

Abstract
The 1,3-dithian-2-yl-methyl (Dim) and its analogous groups including dimethyl-Dim (dM-Dim) can provide a new dimension of orthogonality for carboxylic acid protection. They can be deprotected under nearly neutral oxidative conditions. In this paper, the protection of carboxylic acid with dM-Dim, deprotection of dM-Dim ester with sodium periodate, stability of dM-Dim protected carboxylic acid under acidic and basic conditions, and selective deprotection of dM-Dim protected carboxylic acids in the presence of tertiary butyl and methyl esters are presented.
Keywords: protecting group, carboxylic acid, oxidation, dM-Dim
Introduction
In multistep organic synthesis, the carboxylic acid function is usually protected in the forms of primary, secondary and tertiary alkyl, trityl, methoxymethyl, benzyl and allyl esters. Deprotection is mostly achieved under basic, acidic and hydrogenation conditions.1–3 The silyl groups, which are widely used for the protection of alcohols and can be deprotected with fluoride under nearly neutral conditions, are however less ideal for carboxylic acid protection due to their lability under even mildly acidic and basic aqueous conditions.1–3 For this reason, carboxylic acid protecting groups that can be deprotected under conditions orthogonal to acid, base and hydrogenation conditions would be particularly useful. The 1,3-dithian-2-yl-methyl (Dim) and its analogous groups including dimethyl Dim (dM-Dim), which can be deprotected under nearly neutral oxidative conditions, can be good candidates to serve the purpose (Scheme 1).4–9 These protecting groups meet the basic requirements of being useful in organic synthesis – stable protection and efficient deprotection. Because the H-2 in the Dim and dM-Dim moieties is non-acidic in normal terms of organic chemistry (pKa = ~31), even though there is a tendency for the esters 1 to undergo β-elimination spontaneously, these esters are stable under typical conditions for isolation and purification of organic compounds after reactions. However, during deprotection, once the disulfides in the Dim or dM-Dim moieties are oxidized to sulfoxides or sulfones to give 2, the acidity of H-2 is drastically increased (pKa = ~12). Considering that Fmoc protected amines and 2-cyanoethyl protected phosphates, in which the β hydrogens have a pKa of 22 and 25 respectively, could be deprotected with weak bases such as piperidine10–13 and DBU,14–16 it is easy to imagine that the esters 2 could undergo β-elimination even under neutral conditions giving the deprotected carboxylic acids 3 and the side products 4 (Scheme 1). Recently, we demonstrated the usefulness of the Dim associated function – 1,3-dithian-2-yl-methoxycarbonyl (Dmoc) – as protecting groups and linkers for oligodeoxynucleotide synthesis.17 The protecting groups and linkers were stable under all DNA synthesis conditions and were conveniently cleaved under nearly neutral oxidative conditions. Sensitive functionalities incorporated into the oligodeoxynucleotides such as thioester and α-chloroacetyl survived the conditions. Previously, Kunz and co-workers have reported using Dim as protecting groups in peptide synthesis. In their work, the Dim associated groups were deprotected by oxidation with hydrogen peroxide using ammonium molybdate as a catalyst.7, 8, 18 Later, Kutateladze and co-workers reported that deprotection of Dim esters could be achieved under electrochemical conditions in the absence of a chemical oxidant.19 In this paper, we present the use of a new Dim associated group, the dM-Dim, for carboxylic acid protection, and the use of sodium periodate as the oxidant for deprotection.
Scheme 1.
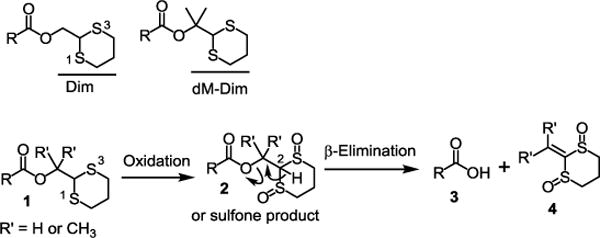
Dim and dM-Dim protected carboxylic acids, and their deprotection under nearly neutral oxidative conditions.
Results and Discussion
We started the studies by identifying suitable conditions to synthesize the dM-Dim carboxylic acid ester 7a from 5 and the acid chloride 6 (Table 1). Compound 5 was readily prepared in high yield by deprotonation of 1,3-dithiane with nBuLi followed by reacting with acetone. To form 7a, many conditions were screened. The one involving using nBuLi to deprotonate 5 and reacting the resulting alkoxide with 6 gave best result (entry 1, Table 1). Other conditions including reacting 5 with 6 in pyridine and other solvents in the presence of DMAP and imidazole, and reacting 5 with benzoic acid in the presence of carboxylic acid activating agents including DCC and EDCI gave less satisfactory results. Using the best conditions identified, the tertiary esters 7b-h were prepared. Compounds 7a-g are aryl carboxylic acid esters, of which 7b-d bear an electron donating group on their aryl ring (entries 2-4), while 7e-f bear an electron withdrawing group on their aryl ring (entries 5-6). Compound 7c is more sterically hindered due to the methyl group ortho to the ester group. Compound 7g represents esters with fused benzene rings. All these esters were formed with modest to good yields with those that bear an electron withdrawing group gave slightly better results. For the formation of tertiary esters of aliphatic acid chlorides such as 6h, one concern was the formation of ketenes via abstraction of the α-hydrogen of acid chloride by the alkoxide.20–24 However, even if this occurs, the ketene could still react with 5 to give the desired ester.25 Therefore, we carefully performed the reaction at low temperature and were able to synthesize 7h in 48% isolated yield (entry 8).
Table 1.
Formation of dM-Dim carboxylic acid esters.a

|
|||
|---|---|---|---|
|
| |||
| Entry | 6 | 7 | Yield b |
| 1 |

|

|
72% |
| 2 |
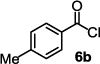
|

|
57% |
| 3 |

|

|
57% |
| 4 |

|

|
58% |
| 5 |

|

|
78% |
| 6 |

|
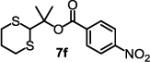
|
63% |
| 7 |

|

|
63% |
| 8 |

|

|
48% |
Reaction conditions: 5 (1.05 equiv.), nBuLi (1 equiv.), THF, −78°C, 30 min; then 6 (1 equiv.), −78°C to rt, 12 h.
Isolated yields.
We next studied suitable conditions for removing the dM-Dim protecting groups. In the literature, the Dim protected carboxylic acid groups were deprotected using hydrogen peroxide as the oxidant. Ammonium molybdate was used as a catalyst for the oxidation.7, 8, 18 The deprotection was also studied under electrochemical conditions.19 We recently used sodium periodate as the oxidation agent for deprotecting Dmoc protected synthetic oligodeoxynucleotides. The reactions were performed at room temperature, and multiple Dmoc groups on the nucleotides were removed efficiently in those studies.17 Because the sodium periodate conditions do not require any transition metal catalyst and special apparatus such as electrochemical cells, these conditions were chosen here for deprotection the dM-Dim protected carboxylic acid groups. The reactions were carried out in the solvent mixture of THF and water to dissolve both the organic substrates and sodium periodate. Five equivalents sodium periodate were used, and the reactions were carried out at room temperature overnight. β-Elimination was then induced with potassium carbonate at room temperature as well. All the dM-Dim ester substrates 7a-h gave good to excellent yields of the corresponding carboxylic acid (Table 2). The reaction of the side product (4 with R′ = Me) from the deprotection, which could potentially serve as a Michael acceptor, with the resulting carboxylic acid was not observed. The steric hindrance from the two methyl groups could be responsible for preventing the reaction. For oxidation, we also tried to use lower equivalent sodium periodate including two equivalents, the reaction was found slower and gave lower yields of carboxylic acids.
Table 2.
Deprotection of dM-Dim protected carboxylic acids.a

|
|||
|---|---|---|---|
|
| |||
| Entry | 7 | 8 | Yield b |
| 1 | 7a |
|
95% |
| 2 | 7b |

|
80% |
| 3 | 7c |

|
80% |
| 4 | 7d |

|
97% |
| 5 | 7e |

|
95% |
| 6 | 7f |

|
95% |
| 7 | 7g |

|
80% |
| 8 | 7h |

|
70% |
Reaction conditions: 7 (1 equiv.), NaIO4 (5 equiv.), THF/H2O (1:1, v/v), rt, 12 h; then K2CO3 (10 equiv.), rt, 1 h.
Isolated yields.
Considering that the dM-Dim protect carboxylic acids such as 7a-h are tertiary alkyl esters, they should behave similarly as tertiary butyl protected carboxylic acids in terms of stability under basic conditions. To demonstrate this potential desirable property, we treated 7a (1 equivalent) and methyl benzoate (1 equivalent) with sodium hydroxide (10 equivalents) in THF at reflux temperature in the same pot (supporting information). The reactions were monitored with TLC. After 2 hours, methyl benzoate was completely converted to benzoate while 7a remained intact as indicated by the absence of alcohol side product from 7a and the presence of 7a. After aqueous workup and flash chromatography, pure 7a and benzoic acid were obtained in 98% and 74% yields, respectively. This result is significant because it shows that methyl protected carboxylic acid groups can be selectively deprotected in the presence of dM-Dim protected ones. We also evaluated the stability of 7a under acidic conditions. Compared with tertiary butyl carboxylates, 7a could be more stable because the electron withdrawing sulfur atoms in the group could destabilize the carbocation that has to be formed during decomposition under acidic conditions. We treated 7a (1 equivalent) and tertiary butyl benzoate (1 equivalent) with TFA at room temperature in the same pot. The reactions were monitored with TLC. After 30 minutes, tertiary butyl benzoate was completely consumed while 7a remained intact. However, when we tried to isolate 7a, the compound decomposed during workup. These results indicated that selective deprotection of tertiary butyl protected carboxylic acid groups in the presence of dM-Dim protected ones is challenging but not impossible.
We next showed that dM-Dim protected carboxylic acid groups can also be selectively deprotected in the presence of methyl and tertiary butyl protected ones. Stirring equal equivalents of 7a and methyl benzoate (both 1 equivalent) in a solution of sodium periodate (5 equivalents) in THF and water (v/v, 1:1) for 8 hours at room temperature followed by a brief contact with potassium carbonate completely deprotected 7a. The methyl benzoate was intact as indicated by TLC. After aqueous workup and flash chromatography, pure benzoic acid and methyl benzoate were obtained in 88% and 98% yields, respectively (supporting information). The same conditions were also applied to 7a and tertiary butyl benzoate. Again, selective deprotection of 7a in the presence of tertiary butyl benzoate was easily achieved, and pure benzoic acid and tertiary butyl benzoate were obtained in 90% and 94% yields, respectively (supporting information). To show that dM-Dim protected carboxylic acid group can be selectively deprotected in the presence of tertiary butyl ester in the same molecule, compound 8 was prepared. Oxidation of the compound under the general oxidation conditions with sodium periodate followed by treating the intermediate with a buffer solution of equal equivalent of acetic acid and sodium acetate gave compound 9 in 87% yield (scheme 2). The use of the buffer solution instead of potassium carbonate as in the general deprotection procedure to induce β-elimination was needed to prevent 9 from cyclizing to succinic anhydride.
Scheme 2.

Selective deprotection of dM-Dmoc protected carboxylic acid in the presence of a tertiary butyl ester.
To determine if a ketene intermediate was indeed formed during the protection of aliphatic carboxylic acids such as during the synthesis of ester 7h, the commercially available enantioenriched (S)-2-methylbutyric acid (10) was converted to the acid chloride 11 (Scheme 3), which was reacted with the lithium salt of 5 to give the dM-Dim protected ester 12 using the general protection procedure. Treating 12 using the general deprotection procedure gave back the compound 10. To determine if 10 was racemized during the protection and deprotection processes via the formation of a ketene intermediate or any other mechanisms, 10 was reacted with the enantioenriched 13 to give compound 14 (Scheme 3). HPLC analysis indicated that there was primarily one diastereoisomer, which must be (S,S)-14 because there was no possible pathway to racemize the amine moiety of the amide in the amide formation process. However, there was a chance that the two potential diastereoisomers existed but they could not be resolved under the HPLC conditions. To find out if this was the case, the commercial enantioenriched (S)-10 and (R)-10 were coupled with (S)-13 using DCC and HOBt to give (S,S)-14 and (R,S)-14, respectively. The two reference diastereoisomers were analyzed under the same HPLC conditions and we found that they were well resolved (see supporting information). These experiments showed that the formation of ketenes during the protection process was unlikely, and racemization of chiral carboxylic acids could be avoided during the entire protection and deprotection processes. Therefore, the dM-Dim protection strategy can be used for the protection of chiral carboxylic acids that possess a potentially racemizable chiral center α to the carbonyl group.
Scheme 3.

dM-Dim protection of chiral aliphatic carboxylic acid.
Finally, we tested if the dM-Dim protection strategy was compatible with substrates that possess the 1,2-diol functionality. We treated equal equivalents of compound 7a and (R,R)-hydrobenzoin with two equivalents of sodium periodate in DCM at room temperature. After only one hour, TLC indicated that the diol was completely consumed while 7a was found intact. The cleavage of 1,2-diol with sodium periodate under mild conditions is consistent with reports in the literature.26–30 The experimental results suggest that if the dM-Dim carboxylic acid protection strategy is used on substrates with a 1,2-diol functionality, the 1,2-diol has to be protected at the time of deprotecting the dM-Dim group if sodium periodate is used for the deprotection.
Conclusion
In conclusion, the dM-Dim group was found suitable for orthogonal carboxylic acid protection. Suitable conditions for the formation of the hindered dM-Dim esters were identified. Deprotection was achieved using sodium periodate followed by potassium carbonate at room temperature. Good to excellent yields of carboxylic acids could be obtained. The protection and deprotection procedures do not cause racemization of chiral carboxylic acids that have a chiral tertiary carbon center α to the carbonyl group. Compared to reported conditions for oxidative removal of Dim,7, 8, 18, 19 the new deprotection procedure does not need any transition metal as catalyst, and can be carried out conveniently without any special apparatus such as electrochemical cells. We also demonstrated that a methyl ester could be selectively hydrolyzed in the presence of a dM-Dim ester under basic conditions, and a tertiary butyl ester could possibly be selectively hydrolyzed in the presence of a dM-Dim ester under acidic conditions. In addition, the dM-Dim carboxylic acid protecting group can be easily removed under oxidative conditions in the presence of methyl and tertiary butyl esters. It is expected that the strategy of using dM-Dim for carboxylic acid protection will find applications in complex multistep synthesis where orthogonal protection is crucial for success.
Supplementary Material
Acknowledgments
Financial support from NIH (R15GM109288), NSF (CHE1111192), PHF Graduate Assistantship (S.S.) and MTU SURF (J.G. and T.W.); the assistance from D. W. Seppala (electronics), J. L. Lutz (NMR), L. R. Mazzoleni (MS), M. Khaksari (MS), and A. Galerneau (MS); and NSF equipment grants (CHE1048655, CHE9512455, AGS1531454); are gratefully acknowledged.
Footnotes
Supporting Information: Experimental details, HPLC profiles of (S,S)-14 and (R,S)-14, and 1H and 13C NMR spectra of new compounds. Supplementary data associated with this article can be found, in the online version, at.
References
- 1.Wuts PGM, Greene TW. Protecting Groups in Organic Synthesis. 4th. John Wiley & Sons; Hoboken, New Jersey: 2007. [Google Scholar]
- 2.Klan P, Solomek T, Bochet CG, Blanc A, Givens R, Rubina M, Popik V, Kostikov A, Wirz J. Chem Rev. 2013;113:119–191. doi: 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jarowicki K, Kocienski P. J Chem Soc Perk T. 2001;1:2109–2135. [Google Scholar]
- 4.Watanabe Y, Ohno Y, Ueno Y, Toru T. J Chem Soc Perk T. 1998;1:1087–1093. [Google Scholar]
- 5.Della Sala G, Labano S, Lattanzi A, Tedesco C, Scettri A. Synthesis-Stuttgart. 2002:505–510. [Google Scholar]
- 6.Kunz H, Barthels R. Chem Ber-Recl. 1982;115:833–845. [Google Scholar]
- 7.Barthels R, Kunz H. Angew Chem Int Ed. 1982;21:292–292. [Google Scholar]
- 8.Waldmann H, Kunz H. J Org Chem. 1988;53:4172–4175. [Google Scholar]
- 9.Barthels R, Kunz H. Angew Chem. 1982;94:302–302. [Google Scholar]
- 10.Behrendt R, White P, Offer J. J Pept Sci. 2016;22:4–27. doi: 10.1002/psc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isidro-Llobet A, Alvarez M, Albericio F. Chem Rev. 2009;109:2455–2504. doi: 10.1021/cr800323s. [DOI] [PubMed] [Google Scholar]
- 12.Chandrudu S, Simerska P, Toth I. Molecules. 2013;18:4373–4388. doi: 10.3390/molecules18044373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seebach D, Kimmerlin T, Sebesta R, Campo MA, Beck AK. Tetrahedron. 2004;60:7455–7506. [Google Scholar]
- 14.Reese CB. Org Biomol Chem. 2005;3:3851–3868. doi: 10.1039/b510458k. [DOI] [PubMed] [Google Scholar]
- 15.Virta P, Katajisto J, Niittymaki T, Lonnberg H. Tetrahedron. 2003;59:5137–5174. [Google Scholar]
- 16.Reese CB. Tetrahedron. 2002;58:8893–8920. [Google Scholar]
- 17.Lin X, Chen JS, Shahsavari S, Green N, Goyal D, Fang SY. Org Lett. 2016;18:3870–3873. doi: 10.1021/acs.orglett.6b01878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunz H. Chem Ber. 1976;109:3693–3706. [Google Scholar]
- 19.Barnhurst LA, Wan YQ, Kutateladze AG. Org Lett. 2000;2:799–801. doi: 10.1021/ol005537w. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi S, Ohira A, Miyoshi N, Mashio H, Ohgo Y. Tetrahedron Asymmetr. 1994;5:1763–1780. [Google Scholar]
- 21.Richmond E, Duguet N, Slawin AMZ, Lebl T, Smith AD. Org Lett. 2012;14:2762–2765. doi: 10.1021/ol300982f. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura Y, Takeuchi S, Ohira A, Ohgo Y. Tetrahedron Lett. 1996;37:2805–2808. [Google Scholar]
- 23.Naef F, Decorzant R. Tetrahedron. 1986;42:3245–3250. [Google Scholar]
- 24.Douglas JJ, Churchill G, Slawin AMZ, Fox DJ, Smith AD. Chem Eur J. 2015;21:16354–16358. doi: 10.1002/chem.201503308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weidert PJ, Geyer E, Horner L. Liebigs Ann Chem. 1989:533–538. [Google Scholar]
- 26.Spannring P, Yazerski V, Bruijnincx PCA, Weckhuysen BM, Kleingebbink RJM. Chem Eur J. 2013;19:15012–15018. doi: 10.1002/chem.201301371. [DOI] [PubMed] [Google Scholar]
- 27.Crimmins MT, Knight JD, Williams PS, Zhang Y. Org Lett. 2014;16:2458–2461. doi: 10.1021/ol5008422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gracias V, Gasiecki AF, Djuric SW. Tetrahedron Lett. 2005;46:9049–9052. [Google Scholar]
- 29.She J, Lampe JW, Polianski AB, Watson PS. Tetrahedron Lett. 2009;50:298–301. [Google Scholar]
- 30.Yamakawa R, Kiyota R, Taguri T, Ando T. Tetrahedron Lett. 2011;52:5808–5811. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.