identified 38 sites whose frequency of editing was shared between the allotetrapolyploid (L. leucocephala) and the maternal progenitor (L. pulverulenta), 45 shared with the paternal progenitor (L. cruziana), 16 where the polyploid displays an intermediate frequency, and 40 sites where the polyploid displays a transgressive pattern of editing (supplementary table S6, Supplementary Material online). For a number of genes, no sites deviated from expected (i.e., the hybrid and its maternal and paternal contributors did not deviate from one another—atp1, atp6, atp9, ccmFn, cox3, nad4L, nad7, rps14, rps3). A wide range of frequency bias patterns were observed among the other loci. The general patterns are exemplified by the 10 genes with at least 50% of their C-to-U edited sites deviating significantly from the expected value (table 4). These include a gene (nad2) that primarily follows the paternal pattern, three genes (rp15, rps14, sdh4) that mimic the maternal pattern, as well as two with representation from each frequency group (matR, mttB) and one with a primarily transgressive-up frequency in the allotetraploid (rpl2).
Table 4.
Comparison of RNA Edit Frequency Differentiation in L. leucocephala Versus Its Parental Taxa
| atp8 | matR | mttB | nad2 | rps19 | sdh4 | ccmFc | rp15 | rpl2 | rps14 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Intermediate | 0 | 4 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Maternal | 1 | 1 | 8 | 0 | 0 | 2 | 4 | 5 | 0 | 1 |
| Paternal | 0 | 2 | 2 | 12 | 1 | 0 | 4 | 2 | 0 | 0 |
| Trangressive down | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Transgressive up | 0 | 2 | 9 | 1 | 0 | 1 | 4 | 1 | 1 | 0 |
| Not significant | 1 | 1 | 10 | 3 | 1 | 2 | 8 | 3 | 0 | 1 |
| Total | 3 | 11 | 31 | 17 | 2 | 5 | 20 | 11 | 1 | 2 |
Note.—This summary focuses on C-to-U edited sites in annotated CDS regions with at least 50% of C-To-U sites displaying a significant deviation from expected. Values represent the number of sites per gene in the allotetraploid L. leucocephala that fit a particular pattern (i.e., the allotetraploid is similar to the maternal or paternal, is intermediate, or is transgressive).
It is interesting to note that the total sites with the paternal frequency outnumber those with the maternal frequency and that the transgressive “up” frequency far outnumbers the transgressive “down.” With editing bias varying considerably within some of these genes, it is unlikely that these findings are simply the result of PCR bias induced through library preparation, because directional bias toward one pattern would be expected across the entirety of the transcript. Patterns of combined RNA editing frequency may confer some adaptive significance (Gott 2003; Gommans et al. 2009), as combinations of partially edited transcripts prove optimal in different genomic backgrounds, environmental conditions, or tissues. These preliminary findings contradict the idea that cytonuclear interactions in allotetraploids necessarily follow the inheritance pattern of the organelle (Sharbrough et al. 2017). Proteomic work with complementary RNA-seq work are needed on systems such as this to fully understand the potential importance of variation in ratios of an RNA edited nucleotide versus the genomic variants.
Conclusions
The L. trichandra mitochondrial genome broadens our understanding of mt-genome variability and structural complexity in the Leguminosae. The observed structural complexity is primarily associated with dispersed repeat-associated recombination that is common in many angiosperm mitochondrial genomes, but which is not evident in all legumes. Genome size variation was largely associated with intergenic regions of uncharacterized DNA and dispersed repeats larger than 1,000 bp. The holoparasite Lophophytum mt-genome, previously demonstrated to predominantly comprise mimosoid legume protein coding genes rather than those native to Santalales, also shares massive amounts of mimosoid intergenic DNA. These findings are consistent with capture of an entire mimosoid mitochondrial genome(s) during its evolutionary history. Genome wide patterns of RNA editing in Leucaena provide new insights into this important phenomenon. Future work, correlating results from protein sequencing in different tissues and/or conditions in diploids and polyploids will be needed to shed light on the degree to which alternative alleles existing in various forms in partially edited transcripts play a role in the function of mitochondria and plant fitness. Additional legume mt-genomes are needed to more fully understand their complex history and the functional interplay between organellar and mitochondrial genetics.
Supplementary Material
Acknowledgments
We thank Aaron Liston, Sanjuro Jogdeo, Michael Schatz, and Kevin Weitemeyer for bioinformatics advice, Oregon State University’s Center for Genome Research NSF’s CyVerse, Texas Advanced Computing Center, and the Mueller Lab (Boyce Thompson Institute) for computational support, L. Mazzola, J. Bybee, and D. Rivizzigno for sequencing assistance, and two anonymous reviewers for constructive comments and guidance. This material is based on work supported by the National Science Foundation under grant 1238731 (CDB), Swiss National Science Foundation grant 31003A_135522 (CEH). L.K. was supported by the NMSU Howard Hughes Medical Institute undergraduate research program (grant 52008103), and S.O.R. was supported by the NMSU undergraduate National Institutes of Health Maximizing Access to Research Careers (MARC) program (grant GMO7667-39S1).
Literature Cited
- Adams KL, Rosenblueth M, Qiu YL, Palmer JD.. 2001. Multiple losses and transfers to the nucleus of two mitochondrial succinate dehydrogenase genes during angiosperm evolution. Genetics 158(3):1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverson AJ, et al. 2010. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol Biol Evol. 27(6):1436–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverson AJ, Zhuo S, Rice DW, Sloan DB, Palmer JD.. 2011. The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS One 6(1):e16404.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendich AJ. 1993. Reaching for the ring: the study of mitochondrial genome structure. Curr Genet. 24(4):279–290. [DOI] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27(2):573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergthorsson U, Adams KL, Thomason B, Palmer JD.. 2003. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 424(6945):197.. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for illumina Sequence Data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill MJ, Köser CU, Ross NE, Archer JAC.. 2010. Read length and repeat resolution: exploring prokaryote genomes using next-generation sequencing technologies. PLoS One 5(7):e11518.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G.. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinform. 13(1):238.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, et al. 2013. The mitochondrial genome of soybean reveals complex genome structures and gene evolution at intercellular and phylogenetic levels. PLoS One 8(2):e56502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chateigner-Boutin A-L, Small I.. 2010. Plant RNA editing. RNA Biol. 7(2):213–219. [DOI] [PubMed] [Google Scholar]
- Dugas DV, et al. 2015. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci Rep. 5(1):16958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Small I.. 2011. The evolution of RNA editing and pentatricopeptide repeat genes. New Phytol. 191(1):37–47. [DOI] [PubMed] [Google Scholar]
- Gommans WM, Mullen SP, Maas S.. 2009. RNA editing: a driving force for adaptive evolution? BioEssays: news and reviews in molecular, cellular and developmental biology. 31:1137–1145. doi:10.1002/bies.200900045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gott JM. 2003. Expanding genome capacity via RNA editing. C R Biol. 326(10–11):901–908. [DOI] [PubMed] [Google Scholar]
- Govindarajulu R, Hughes CE, Alexander PJ, Bailey CD.. 2011. The complex evolutionary dynamics of ancient and recent polyploidy in Leucaena (Leguminosae; Mimosoideae). Am J Bot. 98(12):2064–2076. [DOI] [PubMed] [Google Scholar]
- Govindarajulu R, Hughes CE, Bailey CD.. 2011. Phylogenetic and population genetic analyses of diploid Leucaena (Leguminosae; Mimosoideae) reveal cryptic species diversity and patterns of divergent allopatric speciation. Am J Bot. 98(12):2049–2063. [DOI] [PubMed] [Google Scholar]
- Grewe F, et al. 2014. Comparative analysis of 11 Brassicales mitochondrial genomes and the mitochondrial transcriptome of Brassica oleracea. Mitochondrion 19:135–143. [DOI] [PubMed] [Google Scholar]
- Grice LF, Degnan BM.. 2015. The origin of the ADAR gene family and animal RNA editing. BMC Evol Biol. 15(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes BT, Sisay AK, Carroll HD, Cahoon AB.. 2014. Deep sequencing of the tobacco mitochondrial transcriptome reveals expressed ORFs and numerous editing sites outside coding regions. BMC Genomics 15(1):31.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualberto JM, Newton KJ.. 2017. Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu Rev Plant Biol. 68(1):225–252. [DOI] [PubMed] [Google Scholar]
- Hansen B. 1980. Balanophoraceae. Flora Neotrop. 23:1–80. [Google Scholar]
- Havird JC, Trapp P, Miller CM, Bazos I, Sloan DB.. 2017. Causes and consequences of rapidly evolving mtDNA in a plant lineage. Genome Biol Evol. 9(2):323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CE. 1998a. Leucaena: a genetic resources handbook. Oxford (UK: ): Oxford Forestry Institute. [Google Scholar]
- Hughes CE. 1998b. Monograph of Leucaena (Leguminosae–Mimosoideae). Syst Bot Monogr. 55:1–244. [Google Scholar]
- Hughes CE, Bailey CD, Harris SA.. 2002. Divergent and reticulate species relationships in Leucaena (Fabaceae) inferred from multiple data sources: insights into polyploid origins and nrDNA polymorphism. Am J Bot. 89(7):1057–1073. [DOI] [PubMed] [Google Scholar]
- Hughes CE, et al. 2007. Serendipitous backyard hybridization and the origin of crops. Proc Natl Acad Sci. 104(36):14389–14394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt M, et al. 2015. Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16(1):294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley MA, et al. 2016. Complex regulation of ADAR-mediated RNA-editing across tissues. BMC Genomics 17(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose M, Sugita M.. 2017. RNA editing and its molecular mechanism in plant organelles. Genes 8:5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorizzo M, et al. 2012. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs MA, Payne SR, Bendich AJ.. 1996. Moving pictures and pulsed-field gel electrophoresis show only linear mitochondrial DNA molecules from yeasts with linear-mapping and circular-mapping mitochondrial genomes. Curr Genet. 30(1):3–11. [DOI] [PubMed] [Google Scholar]
- Kanno A, Nakazono M, Hirai A, Kameya T.. 1997. Maintenance of chloroplast-derived sequences in the mitochondrial DNA of Gramineae. Curr Genet. 32(6):413–419. [DOI] [PubMed] [Google Scholar]
- Kearse M, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, et al. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Phillippy AM.. 2016. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. bioRxiv. doi:10.1101/071282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Newton KJ. 2008. Angiosperm mitochondrial genomes and mutations. Curr Mitochondrion 8:5–14. [DOI] [PubMed] [Google Scholar]
- Kubo T, Yanai Y, Kinoshita T, Mikami T.. 1995. The chloroplast trnP–trnW–petG gene cluster in the mitochondrial genomes of Beta vulgaris, B. trigyna and B. webbiana: evolutionary aspects. Curr Genet. 27(3):285–289. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe S, Browne M, Boudjelas S, De Poorter M.. 2000. 100 of the World's Worst Invasive Alien Species: A selection from the Global Invasive Species Database. Published by the Invasive Species Specialist Group (ISSG) a specialist group of the Species Survival Commission (SSC) of the World Conservation Union (IUCN), 12pp. First published as a special lift out in Aliens 12, December 2000.
- Lowe TM, Eddy SR.. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5):955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LPWG 2017. A new subfamily classification of the Leguminosae based on a taxomomically comprehensive phylogeny. Taxon 44–77. [Google Scholar]
- Lurin C, et al. 2004. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. The Plant Cell 16: 2089–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier UG, et al. 2008. Complex chloroplast RNA metabolism: just debugging the genetic programme? BMC Biol. 6(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JC, Kankowski S, Krestel H, Hetsch F.. 2016. RNA editing—systemic relevance and clue to disease mechanisms? Front Mol Neurosci. 9:124.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, et al. 2010. RNA editing of nuclear transcripts in Arabidopsis thaliana. BMC Genomics 11(Suppl 4):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira S, Valach M, Aoulad-Aissa M, Otto C, Burger G.. 2016. Novel modes of RNA editing in mitochondria. Nucleic Acids Res. 44(10):4907–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mower JP, Sloan DB, Alverson AJ.. 2012. Plant mitochondrial genome diversity: the genomics revolution In: Wendel JF, Greilhuber J, Dolezel J, Leitch IJ, editors. Plant genome diversity volume 1: plant genomes, their residents, and their evolutionary dynamics. Vienna: Springer Vienna; p. 123–144. [Google Scholar]
- Mower JP, et al. 2010. Horizontal acquisition of multiple mitochondrial genes from a parasitic plant followed by gene conversion with host mitochondrial genes. BMC Biol. 8(1):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K, et al. 1992. Gene organization deduced from the complete sequence of liverwort Marchantia polymorpha mitochondrial DNA: a primitive form of plant mitochondrial genome. J Mol Biol. 223(1):1–7. [DOI] [PubMed] [Google Scholar]
- Ogihara Y, et al. 2005. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33(19):6235–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Rodriguez S, Bailey CD.. 2016. Using novel methods to assemble plant mitochondrial genomes: an example from the mimosoid legume genus Leucaena Sequencing, Finishing, and Analysis in the Future Meeting; June 1; Santa Fe (NM: ): Los Alamos National Laboratory. [Google Scholar]
- Palmer JD, Herbo LA.. 1987. Unicircular structure of the Brassica hirta mitochondrial genome. Curr Genet. 11(6–7):565–570. [DOI] [PubMed] [Google Scholar]
- Palmer JD, Shields CR.. 1984. Tripartite structure of the Brassica campestris mitochondrial genome. Nature 307(5950):437. [Google Scholar]
- Picardi E, et al. 2010. Large-scale detection and analysis of RNA editing in grape mtDNA by RNA deep-sequencing. Nucleic Acids Res. 38(14):4755–4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plitmann U. 1993. Pollen tube attrition as related to breeding systems in Brassicaceae. Plant Syst Evol. 188(1–2):65–72. [Google Scholar]
- Rice DW, et al. 2013. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342(6165):1468. [DOI] [PubMed] [Google Scholar]
- Richardson AO, Rice DW, Young GJ, Alverson AJ, Palmer JD.. 2013. The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 11(1):29.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Puerta MV, García LE, Wohlfeiler J, Ceriotti LF.. 2017. Unparalleled replacement of native mitochondrial genes by foreign homologs in a holoparasitic plant. New Phytol. 214(1):376–387. [DOI] [PubMed] [Google Scholar]
- Sharbrough J, Conover JL, Tate JA, Wendel JF, Sloan DB.. 2017. Cytonuclear responses to genome doubling. Am J Bot. 104(9):1277–1280. [DOI] [PubMed] [Google Scholar]
- Skippington E, Barkman TJ, Rice DW, Palmer JD.. 2015. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A 112(27):E3515–E3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB. 2013. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 200(4):978–985. [DOI] [PubMed] [Google Scholar]
- Sloan DB, Alverson AJ, Wu M, Palmer JD, Taylor DR.. 2012. Recent acceleration of plastid sequence and structural evolution coincides with extreme mitochondrial divergence in the angiosperm genus Silene. Genome Biol Evol. 4(3):294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, et al. 2012. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10(1):e1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks RB, Dale RMK.. 1980. Characterization of 3H-labeled supercoiled mitochondrial DNA from tobacco suspension culture cells. Molec. Gen. Genet. 180:351–355.
- Stern DB, Palmer JD.. 1984. Extensive and widespread homologies between mitochondrial DNA and chloroplast DNA in plants. Proc Natl Acad Sci U S A 81(7):1946–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28(5):511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C-C, Lee C-J, Chung Y-T, Sung T-Y, Hsieh M-H.. 2013. Differential regulation of Arabidopsis plastid gene expression and RNA editing in non-photosynthetic tissues. Plant Mol Biol. 82(4–5):375–392. [DOI] [PubMed] [Google Scholar]
- Van Bel M, et al. 2013. TRAPID: an efficient online tool for the functional and comparative analysis of de novoRNA-seq transcriptomes. Genome Biol. 14(12):R134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9(11):e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BL, Anderson RS, Bendich AJ.. 1981. The mitochondrial genome is large and variable in a family of plants (Cucurbitaceae). Cell 25(3):793–803. [DOI] [PubMed] [Google Scholar]
- Warren JM, Simmons MP, Wu Z, Sloan DB.. 2016. Linear plasmids and the rate of sequence evolution in plant mitochondrial genomes. Genome Biol Evol. 8(2):364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Cuthbert JM, Taylor DR, Sloan DB.. 2015. The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc Natl Acad Sci U S A 112(33):10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Stone JD, Štorchová H, Sloan DB.. 2015. High transcript abundance, RNA editing, and small RNAs in intergenic regions within the massive mitochondrial genome of the angiosperm Silene noctiflora. BMC Genomics 16(1):938.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z, et al. 2013. Massive mitochondrial gene transfer in a parasitic flowering plant clade. PLoS Genet. 9(2):e1003265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Harding T, Kamikawa R, Simpson AGB, Roger AJ.. 2017. Mitochondrial genome evolution and a novel RNA editing system in deep-branching Heteroloboseids. Genome Biol Evol. 9(5):1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.