

Abstract
Chemical communication is fundamental for the operation of insect societies. Their diverse vocabulary of chemical signals requires a correspondingly diverse set of chemosensory receptors. Insect olfactory receptors (ORs) are the largest family of chemosensory receptors. The OR family is characterized by frequent expansions of subfamilies, in which duplicated ORs may adapt to detect new signals through positive selection on their amino acid sequence. Ants are an extreme example with ∼400 ORs per genome—the highest number in insects. Presumably, this reflects an increased complexity of chemical communication. Here, we examined gene duplications and positive selection on ant ORs. We reconstructed the hymenopteran OR gene tree, including five ant species, and inferred positive selection along every branch using the branch-site test, a total of 3326 tests. We find more positive selection in branches following species-specific duplications. We identified amino acid sites targeted by positive selection, and mapped them onto a structural model of insect ORs. Seventeen sites were under positive selection in six or more branches, forming two clusters on the extracellular side of the receptor, on either side of a cleft in the structure. This region was previously implicated in ligand activation, suggesting that the concentration of positively selected sites in this region is related to adaptive evolution of ligand binding sites or allosteric transmission of ligand activation. These results provide insights into the specific OR subfamilies and individual residues that facilitated adaptive evolution of olfactory functions, potentially explaining the elaboration of chemical signaling in ant societies.
Keywords: gene duplication, positive selection, protein structure, olfactory receptors, social insects
Introduction
Ants represent an extreme example of social behavior in animals and one of the most ecologically successful animal groups in the ecosystem. Chemical communication is central for the efficient operation and coordination of an ant colony, including distinction between reproductive (queens) and nonreproductive castes (workers), coordination and division of labor among worker groups. Ants also discriminate between nestmates and nonnestmates based on a shared, colony-specific odor, which is comprised of a complex mixture of very long chain cuticular hydrocarbons (Nowbahari et al. 1990; Soroker et al. 1995; Akino et al. 2004; Ozaki et al. 2005; Martin et al. 2008).
Insects detect odors and pheromones by chemosensory receptors. In ants, the olfactory receptor (OR) gene family stands out due to its dramatic expansion to ∼300–400 OR genes in every ant genome (Zhou et al. 2012), the largest number in all studied insects. Although new OR genes may underlie various kinds of olfactory functions, it is commonly suggested that this dramatic expansion is primarily related to the evolution of complex chemical communication systems in ant societies (Smith CR et al. 2011). Ants use large numbers of pheromones, which may be composed of complex mixtures. For example, nestmate recognition is based on mixtures of hydrocarbons displayed on the ants’ cuticle. In the desert ant Cataglyphis niger, a mixture of 55 different cuticular hydrocarbons were identified (Saar et al. 2014) and the composition of this mixture changes rapidly among even closely related species (Martin and Drijfhout 2009). Olfactory perception of pheromones is vital for cooperative functions of the nest, and should therefore be an important fitness factor in ants. Hence, new OR genes with new olfactory functions are expected to evolve together with the evolution of ant social behavior.
Gene duplication plays a central role in evolutionary innovation—a gene is duplicated and the new paralogs evolve novel beneficial functions, possibly via positive selection (Ohno 1970). Gene duplication can be followed by neofunctionalization, where one of the paralogs evolves a new function while the other maintains the original function (Force et al. 1998). Alternatively, in subfunctionalization, a multifunctional gene is duplicated and the paralogs retain different subfunctions of the ancestral gene. In Drosophila, two thirds of gene duplications fit the neofunctionalization model (Assis and Bachtrog 2013). Thus, duplication of ant ORs may underlie the evolution of new olfactory functions. OR duplication may be followed by positive selection on the ligand binding site to recognize new odorants (neofunctionalization), or the duplicated ORs may become more specialized in detecting specific odors instead of one OR that responds to several similar odors (subfunctionalization). Different paralogs may also evolve differential regulation of gene expression or kinetics of ligand activation and deactivation.
ORs are seven-transmembrane domain proteins (Clyne et al. 1999; Vosshall et al. 1999), but in contrast to vertebrate ORs, insect ORs show an inversed membrane orientation—cytoplasmic N-terminus and extracellular C-terminus (Benton et al. 2006; Lundin et al. 2007; Smart et al. 2008). Insect ORs are heterodimers consisting of one of many specific ligand-binding ORs and one conserved coreceptor known as ORCO (Larsson et al. 2004). The heterodimer has been suggested to function as a ligand-gated ion channel, which upon activation leads to depolarization of olfactory sensory neurons (Sato et al. 2008; Wicher et al. 2008). The specific OR dictates ligand selectivity and may also transduce the signal by activating a G protein (Wicher et al. 2008). Relevantly, residues 146–150 in transmembrane helix 3 (TM3) and the adjacent extracellular domain 2 (EC2) of Drosophila melanogaster OR 85 b (DmOR85b) have been shown to contribute to ligand binding (Nichols and Luetje 2010). Which additional residues contribute to ligand binding and receptor activation remains an open question.
Roux et al. (2014) and Engsontia et al. (2015) analyzed the gene phylogeny and inferred positive selection on ant ORs of two and five ant species, respectively. Engostia et al. proposed a hypothesis where species-specific gene expansions evolve under positive selection, as they are associated with olfactory adaptation unique to each lineage. They tested for positive selection in these expansions using the site test of positive selection. However, this test does not compare lineage-specific branches to the nonlineage-specific branches of the tree. Here, we apply the branch-site test, which allows this comparison, and thereby testing the hypothesis of more positive selection in species-specific expansions. We find a statistically significant difference between species specific ORs and nonspecies specific ORs, with slightly more positive selection on species-specific ORs. There is extensive evidence for positive selection in most OR subfamilies, with periods of multiple consecutive gene duplications and positive selection on multiple paralogs. Amino acids targeted by positive selection are primarily concentrated around the putative ligand binding site, which suggests adaptation for the perception of new ligands and elaboration and fine-tuning of the response to existing ligands.
Materials and Methods
OR Gene Sequences
OR sequences were collected from four previously annotated ant genomes (table 1) and we de novo annotated three more genomes: Solenopsis invicta (genome assembly version Si_gnH; NCBI accession AEAQ00000000), S. fugax (Sf_gnA; accession QKQZ00000000), and Monomorium pharaonis (Mp_gnA; accession QKWU00000000). De novo gene annotation was conducted using genBlast (She et al. 2009, 2011) (http://genome.sfu.ca/genblast/; last accessed July 2014), in which translated BLAST hits to exons were grouped to represent putative gene models, while stitching hits at predicted splice site junctions. The previously annotated ORs were used as queries for the BLAST searches. Between 239 and 461 ORs were annotated per species (table 1). Novel gene sequences are available in supplementary information files S1–S9, Supplementary Material online and also deposited in the Figshare database (DOI: 10.6084/m9.figshare.5900275). ORs of the wasp Nasonia vitripennis and the honeybee Apis mellifera were also included in the phylogenetic analysis as outgroups.
Table 1.
Olfactory Receptors of the Species Included in the Study
| Species Names (abbreviation) | Number of ORs | References | |
|---|---|---|---|
| Ants | Linepithema humile (LhOR) | 320 | Smith CD et al. (2011) |
| Pogonomyrmex barbatus (PbOR) | 291 | Smith CR et al. (2011) | |
| Harpegnathos saltator (HsOR) | 461 | Zhou (2012) | |
| Camponotus floridanus (CfOR) | 377 | Zhou (2012) | |
| Solenopsis invicta (SiOR) | 407 | De novo annotation, here | |
| Solenopsis fugax (SfOR) | 314 | De novo annotation, here | |
| Monomorium pharaonis (MpOR) | 239 | De novo annotation, here | |
| Bee | Apis mellifera | 168 | Robertson and Wanner (2006) |
| Wasp | Nasonia vitripennis | 262 | Werren et al. (2010) |
Phylogenetic Analysis
Nucleotide sequences were translated to amino acid sequences and aligned using MAFFT (version 7; accurate variant E-INS-i; Katoh et al. 2005). The OR gene tree was constructed using RAxML (version 8.1.15; Stamatikis 2006) with the PROTCATLG model, the LG rate matrix, and 100 bootstraps repeats. The phylogenetic tree included 2793 sequences and 5583 branches, which is too large for any dN/dS-based positive selection inference method. Thus, the tree was divided to 31 clades (subtrees) at branches with the highest possible bootstrap support, so that the largest clade contained 144 sequences. Clades were defined as monophyletic groups wherever possible, with the exception of clade 28, which is paraphyletic. In a few cases, subtrees were split at low bootstrap branches, which might also lead to subsets of sequences that are not monophyletic clades. However, the downstream analysis (the branch-site test) does not rely on an assumption of monophyly, so this should not lead to artifacts in our inference of positive selection. Due to the very long running time of the branch-site test (17 days for each branch in a tree of 200 sequences) we also restricted the positive selection analysis to the five more closely related ant species, including the four myrmicines: P. barbtus (PbOR), S. invicta (SiOR), M. pharaonis (MpOR), and S. fugax (SfOR); and one formicine: C. floridanus (CfOR). After this reduction, the sequences of each clade were realigned using GUIDENCE (version 2.01; Penn et al. 2010) with the aligner PAGAN (Löytynoja et al. 2012) as a codon alignment by reverse-translating the amino acid alignment. Unreliably aligned residues were masked at a GUIDANCE score cutoff of 0.8 by replacing low-scoring codons with “NNN.” Then, phylogenetic trees were built using RAxML for each clade as above, based on the amino acid alignment.
Positive Selection Inference
Each branch of the gene tree was tested for positive selection using the modified branch-site test (model A; Zhang et al. 2005), as implemented in codeml in the PAML package (version 3.15; Yang and Nielsen 2000). Codeml uses maximum likelihood to estimate the ω parameter, which represents the dN/dS ratio in the subset of sites under positive selection. Positive selection was inferred by comparing a model that allows positive selection (dN/dS > 1) only in one “foreground branch” with a null model that does not allow any positive selection (dN/dS <= 1). Since the compared models are nested, likelihood ratio tests (LRT) were used to reject the null model. Thus, P values were obtained for each tested branch, and adjusted into q values to control the false discovery rate (FDR; Benjamini and Hochberg 1995) using the Bioconductor R package “qvalue” (Bass et al. 2015). For each branch where the LRT is successful at FDR of 10%, specific codons under positive selection were identified based on posterior probability >0.9 for dN/dS > 1.
Mapping Selected Residues to the Protein Structure
Amino acid positions under positive selection in ant ORs were mapped onto a structural model of insect ORs, built by Hopf et al. (2015) using the evolutionary coupling method (Marks et al. 2012), which generated a 3D structure for the sequence of OR85b of Drosophila melanogaster (DmOR85b). To map the positions from the alignments of each of the 31 ant ORs clades to the DmOR85b structure, one PbOR representative from each clade and the DmOR85b sequence were aligned using MAFFT with the FFT-NS-i method. Positions in the ant alignments with posterior probability of 0.9 for positive selection were mapped to the homologous positions in the DmOR85b sequence using a perl script (included in Supplementary Material). Positions in insertions/deletions in either ant or fly sequences were mapped to the nearest amino acid position in the homologous sequence. Then, these amino acid positions were visualized in the 3 D structure of DmOR85b using the molecular visualization software Pymol (version 1.3; DeLano 2002). The transmembrane regions of the structure were predicted based on amino acids with positive scores on the Kyle–Doolittle hydropathy scale.
Results
OR Gene Tree
The OR gene tree was reconstructed based on the amino acid sequences of seven ant species: Harpegnathos saltator, Linepithema humile, Camponotus floridanus, Pogonomyrmex barbatus, Monomorium pharaonis, Solenopsis fugax, and Solenopsis invicta. The jewel wasp Nasonia vitripennis and honeybee Apis mellifera were included as outgroups. The tree includes 2793 sequences and 5583 branches. The ORCO subtree was used as an outgroup to root the tree. This tree is too large for positive selection inference, so we divided it to 31 clades, with no more than 200 sequences in each clade (fig. 1; the full tree is included in the Supplementary Material with distinct coloring of each clade). The hymenopteran OR gene tree consists of many subtrees that include ant, bee, and wasp genes, which represent paralogs that evolved in a common ancestor of these Hymenoptera. Each subtree contains more recently evolved paralogs, including gene duplications before the divergence of ants and bees and many more subsequent ant-specific duplications.
Fig. 1.

—OR gene tree including 2,973 genes from seven ants, honeybee, and jewel wasp. The tree was reconstructed using RAxML based on a MAFFT amino acid alignment and divided into 31 clades for subsequent analyses. Formicidae-specific clades are colored red (only ant sequences), Aculeatan-specific clades are colored purple (include bee sequences), and Hymenopteran clades are colored blue (include wasp sequences). Clades colored white were not tested for positive selection.
Extensive Positive Selection across OR Subfamilies
Due to computational run-time constraints of the branch-site test, positive selection was inferred in each of the 31 clades and only the five most closely related ant species were included in the analysis (see Materials and Methods). The reduced clades included 1706 sequences in total. A test for positive selection was applied to each branch of each clade, 3,326 branches in total. We also required that at least one codon site had a posterior probability >0.9 for dN/dS > 1. After correction for multiple testing (false discovery rate q < 0.1), this analysis identified 261 branches with evidence for positive selection (table 2). Virtually all clades contain some branches with positive selection and between 1 and 75 amino acid sites under positive selection.
Table 2.
Branches and Sites Inferred to be Under Positive Selection Based on the Branch-Site Test
| Clade | Number of Sequences | Branches with Positive Selection (%) | Number of Sites with P > 0.9 for dN/dS > 1 |
|---|---|---|---|
| Clade1 | 5 | 0 (0.0%) | 0 |
| Clade2 | 21 | 1 (2.6%) | 1 |
| Clade3 | 144 | 22 (7.7%) | 60 |
| Clade4 | 78 | 14 (9.2%) | 45 |
| Clade5 | 123 | 12 (4.9%) | 19 |
| Clade6 | 19 | 2 (5.7%) | 10 |
| Clade7 | 124 | 21 (8.6%) | 44 |
| Clade8 | 39 | 3 (4.0%) | 6 |
| Clade9 | 35 | 4 (6.0%) | 6 |
| Clade10 | 141 | 21 (7.5%) | 75 |
| Clade11 | 10 | 1 (5.9%) | 1 |
| Clade12 | 8 | 0 (0.0%) | 0 |
| Clade13 | 72 | 13 (9.2%) | 25 |
| Clade14 | 36 | 4 (5.8%) | 14 |
| Clade15 | 84 | 12 (7.3%) | 54 |
| Clade16 | 6 | 0 (0.0%) | 0 |
| Clade17 | 15 | 5 (18.5%) | 74 |
| Clade18 | 74 | 9 (6.2%) | 26 |
| Clade19 | 47 | 8 (8.8%) | 23 |
| Clade20 | 27 | 4 (7.8%) | 9 |
| Clade21 | 54 | 16 (15.2%) | 63 |
| Clade22 | 98 | 17 (8.8%) | 52 |
| Clade23 | 56 | 11 (10.1%) | 46 |
| Clade24 | 19 | 2 (5.7%) | 2 |
| Clade25 | 34 | 3 (4.6%) | 5 |
| Clade26 | 36 | 7 (10.1%) | 13 |
| Clade27 | 14 | 1 (4.0%) | 2 |
| Clade28 | 90 | 10 (5.6%) | 21 |
| Clade29 | 81 | 15 (9.4%) | 25 |
| Clade30 | 48 | 7 (7.5%) | 12 |
| Clade31 | 68 | 16 (12.0%) | 34 |
| Total | 1,706 | 261 |
Note.—Positive results are those that passed the LRT (at false discovery rate q < 0.1) and had at least one site with posterior probability >0.9 for dN/dS >1.
A representative subfamily (clade 22) is shown in figure 2a (the other 30 are included as supplementary figs., Supplementary Material online). Fifteen out of 193 branches in this clade (7.8%) were inferred to be under positive selection (false discovery rate q < 0.1). Branch 121 had the largest number of sites under positive selection (13 codon sites). An inspection of the sequence alignment shows that these sites are located in reliable alignment blocks with no obvious alignment problems that could lead to false inference of positive selection (fig. 2c). Randomly chosen branches from additional clades were likewise manually inspected, leading us to estimate that no more than 10% of the branches inferred to be under positive selection might be affected by unreliable alignment.
Fig. 2.

—A representative subfamily from the OR gene tree. (a) Maximum likelihood phylogeny of clade 22. Scale bar represents 0.3 amino acid substitutions per site. Branches inferred to be under positive selection by the branch-site test are colored in red (q value < 0.1). Bootstrap support values are displayed throughout; branch numbers are written above the red branches only. Subtrees of species-specific gene duplications are marked by curly brackets. (b and c) Two sections of the sequence alignment showing positions under positive selection (posterior probability > 0.9) indicated by red arrows for the partitions by branch 9 (b) and branch 121 (c). Species name abbreviations according to table 1.
Seven of the positively selected branches in clade 22 are immediately following a gene duplication event. For example, the root of this clade (branch 165) is a gene duplication event that created two paralogs that were again duplicated, and each of these three duplications was followed by positive selection on at least one of the paralogs (branches 60 and 121). Most of these events preceded the divergence of the lineages represented by the five ant species included in this analysis, that is, before the divergence of Formicinae from Myrmicinae. Subsequently, there were many more duplications, including species-specific duplications (marked by brackets in fig. 2a), but these were generally not followed by positive selection on the species-specific paralogs.
Positive Selection following Species-Specific Expansions
The OR gene tree reveals dramatic expansions in many subfamilies. Many of the duplication events are species-specific, as highlighted for the example of clade 22 in figure 2a. Each of the studied species had numerous species-specific duplications in most of the clades, with the exception of the smallest clades (table 3). To test whether more paralogs are under positive selection after species-specific duplications compared with nonspecies-specific duplications, we counted positively selected branches following species-specific duplications. In total, 81 out of 905 (9.0%) species-specific branches were under positive selection and 171 out of 2602 (6.6%) nonspecies-specific branches were under positive selection, which is a statistically significant difference (two-tailed Fisher’s exact test P value = 0.02).
Table 3.
Positive Selection in Branches of Species-Specific Gene Duplications
|
Camponotus floridanus |
Pogonomyrmex barbatus |
Monomorium pharaonis |
Solenopsis fugax |
Solenopsis invicta |
Nonspecies-Specific Branches |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clade | Br | PS | %PS | Br | PS | %PS | Br | PS | %PS | Br | PS | %PS | Br | PS | %PS | Br | PS | %PS |
| Clade 1 | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 11 | 0 | 0 |
| Clade 2 | 6 | 0 | 0 | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 2 | 0 | — | 31 | 1 | 3 |
| Clade 3 | 32 | 0 | 0 | 12 | 1 | 8 | 0 | 0 | — | 2 | 1 | 50 | 8 | 0 | 0 | 231 | 17 | 7 |
| Clade 4 | 22 | 1 | 5 | 8 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | — | 0 | 0 | — | 117 | 14 | 12 |
| Clade 5 | 20 | 0 | 0 | 6 | 0 | 0 | 6 | 0 | 0 | 6 | 1 | 17 | 12 | 0 | 0 | 193 | 12 | 6 |
| Clade 6 | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 35 | 3 | 9 |
| Clade 7 | 80 | 8 | 10 | 2 | 1 | 50 | 4 | 0 | 0 | 0 | 0 | — | 8 | 2 | 25 | 151 | 11 | 7 |
| Clade 8 | 12 | 1 | 8 | 2 | 1 | 50 | 4 | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 53 | 1 | 2 |
| Clade 9 | 0 | 0 | — | 2 | 0 | 0 | 0 | 0 | — | 8 | 0 | 0 | 22 | 2 | 9 | 35 | 2 | 6 |
| Clade 10 | 20 | 1 | 5 | 2 | 0 | 0 | 4 | 2 | 50 | 6 | 1 | 17 | 32 | 1 | 3 | 215 | 15 | 7 |
| Clade 11 | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 17 | 1 | 6 |
| Clade 12 | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 13 | 0 | 0 |
| Clade 13 | 24 | 3 | 13 | 4 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | — | 8 | 1 | 13 | 101 | 6 | 6 |
| Clade 14 | 8 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | — | 0 | 0 | — | 2 | 0 | 0 | 53 | 4 | 8 |
| Clade 15 | 6 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | — | 0 | 0 | — | 4 | 1 | 25 | 151 | 11 | 7 |
| Clade 16 | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 7 | 0 | 0 |
| Clade 17 | 0 | 0 | — | 0 | 0 | — | 2 | 0 | 0 | 0 | 0 | — | 2 | 1 | 50 | 149 | 4 | 3 |
| Clade 18 | 6 | 1 | 17 | 4 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | — | 2 | 0 | 0 | 131 | 5 | 4 |
| Clade 19 | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 8 | 2 | 25 | 83 | 6 | 7 |
| Clade 20 | 0 | 0 | — | 10 | 0 | 0 | 8 | 2 | 25 | 5 | 0 | 0 | 12 | 0 | 0 | 16 | 2 | 13 |
| Clade 21 | 6 | 2 | 33 | 14 | 0 | 0 | 6 | 1 | 17 | 0 | 0 | — | 0 | 0 | — | 79 | 12 | 15 |
| Clade 22 | 16 | 0 | 0 | 8 | 0 | 0 | 4 | 0 | 0 | 4 | 1 | 25 | 6 | 1 | 17 | 155 | 13 | 8 |
| Clade 23 | 22 | 0 | 0 | 4 | 1 | 25 | 2 | 1 | 50 | 0 | 0 | — | 6 | 1 | 17 | 75 | 9 | 12 |
| Clade 24 | 0 | 0 | — | 0 | 0 | — | 2 | 0 | 0 | 0 | 0 | — | 6 | 1 | 17 | 27 | 1 | 4 |
| Clade 25 | 2 | 0 | 0 | 6 | 0 | 0 | 2 | 0 | 0 | 2 | 1 | 50 | 4 | 0 | 0 | 49 | 2 | 4 |
| Clade 26 | 4 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | — | 0 | 0 | — | 6 | 1 | 17 | 51 | 6 | 12 |
| Clade 27 | 2 | 1 | 50 | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 0 | 0 | — | 23 | 0 | 0 |
| Clade 28 | 14 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | — | 0 | 0 | — | 32 | 4 | 13 | 129 | 6 | 5 |
| Clade 29 | 50 | 8 | 16 | 8 | 2 | 25 | 4 | 0 | 0 | 0 | 0 | — | 32 | 2 | 6 | 65 | 2 | 3 |
| Clade 30 | 10 | 1 | 10 | 8 | 0 | 0 | 0 | 0 | — | 14 | 1 | 7 | 18 | 3 | 17 | 103 | 2 | 2 |
| Clade 31 | 46 | 7 | 15 | 18 | 0 | 0 | 0 | 0 | — | 0 | 0 | — | 16 | 6 | 38 | 53 | 3 | 6 |
| Total | 408 | 34 | 8.3 | 138 | 6 | 4.3 | 60 | 6 | 10 | 49 | 6 | 12 | 250 | 29 | 12 | 2,602 | 171 | 6.6 |
Br, total number of the branches; PS, number of the branches with positive selection; %PS, percentage of branches with positive selection.
Positively Selected Sites in the Context of Structural Domains
We mapped the amino acid positions under positive selection to the 3 D structural model of insect ORs that was built using the evolutionary coupling method (Marks et al. 2012) by Hopf et al. (2015). The number of branches in which an amino acid site was inferred to be under positive selection was counted (table 4) and mapped to the 3 D structure of the protein (fig. 3). Seventeen sites were under positive selection in six or more, and up to twelve branches, suggesting recurrent episodes of adaptive evolution in the same sites. Only two of these sites are located in the large intracellular region. The other 15 sites are located in the transmembrane region, with 11 sites located closer to the extracellular side of the receptor. The latter can be divided into two clusters (marked by ellipses in fig. 3b and d). The seventh transmembrane region (TM7) contains the largest concentration of positively selected sites (six sites). These sites are adjacent to four additional sites in TM5 and TM6, forming a dominant cluster in the transmembrane region that borders on the extracellular region (cluster 1). A second cluster of three positively selected sites (cluster 2) is located in the large second extracellular loop (EC2), in close proximity to TM2 and TM3. The sites of cluster 2 surround a region in TM3 (marked by an arrow in fig. 3b and d) that was shown by mutagenesis experiments to affect ligand activation in a D. melanogaster OR (Nichols and Luetje 2010).
Table 4.
Mapping of Positively Selected Sites to Amino Acid Positions in DmOR85b
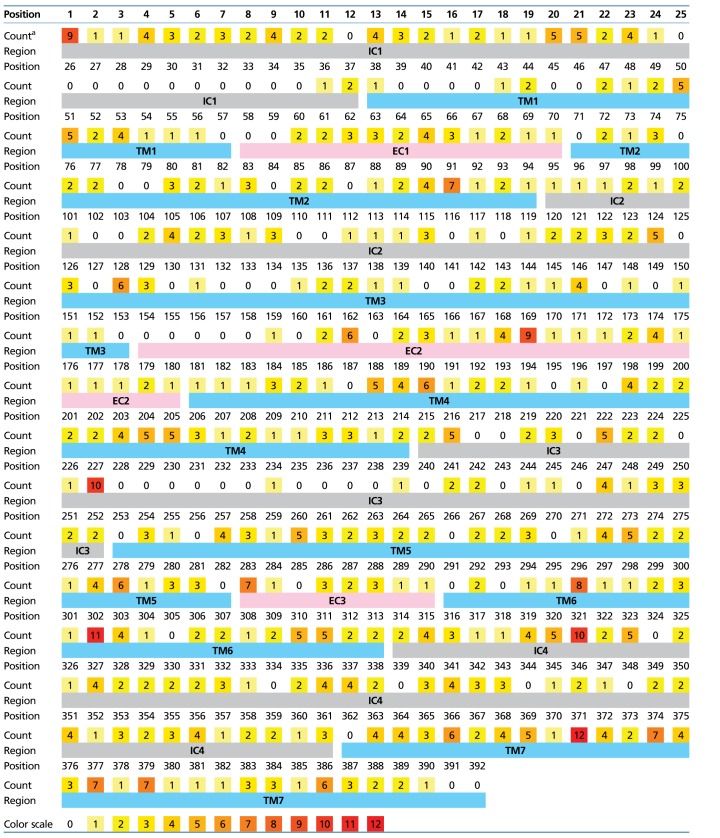 |
Counts of branches with positive selection for each amino acid site, after mapping positions in ant sequences to the homologous positions on the structural model of DmOR85b. Predicted extracellular (EC), intracellular (IC), and transmembrane (TM) regions are marked by shaded boxes.
Fig. 3.

—Mapping of positively selected sites to a 3D structural model of insect ORs. (a) Ribbon diagram of DmOR85b is rainbow colored from the amino terminus (blue) to the carboxy terminus (red), with the seven transmembrane helices numbered. (b) Amino acids under selection in ≥6 branches of the gene tree are shown as spheres and colored orange (between 6 and 8 branches) or red (between 9 and 12 branches). Two clusters of positively selected sites are marked by ellipses. A region in TM3 that affects ligand activation based on the mutagenesis experiments in Drosophila melanogaster is marked by an arrow. (c and d) Top view (from the extracellular side) of (a) and (b), rotated 90° about the X axis.
Discussion
Ants have the largest number of ORs among insects. While the novel olfactory functions coded by these new genes may include various environmental cues such as prey, predators, and vegetation, it is commonly hypothesized that the bulk of these new ORs evolve to mediate the complex pheromonal signaling that evolved in ant societies. The expanded collection of receptors presumably mirrors complex mixtures of pheromones, including cuticular hydrocarbons and other diverse families of compounds. Although it is difficult to estimate the total number of distinct compounds in ant pheromones, this number is likely in the hundreds, if not thousands. Many different classes of chemical compounds have been implicated in ant communication, but only a few were characterized in detail in the same ant species. For example, 55 hydrocarbons were identified in the cuticular lipids of Cataglyphis niger; and this is just the hydrocarbon fraction of the lipids (Saar et al. 2014). Studies in other species identified numerous fatty acids, hydrocarbons, piperidine alkaloids, terpenes, esters, alcohols, and more. Therefore, we hypothesized that multiple OR subfamilies in ants underwent dramatic expansion and adaptive evolution for recognizing new pheromones, through a process of gene duplication and positive selection pressures on key receptor residues that underlie ligand binding and activation. To investigate this hypothesis, a hymenopteran ORs gene tree was reconstructed and the branch-site test for positive selection was applied to each branch of the tree. Relative to previous studies (Roux et al. 2014; Engsontia et al. 2015), this analysis provided a more comprehensive view of ant OR evolution, encompassing a larger sample of ant species and a more detailed survey of every branch in the large gene tree of this superfamily. Our results show that almost every subfamily experienced gene duplications, especially ant-specific duplications, and positive selection was found in most of these subfamilies. Thus, this analysis supports the hypothesis that ant ORs experienced widespread and repeated episodes of adaptive evolution.
The observation of gene duplication events followed by positive selection can be interpreted as alternative evolutionary scenarios (Ohno 1970). Following a duplication, a paralog may lose its function and become a pseudogene (nonfunctionalization); one paralog may retain the ancestral function while the other acquires new functions through adaptive evolution (neofunctionalization); or, the duplication may allow each paralog to evolve more specialized functions, so that the functions of each paralog are a subset of the functions of the ancestral gene (subfunctionalization). Neofunctionalization and subfunctionalization events can be distinguished by the signature of natural selection on paralogs after gene duplication. A duplication followed by an asymmetrical signature of positive selection in one paralog and conservation of the other suggests a neofunctionalization event, while symmetrical signature of positive selection on both paralogs suggests subfunctionalization and specialization. Most OR duplications fit the former scenario of neofunctionalization. For example, branch 9 in clade 22 appears to be a neofunctionalization event, since after duplication this paralog experienced positive selection while the other paralog has a significantly shorter branch (leading to PbOR269 and SiOR449), which is in line with negative selection that maintains a conserved ancestral function (fig. 2a).
The extensive evidence for adaptive evolution in ant ORs parallels the evolutionary dynamics of chemosensory proteins (CSP) and odorant binding protein (OBP). These are two families of small sensillar lymph proteins that solubilize hydrophobic odorants. Kulmuni et al. (2013) showed that ants have a large number of species-specific duplications (especially S. invicta), as well as a higher dN/dS ratio in these species-specific CSP genes relative to nonspecies-specific genes. However, that study did not conduct a test for positive selection. McKenzie et al. (2014) tested for positive selection on some subfamilies of OBPs and CSPs using the site test for positive selection. Their analysis found adaptive evolution in both CSPs and OBPs, suggesting they evolved hand in hand with ORs to produce novel olfactory functions such as response to new pheromones.
The OR gene tree allows dating of gene duplications relative to the speciation of taxonomic groups: recent gene duplication events that created species-specific genes, less recent duplications of genes found in multiple ant species (e.g., all Formicinae), and ancient duplications that preceded the divergence of all ant species included in our analysis. ORs were duplicated throughout evolution of Hymenoptera, yet gene duplications were even more rapid in ants. Engsontia et al. (2015) hypothesized that species-specific duplications are followed by more positive selection pressures than nonspecies-specific duplications. However, that study only applied the site test for positive selection to the subtrees of species-specific expansions, and the branch-site test only to a few selected branches. Thus, their analyses do not provide a comprehensive comparison between branches in the species-specific expansions and the rest of the tree. The analyses in the present study applied the branch-site test to the majority of branches of the full gene tree. Our comparison shows a statistically significant difference in the proportion of branches with positive selection, with more positive selection in the species-specific expansions relative to nonspecies-specific branches. However, the magnitude of this difference is not large—9.0% versus 6.6% (table 3). Ergo, neofunctionalization of ORs proceeded both before the divergences of ant subfamilies/genera and after, in specific lineages.
Conversely, our findings are in agreement and complementary to the gene births and deaths analysis by Engsontia et al. Their results show that clades J (equivalent to clade 17 in our tree), L (clades 12–15), U (clades 9 and 10), V (clades 4 and 5), and 9-exon (clades 21–31) underwent substantial expansion. In our branch-site test analyses these are among the clades with the highest number of positively selected branches. In other clades that had little or no expansion, very few or no branches show evidence for positive selection, for example: clade B (equivalent to clade 16 in our tree), C (clade 1), and K (clade 11). In this respect, both types of analyses highlight specific ant OR subfamilies that underwent adaptive radiation.
Our results also shed light on the functional relevance of positive selection in ORs. Mapping sites under positive selection to the 3 D protein structure showed that positive selection targets mainly the extracellular-facing portions of the receptor. Two clusters of positive selection stand out. One is located around a TM3 region that was shown to affect ligand activation in a mutagenesis study of a D. melanogaster OR, indicating its proximity to the ligand binding site (Nichols and Luetje 2010). The second and larger cluster of positively selected sites is located across a cleft in the extracellular face of the receptor that separates TM2, TM3, and EC2 from TM5 and TM6. According to the structural model this cleft is accessible from the extracellular side of the receptor. Thus, our results are consistent with a binding site located between TM2/3 and TM5/6, and suggest that the most positively selected regions of ORs are related to ligand binding or receptor activation following ligand binding. These sites may play roles in ligand-binding specificity of ant ORs or in receptor activation in response to ligand binding. Following the extensive duplications of ant ORs, adaptive evolution of these sites may have modified receptor specificities toward new odorants and pheromones and/or modulated olfactory response in different paralogs. Multiple paralogs may have also allowed the elaboration of a richer repertoire of responses to existing pheromones, such as the complex mixtures of cuticular hydrocarbons that display variation in the relative proportion of many structurally related compounds.
In conclusion, our analyses revealed extensive evidence for positive selection on the ant ORs following many gene duplication events, typically fitting the signature of neofunctionalization. The distribution of selected sites on the protein structure suggests that ligand binding is the main target of positive selection. Thus, this study supports the hypothesis that the dramatic expansion of the OR superfamily in ants and the adaptive evolution of novel olfactory functions allowed recognition of an expanded collection of olfactory cues, potentially facilitating more complex chemical communication using a larger vocabulary. This work lays the ground for further detailed investigations both into specific subfamilies and periods of adaptive evolution in ants as well as experimental investigation of OR functional sites. Such investigations can be guided by the sites identified here as targets of positive selection.
Supplementary Material
Acknowledgments
All computations were conducted on the Hive computer cluster of the Faculty of Natural Science at the University of Haifa. E.P. was supported by grants from the Israel Science Foundation (grant numbers 646/15, 2140/15, 2155/15) and the US-Israel Binational Science Foundation (grant 2013408). M.K. was supported by grants from the Israel Science Foundation (grant numbers 1454/13, 1959/13, 2155/15)
Literature Cited
- Akino T, Yamamura K, Wakamura S, Yamaoka R.. 2004. Direct behavioral evidence for hydrocarbons as nestmate recognition cues in Formica japonica (Hymenoptera: formicidae). Applied Entomology and Zoology 39(3):381–387. [Google Scholar]
- Assis R, Bachtrog D.. 2013. Neofunctionalization of young duplicate genes in Drosophila. Proc Natl Acad Sci USA. 110(43):17409–17414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attrill H, et al. 2016. FlyBase: establishing a Gene Group resource for Drosophila melanogaster. Nucleic Acid Res. 44(D1):D786–D792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass JD, Dabney A, Robinson D. (2015). qvalue: Q-value estimation for false discovery rate control R package version 2.10.0. Available from: http://github.com/jdstorey/qvalue.
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 57:289–300. [Google Scholar]
- Benton R, Sachse S, Michnick SW, Vosshall LB.. 2006. Atypical membrane topology and heteromeric function of Drosophila odorant receptors in vivo. PLoS Biol. 4(2):e20.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyne PJ, et al. 1999. A novel family of divergent seven-transmembrane proteins: candidate odorant receptors in Drosophila. Neuron 22(2):327–338. [DOI] [PubMed] [Google Scholar]
- DeLano WL. (2002). The PyMOL molecular graphics system. Available from: http://pymol.org.
- Engsontia P, Sangket U, Robertson HM, Satasook C.. 2015. Diversification of the ant odorant receptor gene family and positive selection on candidate cuticular hydrocarbon receptors. BMC Res Notes 8:380.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force A, et al. 1998. Preservation of duplicate genes by complementary. Degenerative mutations. Genetics 151:1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf TA, et al. 2015. Amino acid coevolution reveals three-dimensional structure and functional domains of insect odorant receptors. Nat Commun. 6(1):6077.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T.. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33(2):511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmuni J, Wurm Y, Pamilo P.. 2013. Comparative genomics of chemosensory protein genes reveals rapid evolution and positive selection in ant-specific duplicates. Heredity 110(6):538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson MC, et al. 2004. Or83b encodes a broadly expressed odorant receptor essential for Drosophila olfaction. Neuron 43(5):703–714. [DOI] [PubMed] [Google Scholar]
- Löytynoja A, Vilella AJ, Goldman N.. 2012. Accurate extension of multiple sequence alignments using a phylogeny-aware graph algorithm. Bioinformatics 28(13):1684–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin C, et al. 2007. Membrane topology of the Drosophila OR83b odorant receptor. FEBS letters 581:5601–5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks DS, Hopf TA, Sander C. 2012. Protein structure prediction from sequence variation. Nature biotechnology 30:1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Drijfhout FP.. 2009. A review of ant cuticular hydrocarbons. J Chem Ecol. 35(10):1151–1161. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Vitikainen E, Helanterä H, Drijfhout FP.. 2008. Chemical basis of nest-mate discrimination in the ant Formica exsecta. Proc R Soc B 275(1640):1271–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie SK, Oxley PR, Kronauer DJC.. 2014. Comparative genomics and transcriptomics in ants provide new insights into the evolution and function of odorant binding and chemosensory proteins. BMC Genomics 15:718.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols AS, Luetje CW.. 2010. Transmembrane segment 3 of Drosophila melanogaster odorant receptor subunit 85b contributes to ligand-receptor interaction. J Biol Chem. 285(16):11854–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowbahari E, et al. 1990. Individual, geographical and experimental variation of cuticular hydrocarbons of the ant Cataglyphis cursor (Hymenoptera, Formicidae) – their use in nest and subspecies recognition. Biochem Syst Ecol. 18(1):63–73. [Google Scholar]
- Ohno S. Evolution by gene duplication. New York: Springer Science & Business Media. 1970. [Google Scholar]
- Ozaki M, et al. 2005. Ant nestmate and non-nestmate discrimination by a chemosensory sensillum. Science 309(5732):311–314. [DOI] [PubMed] [Google Scholar]
- Penn O, Privman E, Landan G, Graur D, Pupko T.. 2010. An alignment confidence score capturing robustness to guide tree uncertainty. Mol Biol Evol. 27(8):1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HM, Wanner KW.. 2006. The chemoreceptor superfamily in the honey bee Apis mellifera: expansion of the odorant, but not gustatory, receptor family. Genome Res. 16:1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux J, et al. 2014. Patterns of positive selection in seven ant genomes. Mol Biol Evol. 31(7):1661–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saar M, Leniaud L, Aron S, Hefetz A.. 2014. At the brink of supercoloniality: genetic, behavioral, and chemical assessments of population structure of the desert ant Cataglyphis niger. Front Ecol Evol. 2:13. [Google Scholar]
- Sato K, et al. 2008. Insect olfactory receptors are heteromeric ligand-gated ion channels. Nature 452(7190):1002–1006. [DOI] [PubMed] [Google Scholar]
- She R, Chu JS, Wang K, Pei J, Chen N.. 2009. GenBlastA: enabling BLAST to identify homologous gene sequences. Genome Res 19:143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She R, et al. 2011. genBlastG: using BLAST searches to build homologous gene models. Bioinformatics 27:2141–2143. [DOI] [PubMed] [Google Scholar]
- Smart R, et al. 2008. Drosophila odorant receptors are novel seven transmembrane domain proteins that can signal independently of heterotrimeric G proteins. Insect Biochem Mol Biol. 38(8):770–780. [DOI] [PubMed] [Google Scholar]
- Smith CD, et al. 2011. Draft genome of the globally widespread and invasive Argentine ant (Linepithema humile). Proc Natl Acad Sci USA. 108(14):5667–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CR, et al. 2011. Draft genome of the red harvester ant Pogonomyrmex barbatus. Proc Natl Acad Sci USA. 108(14):5667–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroker V, Vienne C, Hefetz A.. 1995. Hydrocarbon dynamics within and between nestmates in Cataglyphis niger (hymenoptera: formicidae). J Chem Ecol. 21(3):365–378. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22(21):2688–2690. [DOI] [PubMed] [Google Scholar]
- Vosshall LB, Amrein H, Morozov PS, Rzhetsky A, Axel R.. 1999. A spatial map of olfactory receptor expression in the Drosophila antenna. Cell 96(5):725–736. [DOI] [PubMed] [Google Scholar]
- Werren JH, et al. 2010. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327:343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicher D, et al. 2008. Drosophila odorant receptors are both ligand-gated and cyclic-nucleotide-activated cation channels. Nature 452(7190):1007–1011. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R.. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 17(1):32–43. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z.. 2005. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 22(12):2472–2479. [DOI] [PubMed] [Google Scholar]
- Zhou X, et al. 2012. Phylogenetic and transcriptomic analysis of chemosensory receptors in a pair of divergent ant species reveals sex-specific signatures of odor coding. PLoS Genet. 8(8):e1002930.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.