

Abstract
T cells are a heterogeneous population of cells that differ in their differentiation stages. Functional states are reflected in the epigenome that confers stability in cellular identity and is therefore important for naïve as well as memory T cell function. In many cellular systems, changes in chromatin structure due to alterations in histone expression, histone modifications and DNA methylation are characteristic of the aging process and cause or at least contribute to cellular dysfunction in senescence. Here, we review the epigenetic changes in T cells that occur with age and discuss them in the context of canonical epigenetic marks in aging model systems as well as recent findings of chromatin accessibility changes in T cell differentiation. Remarkably, transcription factor networks driving T cell differentiation account for many of the age-associated modifications in chromatin structures suggesting that loss of quiescence and activation of differentiation pathways are major components of T cell aging.
Keywords: Immunosenescence, chromatin accessibility, T cell differentiation, histone modification, DNA methylation, transcription factor
Introduction
A distinguishing feature of the T cell system is its enormous plasticity at the single cell level [1]. As naïve T cells, they essentially function as their own stem cells repopulating the system. Homeostatic proliferation of naïve T cells is of particular importance in the human adult and maintains compartment size, when the thymus becomes too involuted to make a relevant contribution to T cell generation [2, 3]. Upon antigen recognition and under the influence of environmental cues, T cells differentiate into multiple lineages of effector and memory T cells. These different functional states of T cells are reflected in the organization of the chromatin that is modified by transcription factor networks downstream of signaling cascades [4]. Ultimately, the epigenome regulates gene transcription by controlling accessibility of transcription factors to enhancer and promoter regions and facilitating their interaction in the three-dimensional space [5, 6]. For naïve T cells, the epigenetic state keeps the cells alive and quiescent while maintaining their ability to proliferate and differentiate when stimulated. Conversely, the chromatin structure provides stability to memory states after the antigen has been eliminated [7]. The epigenetic landscape of chromatin accessibility sites developed in effector T cells is maintained in memory cells for many years and is therefore an important mechanism for immune memory [8].
Any disarray in the epigenetic structure can result in cellular dysfunction. It is therefore reasonable to implicate epigenetic modifications in the pathologies associated with aging, if not even taking the bolder view to see the functionality of the epigenome as a major driver of the aging process including immune aging [9–11]. Here we review how studies of the epigenetic landscape in human T cells are beginning to be informative to understand the mechanisms that drive T cell aging [12, 13]. In particular, we will discuss two not necessarily mutually exclusive explanations for age-associated epigenetic signatures; their resemblance to epigenetic pathways that drive aging in model organisms; and their relationship to epigenetic modifications that are seen with normal T cell differentiation.
The organization of the chromatin
The DNA in eukaryotic cells is packed into chromatin with nucleosomes being the basic unit (Figure 1). Nucleosomes are composed of an octamer of four core histones (H3, H4, H2A and H2B) [14]. The amino-terminal tails of these core histones are subject to a wide range of posttranslational covalent modifications [6, 15]. Histone modifications establish the global chromatin structure and help partition the genome into distinct domains of euchromatin where DNA is potentially accessible for transcription, and heterochromatin of unaccessible DNA. Best characterized modifications are acetylation and methylation at Lys and Arg residues and phosphorylation. Adding extra complexity, methylation can be in the form of mono-, di-, and trimethyl for Lys and mono- and dimethyl for Arg. Posttranslational modification function by disrupting the contact between nucleosomes to unravel the chromatin and increase accessibility as well as by determining the recruitment and binding of enzymes and other non-histone proteins that regulate gene activation or repression. Acetylation has the most potential to unfold chromatin since it neutralizes the basic charge of the lysine. Also, trimethylation of histone 3 lysine 4 (H3K4me3) at promoters is generally associated with transcriptional activity while trimethylation of histone 3 lysine 27 (H3K27me3) with transcriptional repression [6].
Figure 1. Epigenetic control of gene expression.
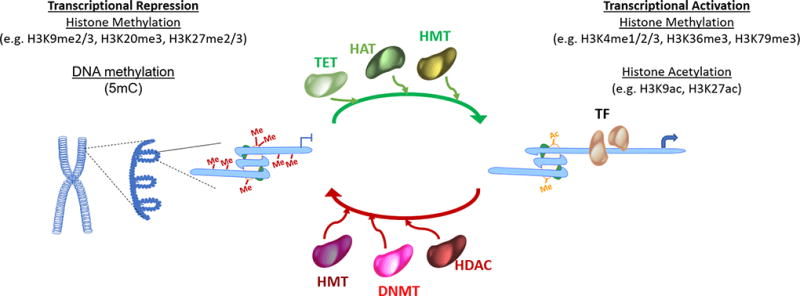
DNA methylation and post-translational histone modifications alter the chromatin structure, thereby facilitation activation or repression of gene transcription.
In addition to histone modification, epigenetic control of transcription is attained through DNA methylation, most frequently on carbon 5 of cytosine in ‘CpG’ dinucleotides (cytosine followed by guanine in the 5′ → 3′ direction). Humans have three functional DNA methyl transferases that transfer the methyl group from S-adenosyl-methionine [16, 17]. DNMT1 is the most abundant DNA methyltransferase in mammalian cells and adds methyl groups to hemimethylated CpG di-nucleotides; its main function is to maintain the methylation during DNA replication. DNMT3A and DNMT3B can methylate hemimethylated and unmethylated CpG, they are critical in establishing methylation states [16, 18]. In humans, 70% to 80% of CpG cytosines are methylated. A notable exception is the CpG island, defined as a DNA region of more than 500 base pairs with at least 50% of GC content. The methylation states of CpG islands, frequently located at promoter regions, are important for regulating gene transcription [17, 19, 20]. Other forms of DNA methylation such as cytosine methylation at non-CpG dinucleotides or 5-hydroxymethylcytosine have been described. Conversely, DNA demethylation is carried out by members of the ten-eleven translocation (TET) family of enzymes [21].
Epigenetic studies in T cells
Epigenetic changes occur with T cell activation and differentiation [22]. The acquisition of effector and memory T cell functions requires not only that genes involved in lineage commitment are turned on but also that genes maintaining stemness are repressed [23–26]. Consequently, DNA methylation as well as DNA demethylation and histone modifications conferring gene activation as well as repression are required for T cells to differentiate [27]. Equally important, the epigenetic code can act as the memory of cellular identity because it can be transmitted from one cell generation to the next [28], obviously crucial for memory T cells that exhibit chromatin accessibility maps similar to effector T cells years after the inciting infection has been cleared [8].
Epigenetic studies of aging so far have mostly focused on global measurements such as histone expression or modification. With the exception for DNA methylation, an impediment for genome-wide profiling of locus-specific changes has been so far the need for high cell numbers. This has been a barrier in particular for human studies and for cell populations as heterogeneous as T cells that include numerous subsets of different states of differentiation, not to speak of the low frequencies of cells sharing the same antigen specificity. In recent years, progress has been accomplished that make such studies increasingly feasible. The ENCODE (Encyclopedia of DNA Elements) Consortium funded by the National Human Genome Research Institute has generated an impressive list of functional elements in the human genome in different cell types that provides a reference point for interpreting human epigenetic data. Equally important, techniques have been developed that can scale down to lower cell numbers. A prime example is the Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq) [29], a method for mapping chromatin accessibility genome-wide that even allows single cell studies [30] and that has been successfully used to track epigenetic changes in antigen-specific T cells after viral infection [8, 31–34]. Chromatin profiling strategies have also been improved to work with smaller cell numbers than conventional chromatin immunoprecipitation assays; Cleavage Under Targets and Release Using Nuclease (CUT&RUN) uses antibody-targeted controlled cleavage by micrococcal nuclease to generate specific protein-DNA complexes for paired-end DNA sequencing [35].
Histone expression in model systems of aging
In a variety of aging models, a loss of core histone protein levels has been observed (Figure 2) [11, 36–38]. The loss in histones appears to be linked to cell division, possibly related to the tight control of histone transcription in the cell cycle with most of the transcription occurring in the G1 to S transition and downregulated at cell cycle exit. Reduced histone expression has been shown in models as diverse as yeast replicative aging, mammalian models of cellular senescence and muscle stem cells from old mice [39]. Replicatively aged yeast showed a global nucleosome loss of ~50% [40] and abnormalities in positioning of existing nucleosomes. Moreover, the epigenome of senescent cells characteristically includes splice variants of canonical histones that are incorporated into nucleosomes independent of replication [41]. The importance of histone dose in the aging process is further supported by the finding that overexpression of histone H3 and H4, but not histone H2A and H2B, extends yeast replicative lifespan [42]. Functional consequences of loss or fuzzy positioning of nucleosomes may reach from transcriptional noise to frank expression of repressed genes [40, 43]. Interestingly, increased transcriptional noise with age appears to be a feature of post-mitotic cells, but not of actively replicating cells like stem cells or naïve T cells [44]. However, age-associated changes in nucleosome structure in senescent cells have been implicated in the transcriptional gene activation resulting in the so-called senescence-associated secretory phenotype, SASP that is characterized by the aberrant production of inflammatory mediators and that contributes to the increased background inflammation in older individuals [45]. Data on histone expression and nucleosome structures with age in T cells are lacking, but will be of interest in particular for terminally differentiated effector T cells (TEMRA) that produce many inflammatory cytokines including those preferentially produced by innate immune cells [46]. It has been suggested that these T cells represent a convergence between innate and adaptive immunity, which may be driven by cellular senescence [47, 48]. So far, the technology to study nucleosome structures in infrequent cell populations has not been available, however, the recent development of ATAC-seq to generate chromatin accessibility maps in infrequent cell population has opened new opportunities and analytical approaches to derive information on nucleosome positioning [49]. Moreover, using ATAC-seq, Ucar et al. found significant chromatin closing with aging of several histone genes (HIST1H3D, HIST1H3E, HIST4H4) consistent with the reduced expression of core histones in model systems of aging [50].
Figure 2. Epigenetic mechanisms in aging.
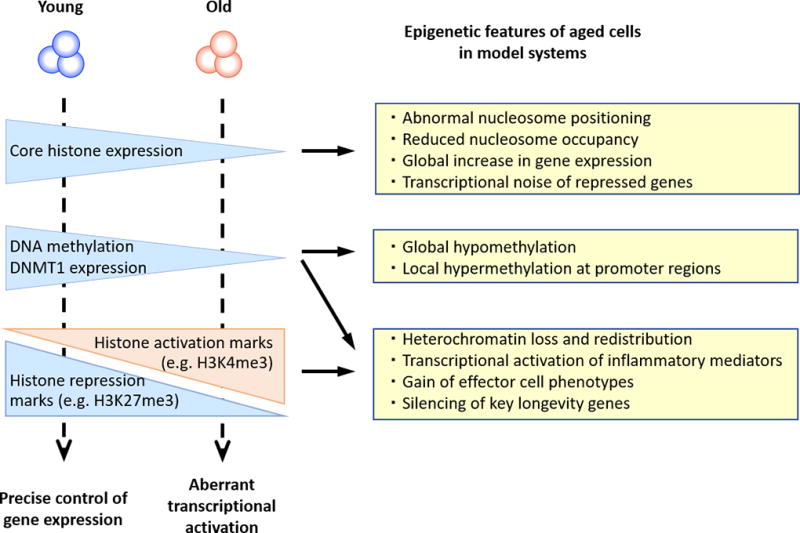
Changes in chromatin structure due to altered histone expression, histone modifications and DNA methylation occur with aging and contribute to cellular dysfunction.
Histone modifications in aging
Age-associated epigenetic changes encompass a broad decrease of heterochromatin, site-specific decrease of heterochromatin, as well as region-specific gains of heterochromatin (Figure 2) [51]. Broadly interpreted, activating histone marks are gained and repressive marks are lost with age [11, 38]. In addition, gene-specific changes in chromatin states regulating expression of key longevity genes occur. Sun and colleagues compared young and old purified murine bone marrow hematopoietic stem cells by RNA-Seq, ChIP-Seq for histone modification and DNA methylation arrays [52]. They found an accumulation of H3K4me3 with age, particularly on genes in which H3K4-trimethylation is already broad and which are associated with stem cell self-renewal, letting the authors to suggest that stem cell aging epigenetically is a loss in differentiation capacity.
Human studies are mostly based on cultured cells that have reached senescence due to excessive replication or cellular stress. Cellular senescence is characterized by a state of cell cycle arrest [53] and characterized by a dynamic and imbalanced chromatin environment that is markedly different from the proliferating state. Many cultured mammalian cells develop senescence-associated heterochromatin foci, regions of highly condensed chromatin associated with heterochromatic histone modifications, heterochromatic proteins, and histone variants [54, 55].
Studies of histone modifications on human T cells directly ex vivo are just in the beginning, as techniques for genomic-wide histone profiling from low cell numbers become available. Chromatin accessibility maps allow a broad assessment. Studies by Ucar et al, mostly on unfractionated peripheral blood mononuclear cells, revealed a systematic loss of chromatin accessibility at the promoters and enhancers of immune module genes and a gain in accessibility in the vicinity of genes that in the ENCODE data base are described as generally repressed or quiescent. Moreover, genes for several histone modifiers (EZH1, SETD7) were found to be less accessible that cause histone modification patterns similar to those seen with cellular aging [50].
Whether T cells reach the state of cellular senescence is difficult to ascertain. Effector T cells are end-differentiated and post-mitotic including the expression of cell cycle inhibitors like p16 and p21 and therefore difficult to distinguish from senescent cells. There is no evidence that human naïve CD4 T cells lose their ability to proliferate and develop features of cellular senescence. Epigenetic patterns associated with differentiation have been mostly examined for naive CD8+ T cells. These studies describe a loss of activation-associated histone modifications, such as H3K4me3 and H3K9ac, and a gain in DNA methylation and H3K27me3 modifications at transcription factors that are associated with stemness or naivety, such as FOXO1, KLF2, LEF1 and TCF7 [22, 25, 56, 57]. Not surprisingly, the opposite histone modification pattern was seen for effector cell-associated transcription factors (EOMES, TBX21 and PRDM1) and functional effector genes (GZMA, GZMB, PRF1, IFNG). Moreover, transcription factor motif analysis showed an enrichment for FOXO1 and TCF1 at memory-specific enhancers and TCF1 at naive-specific enhancers. How this differentiation-associated histone modification pattern is different from aging remains to be seen. However, these data clearly emphasize the need to control for cell population heterogeneity in epigenetic aging studies, in particular for human CD8 T cells that experience a large loss in naïve and a gain in effector T cells with age (Table 1).
Table 1.
Subset-specific Differences of Human CD4 and CD8 T cells with Age
| CD4 T cells | CD8 T cells |
|---|---|
| Circulating naïve cell number decline moderately | Circulating naïve cell number decline markedly |
| Distribution of memory cell subsets is stable | Effector memory and TEMRA cells increase, mostly due to stimulation with latent viruses |
| Central memory cells remain CD45RO positive | Central memory cells revert to CD45RA, masquerading as naïve CD8 T cells |
| Naïve T cell homeostasis dependent on recognition of MHC class II molecules | Naïve T cell homeostasis dependent on recognition of MHC class I molecules |
| Decline in TCR richness in naïve cells by 3–5 fold | Decline in TCR richness in naïve cells by 3–5 fold |
| Minor TCR repertoire oligoclonality in naïve cells | Increased TCR repertoire oligoclonality in naïve cells |
| CpG methylation changes at >10,000 sites | CpG methylation changes at >40,000 sites |
| Minor changes in chromatin accessibility in naïve and central memory cells | Naïve and central memory cells exhibit evidence of progressive differentiation in their chromatin accessibility patterns |
| Normal mitochondrial function (oxygen consumption rates) in naive cells | Impaired mitochondrial function (reduced oxygen consumption rates) in naive cells |
DNA methylation in aging
Due to the availability of assay systems, genome-wide changes in DNA methylation are one of the best characterized epigenetic modifications in aging. Mammalian aging is generally associated with CpG hypomethylation, especially at repetitive regions of the genome in the heterochromatin paralleling the changes in histone modification (Figure 2) [58–61]. This loss may be attributed to a decline in DNMT1 expression with age [18]. It has been proposed that the loss of CpG methylation at repetitive sequences will heighten the risk of genomic instability due to retrotransposition events, although direct evidence in human aging is lacking [34, 51]. In contrast to this general demethylation, DNA methylation arrays have also identified regions of hypermethylation [9, 38]. These occur predominantly at promoter regions and are frequently tissue specific [62]. These observations appear to be also pertinent for T cells. A comparison of CD4 T cells from newborns and centenarians found global decreases in DNA methylation with age, accompanied by heterogeneous DNA methylation in the centenarian genome [61]. The majority of age-related changes occurred in CD8 T cells at CpG sites that correlated with the expression of effector molecules and transcriptional regulator genes with fundamental roles in CD8 T cell differentiation. An increased susceptibility of CD8 T cells to undergo epigenetic changes with age was also observed by Tserel et al who compared the methylome in purified CD4 and CD8 T cells from 50 young and 50 older adults using methylation arrays [63]. The authors identified approximate four times as many differentially methylated CpG sites in CD8 than in CD4 T cells (48,876 vs 12,275). Moreover, they found CpG methylation to be more variable in all CpG island subregions of CD8 T cells from older individuals. In this study, hypermethylation was mostly seen in CpG islands, while hypomethylated CpG sites were located at the border of CpG islands or in the gene body.
This increased age-associated variability in CD8 T cell may indicate that CD8 more than CD4 T cells change with age or it may reflect the increased population heterogeneity seen in CD8 T cells with age. Obviously, the two interpretations are not mutually exclusive. Consistent with the latter interpretation, CD4 TEMRA cells, generally considered to be senescent or end-differentiated, are less frequent than their CD8 counterparts but are similar in having reduced DNMT1 expression and reduced DNA methylation at effector molecules associated with cytotoxic function [64, 65].
To understand whether age-associated DNA methylation is functionally important, Reynolds et al. identified potentially functional age- and cis-gene expression-associated methylation sites (age-eMS) by integrating genome-wide CpG methylation and gene expression profiles from circulating T cells and monocytes from individuals aged 55 to 94 years [66]. None of the age-eMS detected in 227 T-cell samples were detectable in 1,264 monocyte samples suggesting that functional sites are tissue-specific. Age-eMS tended to be hypomethylated with older age, located in predicted enhancers and preferentially linked to expression of antigen processing and presentation genes. In the studies by Tserel et al., age-related hypo- and hypermethylated sites were mostly mapped to regions with repressive histone marks as identified by ENCODE [63]. Accordingly, transcription of many of these genes was low or absent, suggesting that they are not involved in basic cell maintenance. However, because all of these studies were done in quiescent cell, these epigenetic changes could confer differences in responsiveness to stimulation. For a subset of genes that were expressed in CD8 T cells, Tserel et al observed the expected inverse relationship between methylation and transcription. Again, most of the differentially regulated genes were related to T cell differentiation, i.e. gain of effector function (LGALS1, IFNG, CCL5, GZMH) and loss of naïve or central memory state (CCR7, CD27, TCF7, SAT1B) with age, either reflecting changes in CD8 T cell subset distribution or progressive differentiation within naïve and memory subsets with age..
Hannum et al. built a predictive model of the aging methylome from the blood of individuals aged 19 to 101 years and identified a set of 71 CpGs that have a high accuracy of age prediction and occur near genes associated with aging [67]. In a parallel study, Horvath et al. analyzed ~8000 samples representing 51 healthy human tissue and cell types including liver, kidney, immune and brain cells and ~6000 cancer samples and identified DNA methylation at 353 CpGs accurately predicted age [68, 69]. This signature has been coined as the epigenetic clock of aging and is independent of tissue function and stressors throughout lifetime. It is found in cell types with very different levels of replicative stress, setting it completely apart from cellular senescence. The function of these altered DNA methylation sites that appear to be tightly correlated to age remains elusive. By its nature of its tissue non-specificity, the signature should be present in all T cell subsets irrespective of their differentiation state, however, specific studies on functional T cell subsets are lacking. We have explored whether these differential methylation sites coincide with sites that change in chromatin accessibility in CD8 T cells with age [70] and have not found any relationship (unpublished observation).
Differential susceptibility of T cell subsets to undergo chromatin accessibility changes with age
Recent methodology advances have enabled mapping the regulatory chromatin landscapes that control gene expression. In particular, the assay for transposase-accessible chromatin by sequencing (ATAC-seq) can be applied to low cell numbers and even single cells to generate high-fidelity chromatin accessibility profiles and to derive conclusions on transcription factor networks that account for different cell states [29]. The technique has been successfully applied by us and others to define the epigenetic landscape of antigen-specific CD8 T cells as they differentiate from naïve T cells into functional effector and memory cells[8, 31, 32]. Two manuscripts have been recently published that used this technique to describe how the epigenetic landscapes of T cells change with aging and whether these changes allow conclusions on the mechanisms and regulatory transcription factor networks involved [50, 70]. Ucar and colleagues compared chromatin accessibility maps in peripheral blood mononuclear cells (PBMC) of twenty-eight 22 to 40 year-old healthy individuals and twenty-one individuals older than 65 years and identified 12,626 differentially accessible sites (9% of those tested) with approximately an equal number of sites opening or closing with age. Sites with increased accessibility were frequently subject-specific and located at less accessible chromatin region whereas closing sites mapped to promoter and enhancer regions that were shared between individuals. As discussed above, differentially open sites included histone and histone modifiers genes as well as increased accessibility to heterochromatin regions, suggesting that the generic epigenetic aging pathways discussed above for aging model systems also apply to T cells. However, the vast majority of genes in the vicinity of differentially open sites were genes that encode for molecules critical for immune function. Interestingly, PBMC subsets were not equally affected. Ucar et al followed up with an analysis of CD4 and CD8 T cell subset in a smaller cohort and found very few age-associated changes in CD4 T cells. In contrast, CD8 T cells and in particular CD8 memory T cells showed extensive chromatin remodeling and differentially open genes correlated with those found in PBMC. Many genes found to change in accessibility and expression with age were related to T cell differentiation; for example, the age-associated declines in the transcription factors LEF1 and TCF7 and the IL-7 receptor are also a hallmarks of effector T cell differentiation as is the increase in cytotoxic mediators. Whether this age-associated differentiation is driven by a limited set of transcription factors is unknown. One possible candidate is FOXO1 that controls expression of many of the genes associated with stemness, including IL7R. FOXO1 deficiency in mice is associated with a failure to maintain naïve T cells or to generate memory cells [71–73]. In preliminary studies, we have identified miRNAs that change with age. Pathway analysis of the genes regulated by these miRNAs have identified enrichment for the FOXO1 pathway [74].
The differential susceptibility of CD4 and CD8 T cells to age-associated epigenetic changes reflects the different biology of these cell subsets (Table 1). The age-associated loss in naïve T cells is much more dramatic for CD8 T cells [75, 76]. In fact, the loss in circulating naive CD8 T cells was the major age-associated immunological marker of all variables tested in a comprehensive immune analysis of 243 healthy individuals [77]. Also, the increase in effector T cells and in particular TEMRA cells is characteristic for CD8 T cells, but very minor for CD4 T cells. Moreover, the phenotypically defined naïve CD8 T cell compartment in older individuals include increased frequencies of clonally expanded populations that may represent either true or virtual memory cells and that is rarely seen for naïve CD4 T cells [78, 79]. The mechanism underlying these different subset behaviors, which is characteristic for human but not murine T cell aging, are unknown.
Epigenetic signatures of cellular differentiation in CD8 T cell aging
We performed a more detailed analysis of CD8 T cell aging to identify transcription factor networks that correlate with chromatin accessibility landscapes [70]. In these studies, we compared naïve, central memory and effector memory CD8 T cells in young and older healthy adults. TEMRA were excluded because this state is highly dependent on CMV infection rather than on age. We excluded all peaks that were only found in one sample, therefore focusing on functional sites, and we excluded sex chromosomes, which have been shown to account for much of inter-individual variation [80]. Hierarchical clustering of open sites segregated the three T cell subsets without clear impact of age, demonstrating that variability related to differentiation states by far outweighed that of age. These data re-emphasize the need for separately analyze subpopulations, in particular because the subset composition of CD8 T cells change with age. However, principal component analysis showed segregation by differentiation state as well as age in that old naïve and central memory T cells appeared to be more differentiated than those from young individuals are. Direct comparison identified age-related differences for naïve and central memory CD8 T cells, but not for effector T cells. Transcription factor motif analysis of differentially open sites identified bZIP family motifs enriched at sites more open and ETS family members at sites more closed in aged compared to young naïve and central memory cells, a transcription factor motif pattern that largely resembles that seen with differentiation from naïve to memory T cells. Interestingly, T-box family members such as TBX21 and EOMES, also hallmarks of differentiated cells, did not come up in the age analysis, either because the age-associated differentiation is incomplete or because our analysis was not sufficiently powered to detect an enrichment. Together with the findings by Ucar et al that TCF/LEF1 accessibility and FOXO-dependent IL-7R expression is lost with aging, our data suggest that one major dimension of CD8 T cell subset aging is progressive differentiation, i.e., naïve T cells have lost complete stemness and central memory T cells assume features of effector T cells with age. Single cell analysis will have to be performed to show whether this signature reflects increasing cell heterogeneity within each subset. Contamination with age of memory T cells that are phenotypically naïve may contribute [8, 78, 81], but are unlikely the only reason for this phenomenon. More likely, CD8 T cells enter progressive differentiation with age in absence of an exogenous antigenic driver, reminiscent of virtual memory T cells in the mouse. As Ucar et al have shown and unpublished results from our lab confirm, the age-associated differentiation is a feature of CD8 T cells and much less prominent in CD4 T cells [50]. In conclusion, T cell aging involves gene regulatory pathways that drive T cell differentiation and the increased susceptibility of CD8 T cells to age-associated changes appears to be a heightened inducibility of these pathways (Figure 3). This shift to differentiation is also seen at the transcriptome level, although less robust than with the epigenetic markers. Obviously, the relationship between chromatin accessibility and transcriptome is complex and not linear. It is one of the characteristic features of T cells that effector genes are poised in memory cells but not actively transcribed. However, we have found an increased transcription of BATF target genes in older naïve CD8 T cells consistent with increased accessibility to bZIP motifs [70].
Figure 3. The chromatin landscape in CD8 T cell differentiation and aging.
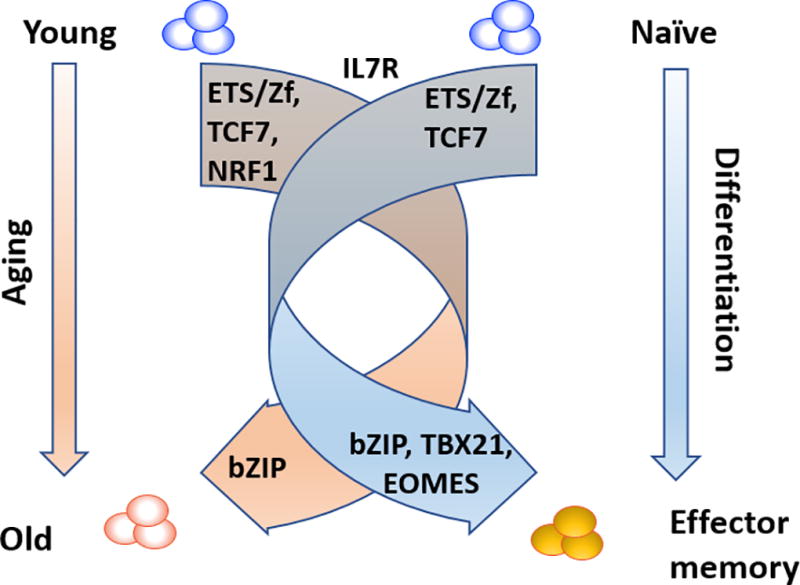
T cell differentiation is associated with major changes in chromatin accessibility. Age-associated changes are smaller by an order of magnitude, but exhibit similar patterns suggesting that transcription factor networks involved in T cell differentiation also drive T cell aging.
A second dimension of CD8 naïve and central memory T cell aging in these studies was a reduced chromatin accessibility at promoters that frequently involved an NRF1 binding site [70]. Interestingly, the NRF1 motif includes CpG sequences and knock-down experiments of DNMT1 have shown that NRF1 binding at selected promoters is more sensitive to DNA methylation than that of other transcription factors [82]. It is therefore possible that the reduced accessibility is related to the increased CpG methylation that is seen at selected promoters with age [63]. NRF1-binding sites mostly locate to promoters and not enhancers consistent with its role in regulating basic cell functions. In particular, NRF1 controls the expression of many mitochondrial genes, including the genes of the mitochondrial respiratory chain (MRC) [83–85]. Indeed, in RNA-seq experiments we found reduced expression of MRC genes with age. Moreover, aged naïve CD8, but not CD4 T cells have impaired mitochondrial function as seen in reduced oxygen consumption rates in Seahorse assays. In addition to changes in homeostatic pathways associated with T cell differentiation, such metabolic changes could also contribute to the accelerated loss of naïve CD8 T cells seen with age.
Concluding Remarks
Over the last decade, it has become clear that life span is at least in part epigenetically determined. Changes in the epigenome have a large influence on the aging process. Age-associated changes in the level of histone protein concentrations as well as in DNA methylation and histone modifications alter chromatin structure and local accessibility. Most recently, new technological developments have improved our ability to perform genome-wide gene-specific epigenetic studies in infrequent cell populations such as T cell subsets. In particular, chromatin accessibility mapping by ATAC-seq has been informative to define the epigenetic state of naïve T cells vs effector or memory T cells and follow the chromatin changes that occur in antigen-specific T cells when they are activated in a viral infection and differentiate into various effector T cells and memory T cells. These studies have led to the recognition that T cells undergo huge changes in chromatin structure with more than 20% of all accessible sites either opening or closing. Since T cell population compositions change with age, these large differences need to be controlled for when studying the influence of age on the T cell epigenome. Initial ATAC-seq studies on T cell subpopulations have provided evidence that T cells also acquire some of the key epigenetic changes thought to be causative for aging in model systems such as the downregulation of repressive marks; however, typical epigenetic marks of cellular senescence are largely elusive in T cells. In contrast, signatures that are characteristic for the differentiation process are gained with age and outnumber age-specific marks. Interestingly, there is a large difference in CD4 and CD8 T cell subsets to acquire these signatures, consistent with the observations that CD8 T cells are more susceptible to undergo age-associated functional changes (Table 1). Obviously, heterogeneity within phenotypically defined population need to be further studied. However, these data raise the interesting concept that the major aging process in T cells is the aberrant activation of transcription factor networks that generally drive the differentiation process after T cell activation, i.e., the major component of T cell aging is the failure to maintain stemness and the entering of a differentiation process.
Summary Statement.
The aging process in T cells is characterized by changes in chromatin structures, many of which resemble epigenetic modifications seen with differentiation.
Acknowledgments
This work was supported by the National Institutes of Health (R01 AR042527, R01 HL117913, R01 AI108906 and P01 HL129941 to CMW and R01 AI108891, R01 AG045779, U19 AI057266, R01 AI129191, and I01 BX001669 to JJG). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- ATAC-seq
Assay for Transposase Accessible Chromatin with high-throughput sequencing
- bZIP
Basic Leucine Zipper Domain
- CpG
Cytosine nucleotide followed by a guanine nucleotide in the linear sequence of bases along its 5' → 3' direction
- CMV
Cytomegalovirus
- DNMT
DNA methyltransferase
- ENCODE
Encyclopedia of DNA Elements
- EOMES
Eomesodermin
- ETS
E26 transformation-specific
- FOXO
Forkhead box O
- GZMA
Granzyme A
- GZMB
Granzyme B
- HAT
Histone acetyltransferase
- HDAC
Histone deacetylase
- HMT
histone methyltransferase
- IFNG
Interferon gamma
- KLF2
Krüppel-like Factor 2
- LEF
Lymphoid enhancer-binding factor
- MRC
mitochondrial respiratory chain
- NRF1
Nuclear respiratory factor 1
- PBMC
peripheral blood mononuclear cell
- PRDM1
PR domain zinc finger protein 1
- PRF1
Perforin-1
- SASP
Senescence-Associated Secretory Phenotype
- SETD7
SET Domain Containing Lysine Methyltransferase 7
- TBX21
T-Box 21
- TEMRA
T effector cell expressing CD45RA
- TET
ten-eleven translocation
- TF
transcription factor
Footnotes
The authors declare no competing financial interests.
References
- 1.DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol. 2016;16:149–63. doi: 10.1038/nri.2015.18. [DOI] [PubMed] [Google Scholar]
- 2.Boyman O, Letourneau S, Krieg C, Sprent J. Homeostatic proliferation and survival of naive and memory T cells. Eur J Immunol. 2009;39:2088–94. doi: 10.1002/eji.200939444. [DOI] [PubMed] [Google Scholar]
- 3.den Braber I, Mugwagwa T, Vrisekoop N, Westera L, Mogling R, de Boer AB, Willems N, Schrijver EH, Spierenburg G, Gaiser K, Mul E, Otto SA, Ruiter AF, Ackermans MT, Miedema F, Borghans JA, de Boer RJ, Tesselaar K. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity. 2012;36:288–97. doi: 10.1016/j.immuni.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Rothenberg EV. The chromatin landscape and transcription factors in T cell programming. Trends in immunology. 2014;35:195–204. doi: 10.1016/j.it.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brettingham-Moore KH, Taberlay PC, Holloway AF. Interplay between Transcription Factors and the Epigenome: Insight from the Role of RUNX1 in Leukemia. Front Immunol. 2015;6:499. doi: 10.3389/fimmu.2015.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Weng NP, Araki Y, Subedi K. The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation. Nat Rev Immunol. 2012;12:306–15. doi: 10.1038/nri3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, Youngblood BA, Abdelsamed HA, McGuire DJ, Cohen KW, Alexe G, Nagar S, McCausland MM, Gupta S, Tata P, Haining WN, McElrath MJ, Zhang D, Hu B, Greenleaf WJ, Goronzy JJ, Mulligan MJ, Hellerstein M, Ahmed R. Origin and differentiation of human memory CD8 T cells after vaccination. Nature. 2017;552:362–367. doi: 10.1038/nature24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jasiulionis MG. Abnormal Epigenetic Regulation of Immune System during Aging. Front Immunol. 2018;9:197. doi: 10.3389/fimmu.2018.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ucar D, Benayoun BA. Aging Epigenetics: Changes and Challenges. In: Moskalev A, Vaiserman AM, editors. Epigenetics of Aging and Longevity. Vol. 4. Elsevier Inc; 2017. pp. 3–32. [Google Scholar]
- 12.Goronzy JJ, Weyand CM. Successful and Maladaptive T Cell Aging. Immunity. 2017;46:364–378. doi: 10.1016/j.immuni.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikolich-Zugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. 2018;19:10–19. doi: 10.1038/s41590-017-0006-x. [DOI] [PubMed] [Google Scholar]
- 14.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 15.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–28. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19:81–92. doi: 10.1038/nrg.2017.80. [DOI] [PubMed] [Google Scholar]
- 17.Zampieri M, Ciccarone F, Calabrese R, Franceschi C, Burkle A, Caiafa P. Reconfiguration of DNA methylation in aging. Mech Ageing Dev. 2015;151:60–70. doi: 10.1016/j.mad.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Ciccarone F, Malavolta M, Calabrese R, Guastafierro T, Bacalini MG, Reale A, Franceschi C, Capri M, Hervonen A, Hurme M, Grubeck-Loebenstein B, Koller B, Bernhardt J, Schn C, Slagboom PE, Toussaint O, Sikora E, Gonos ES, Breusing N, Grune T, Jansen E, Dolle M, Moreno-Villanueva M, Sindlinger T, Burkle A, Zampieri M, Caiafa P. Age-dependent expression of DNMT1 and DNMT3B in PBMCs from a large European population enrolled in the MARK-AGE study. Aging Cell. 2016;15:755–65. doi: 10.1111/acel.12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu H, Caffo B, Jaffee HA, Irizarry RA, Feinberg AP. Redefining CpG islands using hidden Markov models. Biostatistics. 2010;11:499–514. doi: 10.1093/biostatistics/kxq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreira HJ, Esteller M. CpG Islands in Cancer: Heads, Tails, and Sides. Methods Mol Biol. 2018;1766:49–80. doi: 10.1007/978-1-4939-7768-0_4. [DOI] [PubMed] [Google Scholar]
- 21.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733–50. doi: 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol. 2018 doi: 10.1038/nri.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henning AN, Klebanoff CA, Restifo NP. Silencing stemness in T cell differentiation. Science. 2018;359:163–164. doi: 10.1126/science.aar5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ladle BH, Li KP, Phillips MJ, Pucsek AB, Haile A, Powell JD, Jaffee EM, Hildeman DA, Gamper CJ. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proc Natl Acad Sci U S A. 2016;113:10631–6. doi: 10.1073/pnas.1524490113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray SM, Amezquita RA, Guan T, Kleinstein SH, Kaech SM. Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8(+) T Cell Terminal Differentiation and Loss of Multipotency. Immunity. 2017;46:596–608. doi: 10.1016/j.immuni.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall JJ, Quivy JP, Almouzni G, Amigorena S. The epigenetic control of stemness in CD8(+) T cell fate commitment. Science. 2018;359:177–186. doi: 10.1126/science.aah6499. [DOI] [PubMed] [Google Scholar]
- 27.Abdelsamed HA, Moustaki A, Fan Y, Dogra P, Ghoneim HE, Zebley CC, Triplett BM, Sekaly RP, Youngblood B. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med. 2017;214:1593–1606. doi: 10.1084/jem.20161760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moazed D. Mechanisms for the inheritance of chromatin states. Cell. 2011;146:510–8. doi: 10.1016/j.cell.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–8. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–90. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott-Browne JP, Lopez-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, Pereira RM. Dynamic Changes in Chromatin Accessibility Occur in CD8+ T Cells Responding to Viral Infection. Immunity. 2016;45:1327–1340. doi: 10.1016/j.immuni.2016.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, Tonnerre P, Chung RT, Tully DC, Allen TM, Frahm N, Lauer GM, Wherry EJ, Yosef N, Haining WN. The epigenetic landscape of T cell exhaustion. Science. 2016;354:1165–1169. doi: 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, Barnitz RA, Bartman C, Bengsch B, Huang AC, Schenkel JM, Vahedi G, Haining WN, Berger SL, Wherry EJ. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rendeiro AF, Schmidl C, Strefford JC, Walewska R, Davis Z, Farlik M, Oscier D, Bock C. Chromatin accessibility maps of chronic lymphocytic leukaemia identify subtype-specific epigenome signatures and transcription regulatory networks. Nat Commun. 2016;7:11938. doi: 10.1038/ncomms11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skene PJ, Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. 2017;6:e21856. doi: 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802–7. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol. 2010;17:1218–25. doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sen P, Shah PP, Nativio R, Berger SL. Epigenetic Mechanisms of Longevity and Aging. Cell. 2016;166:822–839. doi: 10.1016/j.cell.2016.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu L, Cheung TH, Charville GW, Hurgo BM, Leavitt T, Shih J, Brunet A, Rando TA. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4:189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu Z, Chen K, Xia Z, Chavez M, Pal S, Seol JH, Chen CC, Li W, Tyler JK. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014;28:396–408. doi: 10.1101/gad.233221.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, Hewitt RN, Anannya O, McBryan T, Manoharan I, van Tuyn J, Morrice N, Pchelintsev NA, Ivanov A, Brock C, Drotar ME, Nixon C, Clark W, Sansom OJ, Anderson KI, King A, Blyth K, Adams PD. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014;28:2712–25. doi: 10.1101/gad.247528.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, Tyler JK. Elevated histone expression promotes life span extension. Mol Cell. 2010;39:724–35. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Celona B, Weiner A, Di Felice F, Mancuso FM, Cesarini E, Rossi RL, Gregory L, Baban D, Rossetti G, Grianti P, Pagani M, Bonaldi T, Ragoussis J, Friedman N, Camilloni G, Bianchi ME, Agresti A. Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol. 2011;9:e1001086. doi: 10.1371/journal.pbio.1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warren LA, Rossi DJ, Schiebinger GR, Weissman IL, Kim SK, Quake SR. Transcriptional instability is not a universal attribute of aging. Aging Cell. 2007;6:775–82. doi: 10.1111/j.1474-9726.2007.00337.x. [DOI] [PubMed] [Google Scholar]
- 45.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodriguez RM, Suarez-Alvarez B, Lavin JL, Mosen-Ansorena D, Baragano Raneros A, Marquez-Kisinousky L, Aransay AM, Lopez-Larrea C. Epigenetic Networks Regulate the Transcriptional Program in Memory and Terminally Differentiated CD8+ T Cells. J Immunol. 2017;198:937–949. doi: 10.4049/jimmunol.1601102. [DOI] [PubMed] [Google Scholar]
- 47.Pereira BI, Akbar AN. Convergence of Innate and Adaptive Immunity during Human Aging. Front Immunol. 2016;7:445. doi: 10.3389/fimmu.2016.00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Warrington KJ, Takemura S, Goronzy JJ, Weyand CM. CD4+,CD28− T cells in rheumatoid arthritis patients combine features of the innate and adaptive immune systems. Arthritis Rheum. 2001;44:13–20. doi: 10.1002/1529-0131(200101)44:1<13::AID-ANR3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 49.Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 2015;25:1757–70. doi: 10.1101/gr.192294.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ucar D, Marquez EJ, Chung CH, Marches R, Rossi RJ, Uyar A, Wu TC, George J, Stitzel ML, Palucka AK, Kuchel GA, Banchereau J. The chromatin accessibility signature of human immune aging stems from CD8(+) T cells. J Exp Med. 2017;214:3123–3144. doi: 10.1084/jem.20170416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pal S, Tyler JK. Epigenetics and aging. Sci Adv. 2016;2:e1600584. doi: 10.1126/sciadv.1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, Gu H, Bock C, Meissner A, Gottgens B, Darlington GJ, Li W, Goodell MA. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–88. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–46. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll T, Stark R, Janky R, Narita M, Xue L, Chicas A, Nunez S, Janknecht R, Hayashi-Takanaka Y, Wilson MD, Marshall A, Odom DT, Babu MM, Bazett-Jones DP, Tavare S, Edwards PA, Lowe SW, Kimura H, Gilbert DM, Narita M. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell. 2012;47:203–14. doi: 10.1016/j.molcel.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang R, Adams PD. Heterochromatin and its relationship to cell senescence and cancer therapy. Cell Cycle. 2007;6:784–9. doi: 10.4161/cc.6.7.4079. [DOI] [PubMed] [Google Scholar]
- 56.Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, Li Y, Phillips FC, Maina PK, Qi HH, Liu C, Zhu J, Pope RM, Musselman CA, Zeng C, Peng W, Xue HH. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat Immunol. 2016;17:695–703. doi: 10.1038/ni.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roychoudhuri R, Clever D, Li P, Wakabayashi Y, Quinn KM, Klebanoff CA, Ji Y, Sukumar M, Eil RL, Yu Z, Spolski R, Palmer DC, Pan JH, Patel SJ, Macallan DC, Fabozzi G, Shih HY, Kanno Y, Muto A, Zhu J, Gattinoni L, O'Shea JJ, Okkenhaug K, Igarashi K, Leonard WJ, Restifo NP. BACH2 regulates CD8(+) T cell differentiation by controlling access of AP-1 factors to enhancers. Nat Immunol. 2016;17:851–860. doi: 10.1038/ni.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bocklandt S, Lin W, Sehl ME, Sanchez FJ, Sinsheimer JS, Horvath S, Vilain E. Epigenetic predictor of age. PLoS One. 2011;6:e14821. doi: 10.1371/journal.pone.0014821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Acevedo N, Reinius LE, Vitezic M, Fortino V, Soderhall C, Honkanen H, Veijola R, Simell O, Toppari J, Ilonen J, Knip M, Scheynius A, Hyoty H, Greco D, Kere J. Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes. Clin Epigenetics. 2015;7:34. doi: 10.1186/s13148-015-0064-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petkovich DA, Podolskiy DI, Lobanov AV, Lee SG, Miller RA, Gladyshev VN. Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions. Cell Metab. 2017;25:954–960. e6. doi: 10.1016/j.cmet.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, Puca AA, Sayols S, Pujana MA, Serra-Musach J, Iglesias-Platas I, Formiga F, Fernandez AF, Fraga MF, Heath SC, Valencia A, Gut IG, Wang J, Esteller M. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A. 2012;109:10522–7. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, Myers RM, Absher D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013;14:R102. doi: 10.1186/gb-2013-14-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tserel L, Kolde R, Limbach M, Tretyakov K, Kasela S, Kisand K, Saare M, Vilo J, Metspalu A, Milani L, Peterson P. Age-related profiling of DNA methylation in CD8+ T cells reveals changes in immune response and transcriptional regulator genes. Sci Rep. 2015;5:13107. doi: 10.1038/srep13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y, Chen Y, Richardson B. Decreased DNA methyltransferase levels contribute to abnormal gene expression in "senescent" CD4(+)CD28(−) T cells. Clin Immunol. 2009;132:257–65. doi: 10.1016/j.clim.2009.03.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suarez-Alvarez B, Rodriguez RM, Schlangen K, Raneros AB, Marquez-Kisinousky L, Fernandez AF, Diaz-Corte C, Aransay AM, Lopez-Larrea C. Phenotypic characteristics of aged CD4(+) CD28(null) T lymphocytes are determined by changes in the whole-genome DNA methylation pattern. Aging Cell. 2017;16:293–303. doi: 10.1111/acel.12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reynolds LM, Taylor JR, Ding J, Lohman K, Johnson C, Siscovick D, Burke G, Post W, Shea S, Jacobs DR, Jr, Stunnenberg H, Kritchevsky SB, Hoeschele I, McCall CE, Herrington D, Tracy RP, Liu Y. Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat Commun. 2014;5:5366. doi: 10.1038/ncomms6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, Friend S, Ideker T, Zhang K. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, Pattie A, Corley J, Murphy L, Martin NG, Montgomery GW, Feinberg AP, Fallin MD, Multhaup ML, Jaffe AE, Joehanes R, Schwartz J, Just AC, Lunetta KL, Murabito JM, Starr JM, Horvath S, Baccarelli AA, Levy D, Visscher PM, Wray NR, Deary IJ. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16:25. doi: 10.1186/s13059-015-0584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, Buenrostro JD, Weyand CM, Greenleaf WJ, Goronzy JJ. Epigenomics of human CD8 T cell differentiation and aging. Sci Immunol. 2017;2:eaag0192. doi: 10.1126/sciimmunol.aag0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, Hedrick SM. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–84. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med. 2013;210:1189–200. doi: 10.1084/jem.20130392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Delpoux A, Lai CY, Hedrick SM, Doedens AL. FOXO1 opposition of CD8(+) T cell effector programming confers early memory properties and phenotypic diversity. Proc Natl Acad Sci U S A. 2017;114:E8865–E8874. doi: 10.1073/pnas.1618916114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gustafson CE, Cavanagh MM, Jin J, Weyand CM, Goronzy JJ. MicroRNA Networks Modulate FOXO1 Activity in CD8 T cell Aging. doi: 10.1111/acel.12879. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Czesnikiewicz-Guzik M, Lee WW, Cui D, Hiruma Y, Lamar DL, Yang ZZ, Ouslander JG, Weyand CM, Goronzy JJ. T cell subset-specific susceptibility to aging. Clin Immunol. 2008;127:107–18. doi: 10.1016/j.clim.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wertheimer AM, Bennett MS, Park B, Uhrlaub JL, Martinez C, Pulko V, Currier NL, Nikolich-Zugich D, Kaye J, Nikolich-Zugich J. Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J Immunol. 2014;192:2143–55. doi: 10.4049/jimmunol.1301721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Whiting CC, Siebert J, Newman AM, Du HW, Alizadeh AA, Goronzy J, Weyand CM, Krishnan E, Fathman CG, Maecker HT. Large-Scale and Comprehensive Immune Profiling and Functional Analysis of Normal Human Aging. PLoS One. 2015;10:e0133627. doi: 10.1371/journal.pone.0133627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pulko V, Davies JS, Martinez C, Lanteri MC, Busch MP, Diamond MS, Knox K, Bush EC, Sims PA, Sinari S, Billheimer D, Haddad EK, Murray KO, Wertheimer AM, Nikolich-Zugich J. Human memory T cells with a naive phenotype accumulate with aging and respond to persistent viruses. Nat Immunol. 2016;17:966–75. doi: 10.1038/ni.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY, Olshen RA, Weyand CM, Boyd SD, Goronzy JJ. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci U S A. 2014;111:13139–44. doi: 10.1073/pnas.1409155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qu K, Zaba LC, Giresi PG, Li R, Longmire M, Kim YH, Greenleaf WJ, Chang HY. Individuality and variation of personal regulomes in primary human T cells. Cell Syst. 2015;1:51–61. doi: 10.1016/j.cels.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fuertes Marraco SA, Soneson C, Cagnon L, Gannon PO, Allard M, Abed Maillard S, Montandon N, Rufer N, Waldvogel S, Delorenzi M, Speiser DE. Long-lasting stem cell-like memory CD8+ T cells with a naive-like profile upon yellow fever vaccination. Sci Transl Med. 2015;7:282ra48. doi: 10.1126/scitranslmed.aaa3700. [DOI] [PubMed] [Google Scholar]
- 82.Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schubeler D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015;528:575–9. doi: 10.1038/nature16462. [DOI] [PubMed] [Google Scholar]
- 83.Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol. 2005;25:1354–66. doi: 10.1128/MCB.25.4.1354-1366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Jr, Kelly DP. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 85.Scarpulla RC. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann N Y Acad Sci. 2008;1147:321–34. doi: 10.1196/annals.1427.006. [DOI] [PMC free article] [PubMed] [Google Scholar]