

Abstract
Chronic rhinosinusitis with nasal polyps (CRSwNP) is an inflammatory disease with an unknown etiology. Recent studies have implicated the complement system as a potential modulator of disease immunopathology. We preformed proteomic pathway enrichment analysis of differentially increased proteins, and found an enrichment of complement cascade pathways in the nasal mucus of individuals with CRSwNP as compared to control subjects. Sinonasal mucus levels of C3 correlated with worse subjective disease severity, whereas no significant difference in systemic C3 levels could be determined in plasma samples. Given that human sinonasal epithelial cells were the predominate sinonasal source of C3 and C3a staining, we focused on their role in in-vitro studies. Baseline intracellular C3 levels were higher in CRSwNP cells, and following exposure to Aspergillus fumigatus (Af) extract they released significantly more C3 and C3a. Inhibition of C3aR signaling led to a decrease in Af-induced C3 and C3a release, both in-vitro and in-vivo. Finally, we found in-vivo that C3aR deficiency or inhibition significantly reduced inflammation and CRS development in a mouse model of Af induced CRS. These findings demonstrate that local sinonasal complement activation correlates with subjective disease severity, and that local C3aR antagonism significantly ameliorates Af induced CRS in a rodent model.
Keywords: Complement, sinusitis, epithelial cell, inflammation, animal models, nasal polyp, aspergillus
Introduction
Chronic rhinosinusitis (CRS) results in billions of dollars in healthcare expenditures each year1. Patients with CRS have abnormal immune responses triggered by a variety of infectious agents, airborne toxins and fungi.1–3 The respiratory epithelium is the first point of contact with the outside world and as such epithelial cells serve as relay stations capable of amplifying or augmenting cues received from external stimuli to nearby immune cells located in the sinus mucosa. Upon stimulation, epithelial cells secrete factors that are capable of regulating inflammation, and promoting CRS development.4 Recent reviews have described the important regulatory roles of the sinonasal mucosal epithelium in modulating mucociliary clearance, pathogen recognition, and production of innate immune mediators which are designed to cope with local environmental challenges and further orchestrate adaptive immune responses when needed.5, 6 Breakdown, dysfunction or uncontrolled activation of any of these pathways can lead to overstimulation of the adaptive immune response, resulting in the pathogenic inflammation characteristic of CRS.5–7
Previous studies by our laboratory and others have demonstrated that the complement system, as shown by increased complement deposition and gene transcription indicating that the complement system is activated and upregulated in the mucosa of patients with CRS with nasal polyps (CRSwNP).8–10, 12–15 The complement cascade consists of three separate but overlapping pathways: the classical, alternative, and mannose-binding lectin (MBL) pathways. These pathways differ in how complement activation is initiated. Classical pathway activation is initiated by C1q binding to antibody. The alternative pathway is activated by the generation of C3b via hydrolysis or as a by-product of the classical or MBL pathways. The MBL pathway is activated, as its name indicates, when MBL interacts with carbohydrates on the surface of pathogens. Post-activation, the classical and MBL pathways are largely similar, but the true convergence point of all three pathways occurs at the formation of a C3 convertase. Thereafter, the complement system serves to elicit inflammatory responses, driving leukocyte chemotaxis (C3a, C5a), assisting phagocytosis/opsonization (C3b), and lysing cells via the formation of the membrane attack complex (MAC; C5b-C9)(see reviews16, 17 for an in depth overview of the complement system). These traditional roles are well described, but recent research has brought to light a variety of additional functions of the complement system and has served to highlight the complex role complement plays in immunologic homeostasis. Complement is viewed as a systemic serum effector system, with the liver producing the majority of soluble complement proteins.18, 19 Although liver-generated, circulating C3 and C5 are required for the detection and removal of pathogens,18, 19 an emerging paradigm suggests that local cell-derived complement activation fragments are key in driving and modulating adaptive T-cell immunity.20–22 Studies in organ transplantation and T-cell biology have determined that local cell specific complement synthesis and release plays a key role in shaping immune responses in the local microenvironment.23–26
Complement has been shown to be produced and activated by airway epithelium after stimulation by environment factors such as cigarette smoke and particulate matter, and has been repeatedly proposed as a contributing factor in the pathogenic inflammation of CRS.27–29 In addition to increased activation of complement components, such as, C1, C3, C5, and C5b-9, complement receptors, such as C3aR and C5aR,30 the expression of membrane regulators of complement, decay-accelerating factor and CD59, have also been demonstrated on epithelial cells.31–35 Therefore, excessive complement activation, local complement production, complement receptor signaling, and insufficient complement regulation could contribute to CRS development. However, the underlying importance and scope of complement activation, particularly C3 activation, in regulating the inflammation associated with CRSwNP has not been extensively studied.
In this study we investigate the expression and cellular localization of complement component C3, and their relationship with human subjective disease severity. We also evaluate the effects of systemic and sinonasal delivery of a complement receptor antagonist on CRS inflammation and epithelial injury in a murine model. We show that complement is elevated in the local sinonasal microenvironment, produced and stored intracellularly at supranormal levels in CRSwNP epithelial cells, and that C3a receptor antagonism modulates CRS development following systemic or local sinonasal delivery in a rodent model of CRS-like inflammation.
Materials & Methods
Patients enrollment, inclusion and exclusion criteria
Sinus tissue and nasal mucus was collected at the time of endoscopic sinus surgery. Inclusion criteria include CRSwNP patients that met the diagnostic criteria outlined by the European Position Paper on Rhinosinusitis and Nasal Polyps 2012.36 Control sinus tissue was collected from the uncinate process of subjects who were undergoing surgery for repair of cerebrospinal fluid leak repair or removal of non-hormone secreting pituitary tumor. Exclusion criteria include CRS without nasal polyps, active and former smokers, use of oral steroids or immunomodulatory agents within the preceding 30 days, other immunologic, renal, gastrointestinal, endocrine or skeletal disorders or pregnancy. Sinus mucosa was processed and used to establishment human sinonasal epithelial cell (HSNEC) lines or single cell suspensions (SCS) as previously described.3, 37–39 Sinonasal outcome test-22 question (SNOT22) quality of life questionnaires were completed by patients prior to surgery, as previously described.40
Nasal mucus procurement and proteomics screening
Nasal mucus was collected as previously described.41–44 Please see supplemental methods for detailed information on proteomics screening.
In-vitro treatments and related measures
SCS were plated at density of 1×105 cells/well in 96 well plates in BEGM. Aspergillus fumigatus (Af) was obtained from Greer Laboratories (Lenoir, NC) and was a combination of mycelial extract and culture filtrate. Af is a ubiquitous fungal antigen, is the most common fungi in the airway,45, 46 and the most strongly associated with exacerbations in respiratory diseases.47 We have previously established an optimal dose of 2 μg/ml (range 0.1 – 20 μg/ml) which is consistent with other reported in-vitro doses48, 49 and have determined that this dose does not induce cell death, as determined by LDH assay. Af was determined to be endotoxin free (<0.06 EU/ml) by Limulus Amebocyte Lysate (LAL) assay.39 2 μg /ml Af. control (PBS) and/or C3a receptor antagonist (C3aRA) (Calbiochem/EMD Millipore, Burlington, MA)was added to cells for 6 hours after which samples were stained for C3 and C3a as previously described.24 A DMSO vehicle control was also included and found to have no impact on any of the outcomes described herein (data not shown). Samples were assayed immediately using a Guava 8HT flow and analyzed with FCS Express 4.0. Dead cells (7AAD positive) were excluded from the final data analysis. Marker gates were set using matched isotype controls.
Sinonasal explant tissue from control subjects and CRSwNP patients will be used as the source of all primary HSNEC using our previously described methods.3, 39, 50, 51 Briefly, HSNECs were cultured and treated in serum-free, BEGM, used at passage 2, and were in culture for 10 - 15 days from the time the sample left the patient to the time the experiment ended. Cultures were established, maintained, purity confirmed and treated with Af as we have previously described.38, 39, 52 Twenty-four hours post treatment, protein levels, C3 (Genway, San Diego, CA), C3a (BD Biosciences, San Jose, CA), C1q, C4, factor B, C5 (Abcam, USA), and C5b-9 (Quidel, USA) were quantified by ELISA.
Quantitative RT-PCR
Total RNA from control and CRSwNP HSNECs was isolated using a RNeasy MiniKit (Qiagen) according to the manufacturer’s instructions. cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad) and mRNA expression of target genes C1q, C4, C3, C5, fB, and C9 were determined using MasterCycler RealPlex2 PCR System (Eppendorf). Primers used are listed in Supplemental Table I.
Murine Model of Aspergillus Fumigatus induced CRS
The murine model of CRS as described by Khalid, et al., (2008) was employed53 utilizing Balb/c and Balb/c C3aR−/− mice (Jackson Laboratories, USA). Eight week old mice received an intraperitoneal challenge of Af extract. One week later mice received intranasal Af extract 3 times per week for a total of 4 weeks, at which time they were sacrificed (n=6). Unsensitized mice received PBS intranasally 3 times per week for a total of 4 weeks and were used as controls (n=6). To assess the therapeutic utility of C3a receptor signaling inhibition, sub-groups of mice (n=6/group) were treated with C3aR antagonist (C3aRA, Calbiochem, USA), by either intra nasal (i.n, local) or intra peritoneal (i.p, systemic) route with 1 mg/kg/every three days for 4 weeks starting the day after intranasal Af inoculation began.
Histopathology
Snouts were harvested, fixed in 4% buffered formalin, decalcified, embedded in paraffin, and sectioned at 5μm, and stepped serial sections from all groups were graded as previously described.53 Every fifth section containing the vomeronasal organ and nasal-associated lymphoid tissue was examined and graded, using a total of 5 sections/animal. To characterize the pathology and inflammatory cells, several stains were used, including hematoxylin and eosin (H&E) for overall morphology and inflammation, Chromotrope 2R for eosinophils, and Periodic acid Schiff’s alcian blue stain for mucus hyperplasia. Two sections with similar sinus cavities were chosen and examined by an independent researcher blinded to the experimental groups. The numbers of eosinophils, and mast cells were counted under 10 high-power fields from two independent sections, and the average value from four different areas was used for between-group comparisons. The maximal mucosal thickness was measured at the transition zone of the olfactory and respiratory epithelia using an image analysis system (Zeiss Axiovision, USA) using two sections/animal. Epithelial cell goblet cell hyperplasia was quantified using Periodic acid shiff’s alcian blue stain and quantified by counting the total number of cells and the total number of mucus containing goblet cells/unit length of mucosa using two sections/animal as previously described.53, 54
Sinonasal Lavage Cytokine Analysis
A 22G catheter was inserted into the tracheal opening in the direction of the choana after a partial tracheal resection under anesthesia, and lavage performed as previously described.54–56 Complement C3a anaphylatoxin, CCL20, GM-CSF, KC, IL-4, IL-5, and IL-13 were measured by ELISA (R&D systems, Minneapolis, MN).
Statistical Analysis
Statistical analysis was conducted using GraphPad Prism 6.0 software (La Jolla, CA). Descriptive statistics were used to characterize patient demographic data. A D'Agostino & Pearson omnibus test was used to determine if data sets were normally distrusted. A chi-square test was used to determine if there were statistically significant difference in the population composition with regards to gender and race. An unpaired t-test was used determine if there were differences in age between controls and patients with CRSwNP. For data in which all groups were normally distributed, a one-way ANOVA with post-hoc, unpaired student t-test was used. For analysis where all data sets were not normally distributed, a Mann-Whitney test was used to determine statistically significant differences between indicated groups. A Spearman correlation analysis was used to determine if a significant correlation existed between patient-matched nasal mucus and plasma C3 and C3a levels.
Results
Sinonasal, but not circulating, C3 levels correlate with CRSwNP subjective disease severity
Previous studies have shown increased complement fragment C4d, C3d, and C5b-9 deposition, complement 3 receptor expression, and increased complement gene expression (C3, C5 and fB), in the sinonasal mucosa of patients with CRSwNP.8, 9, 30, 57 Given these findings we assessed whether plasma C3 concentrations were altered in CRSwNP patients, and whether C3 levels correlated with subjective disease severity symptoms. As shown in Table I, there were no statistically significant differences between control and CRSwNP patient cohorts with regard to age, race or gender. We observed no significant difference in plasma levels of C3 between controls and patients with CRSwNP (Fig. 1A), and could determine no association between C3 levels and SNOT22 scores (Fig. 1B).
Table I.
Patient Demographics
| A. Plasma Samples | |||
|---|---|---|---|
|
| |||
| Control (n = 7) | CRSwNP (n = 18) | p Value | |
| Sex, no. (%) | 0.9428 | ||
| Men | 3 (42.9) | 8 (44.4) | |
| Women | 4 (57.1) | 10 (55.6) | |
| Age, mean ± SD | 47.4 ± 23.6 | 40.2 ± 18.5 | 0.1629 |
| Race, no. (%) | 0.4090 | ||
| White | 3 (42.9) | 11 (61.1) | |
| African American | 4 (57.1) | 7 (38.9) | |
| Other | 0 (0) | 0 (0) | |
| SNOT22 Score, mean ± SD | – | 49.3 ± 27.8 [14] | – |
|
| |||
| B. Nasal Mucus Samples | |||
|
| |||
| Control (n = 6) | CRSwNP (n = 11) | p Value | |
|
| |||
| Sex, no. (%) | 0.7933 | ||
| Men | 4 (66.7) | 8 (72.72) | |
| Women | 2 (33.3) | 3 (27.3) | |
| Age, mean ± SD | 35.5 ± 14.5 | 42.45 ± 21.92 | 0.4985 |
| Race, no. (%) | 0.6905 | ||
| White | 4 (66.7) | 7 (63.64) | |
| African American | 2 (33.3) | 3 (27.27) | |
| Other | 0 (0) | 1 (9.1) | |
| SNOT22 Score, mean ± SD | – | 49.625 ± 20.27 [8] | – |
SNOT22 = sinonasal outcome test-22. SD = standard deviation. [n] = number of patients with available SNOT22 scores.
Figure 1. Local sinonasal complement protein expression is associated with increased subjective disease severity.
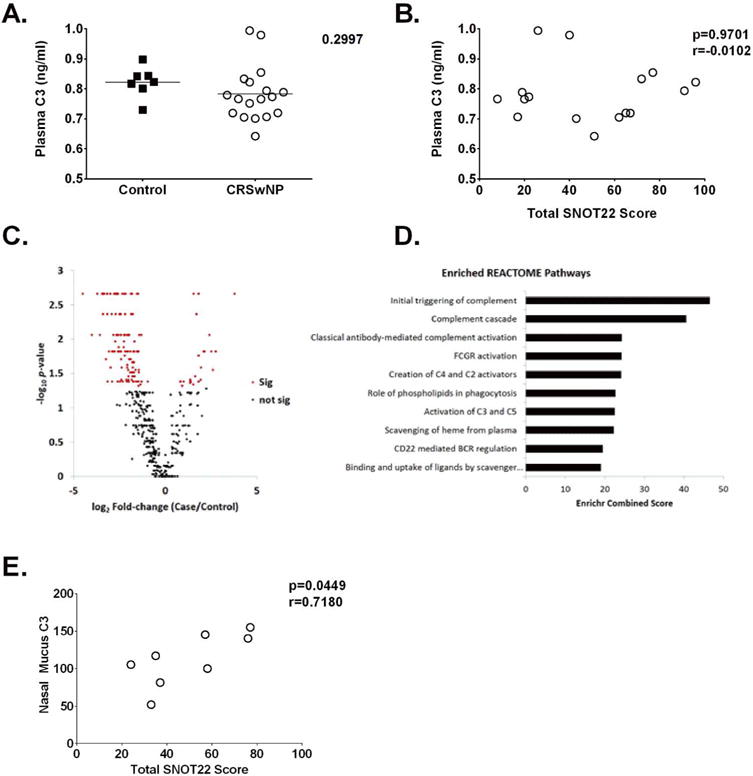
A. Quantification of systemic circulating plasma C3 concentrations in controls and patients with CRSwNP. B. Correlation analysis between circulating C3 and subjective disease severity as measured by the sinonasal outcome test-22 (SNOT22) C. Volcano plot of shotgun proteomics results. Normalized spectral counts were used to compare protein abundance between CRSwNP and control mucus samples. The majority of the 466 identified proteins were decreased in CRSwNP (77.3%). D. Top 10 Enriched REACTOME Pathways. Using just the differentially abundant proteins that were increased in CRSwNP, pathway enrichment was performed against the REACTOME database. The top 10 pathways based on combined score are shown. E. Pearson correlation analysis demonstrating a positive correlation between nasal mucus levels of C3 and subjective disease severity in patients with CRSwNP. NSc = normalized spectral counts.
Recently there has been a growing emphasis on the role of locally produced effector molecules in disease modulation. Therefore, we next utilized shotgun-proteomics to characterize significant protein pathways differentially expressed in CRSwNP by comparing nasal mucus samples from control patients to those with CRSwNP, resulting in the identification of 466 proteins (FDR < 5%). Differentially abundant proteins were determined using normalized spectral counts and 146 were significantly different, with 24 increased in CRSwNP and 122 decreased (Fig. 1C). Since we were specifically interested in proteins that were increased in CRSwNP, we used these 24 proteins to perform pathway enrichment analysis against the REACTOME database (see supplemental Table II). The top two identified pathways were Initial Triggering of Complement and Complement Cascade (Fig. 1D), both of which included C3, C4a, and complement factor B, which were increased 1.8, 6.7 and 3.8-fold in CRSwNP, respectively. Moreover, while there was no association between plasma levels of C3 and disease severity, conversely there was a positive correlation between nasal mucus C3 levels and poorer subjective disease severity in patients with CRSwNP (Fig. 1E). Together these data point towards the importance of the complement system in the local sinonasal microenvironment of CRSwNP patients.
HSNEC from patients with CRSwNP have heightened intracellular stores and increased secretion of C3 & C3a
Given our studies demonstrating associations between disease severity and sinonasal mucus C3 concentrations, we next investigated whether C3 and C3a was present locally within sinonasal tissues. Using single cell suspensions of sinonasal tissue explants prepared from CRSwNP and control subjects, we demonstrated that both control and CRSwNP samples contained C3 and C3a positive cells, and that C3 and C3a levels were elevated in CRSwNP as compared to controls (Fig. 2A and B). Furthermore, HSNECs were identified as the predominant cell type staining for C3 and C3a, as determined by positive staining for the epithelial specific marker, EpCam. To further characterize this increase in intracellular C3 and determine the source of C3 we performed qRT-PCR studies on HSNECs from control and CRSwNP patients. mRNA levels of key complement factors C1q, C4, C3, C5, fB, and C9 were analyzed. In keeping with previous studies of epithelial cells of the respiratory system58 we could determine no C9 expression in HSNECs from either group. In support of our flow cytometry data we demonstrate increased C3 gene expression in HSNECs of CRSwNP, as compared controls, along with increases in other complement components C4, C5, and fB. We were unable to detect any gene expression for C1q in HSNECs.
Figure 2. Sinonasal tissue from patients with CRSwNP have heightened levels of C3 and C3a.
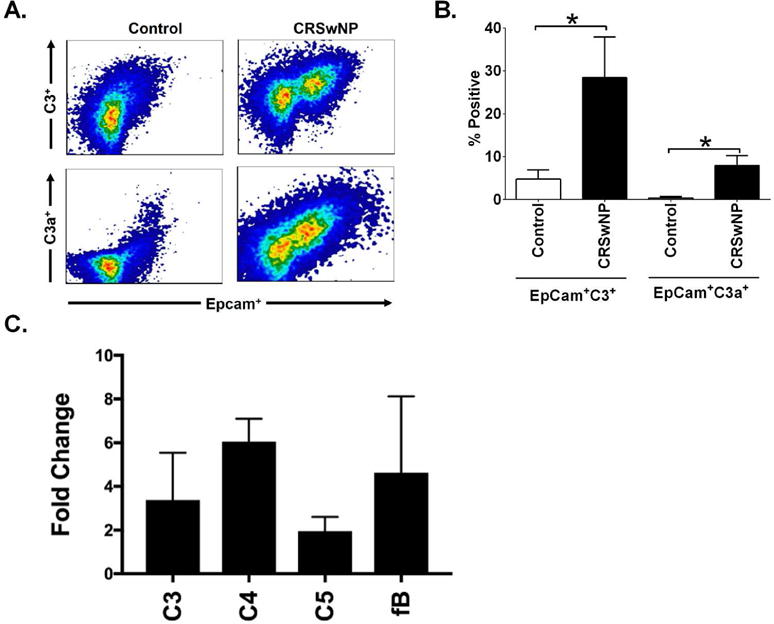
(A). Single cell suspensions were prepared by mechanical separation and stained for the epithelial cell marker EpCAM. Following permeabilization cells were immunostained for C3 and C3a. Note that human sinonasal epithelial cells were the predominate cell type staining positively for C3 and C3a. (B). Quantification of the percent of viable cells double positive for EpCam and C3 or C3a, note the significant increases seen in EpCam+ C3 and C3a expression sinonasal epithelial cells in patients with CRSwNP. n=4/group. Statistics shown are unpaired student t test between indicated groups. *p<0.05 (C). Complement mRNA levels were measured in control and CRSwNP HSNECs by qRT-PCR. Relative HSNEC expression of C3, C4, C5, and fB was assessed and expressed as a fold change in CRSwNP as compared to control HSNEC expression. Note the increase in C3, C4, C5, fB and fH. No gene expression for C1 or C9 could be detected in HSNECs from either group (data not shown). n=5-8/group.
To determine whether the increase in C3 and C3a noted was associated with increased surface or intracellular stores of C3/C3a, we performed flow cytometry on HSNECs, cultured in serum free conditions, from control and CRSwNP patients using samples that were permeabilized and non-permeabilized, as previously described24. Similar to the results observed in our sinonasal explant studies, and keeping with our previously reported RT-PCR data,9 we observed higher C3 levels in HSNEC from patients with CRSwNP than those from controls (Fig. 3A), and determined that 94% of the C3 that was identified was intracellularly located. C3 was present at significantly lower levels in control samples, but similar to CRSwNP, the majority of C3 present (84%) was intracellularly located. C3a levels were similar between control and CRSwNP HSNEC, and were found in similar amounts, on the cell surface and within the intracellular compartment (Fig. 3B).
Figure 3. CRSwNP-derived HSNEC have a heightened release of C3 and C3a upon Aspergillus Fumigatus (Af) extract challenge.
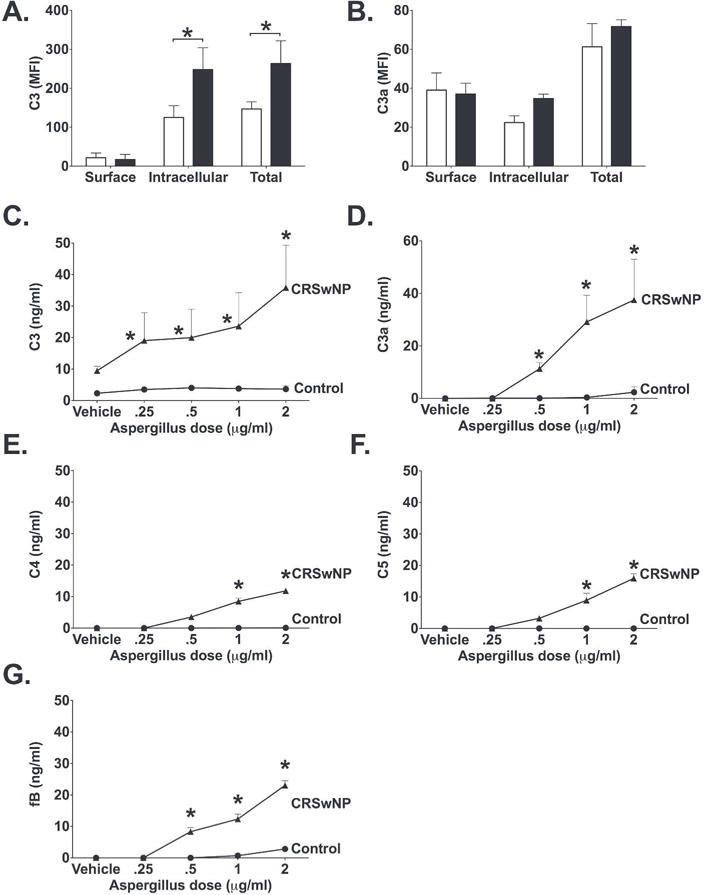
Surface, intracellular and total C3 (A) and C3a (B) levels in unstimulated HSNEC show that CRSwNP HSNEC have increased intracellular stores of C3 as compared to those from control subjects. * p<0.05 between indicated groups. ELISA measurement of (C) C3, (D) C3a, (E) C4, (F) C5, and (G) fB in HSNEC culture supernatants following exposure to increasing doses of Af extract. Note that CRSwNP cells had significantly more C3, C3a, C4, C5 and fB as compared to control. Values shown are mean+SD. *p<0.05 vs dose-matched control. n=4/group.
To investigate whether these increased intracellular stores were due to a failure to release C3 into the surrounding tissues or to increased synthesis by CRSwNP epithelial cells, we measured C3 concentrations in the media supernatants within our culture system, which contained no serum. We showed that at baseline, C3 media concentrations were significantly elevated in CRSwNP HSNEC culture media as compared to controls epithelial cells (Fig. 3C). Following 24 hours of stimulation with the ubiquitous fungal allergen Af, there was a dose dependent increase in the secretion of C3 and C3a by CRSwNP cells, as compared to controls (Fig. 3C). C3a levels were undetectable in the supernatants of unstimulated CRSwNP and control HSNEC cultures (Fig. 3D). Again, upon Af stimulation there was a significant increase in C3a concentrations within the supernatant of CRSwNP as compared to controls (Fig. 3D). Stimulation of HSNEC from control patients with Af had little to no impact on C3 or C3a release. Further analysis of key complement pathway proteins, C1q (classical pathway), C4 (Classical and Lectin pathways), factor B (alternative pathway), C5 and sC5b-9 (terminal pathway) demonstrated dose dependent increases in C4, fB and C5 (Fig. 3E, F, and G). C1q and sC5b-9 were undetectable in the culture supernatant, which is not unsurprising given our mRNA expression analysis. Together these data suggest that CRSwNP HSNEC have an increased complement activity phenotype and have the ability to secrete increased amounts of C3 into the microenvironment, both at baseline, as well as post Af stimulation, which may explain the increased C3 mucus concentrations noted in our proteomic and ELISA studies.
Human T-cell studies have suggested that intracellular tonic levels of C3a are required for T-cell survival and function,24 with stores of intracellular complement maintained via an autocrine loop induced by C3a ligation to its receptor (C3aR). We therefore examined the expression of C3aR on control and CRSwNP HSNECs. We demonstrated that C3aR expression is elevated in CRSwNP patients at baseline as compared to controls (Fig. 4A and B). Given this data, we next examined if inhibition of C3a signaling could diminish Af-induced increases in HSNEC C3 release. While C3aRA had no statistically significant impact on baseline HSNEC C3 secretion, it was able to significantly reduce Af stimulated C3 release to near control levels (Fig. 4C).
Figure 4. Human sinonasal epithelial cells (HSNEC) from patients with CRSwNP have elevated expression of C3aR.
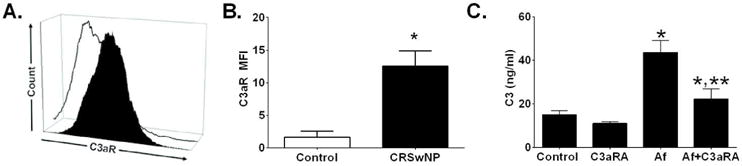
(A) Representative histogram showing epithelial surface C3aR expression by control HSNEC (open histogram) and HSNECs from patients with CRSwNP (black histogram). (B) Quantification of C3aR expression. Note the increase of C3aR expression in CRSwNP as compared to control epithelial cells. (C) Inhibition of C3a signaling with a C3a receptor antagonist (C3aRA), blocks Af-induced increases in HSNEC secretion of C3. *p<0.05 vs control. ** p<0.05 vs Af treatment. For A & B, n=3/group (C) Mean ± SD from experiments with four different control HSNEC cultures.
Af stimulation induces C3 activation in-vivo.
We next assessed the impact of Af stimulation on complement activation in-vivo to determine whether Af could promote localized sinonasal complement activation. Utilizing mice with developed CRS that had been allergen sensitized for 4 weeks with Af, we analyzed sinonasal C3 complement activation. Immunohistochemical studies, with an antibody to the long-lived C3 cleavage fragment C3d, showed that direct intranasal exposure of Af lead to significant C3d deposits within the microvasculature and on epithelial cells (Fig. 5A), consistent with that seen in human studies.9, 15, 57 C3d deposition was not evident in saline control-challenged mice (Fig. 5B).
Figure 5. Intranasal Aspergillus Fumigatus (Af) extract challenge activates complement and promotes inflammation within the sinonasal cavity, and can be ameliorated by C3a receptor antagonism (C3aRA).
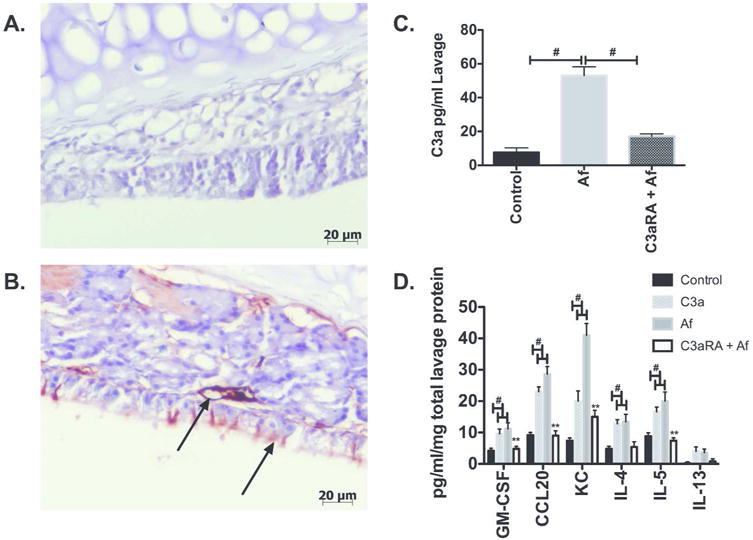
Balb/c mice that received 4 weeks of Af allergen challenge and were confirmed to have CRS-like inflammation were challenged with Af intranasally to investigate dynamic complement activation. Seventy-two hours post final Af intranasal challenge heads were isolated for immunohistochemistry detection of C3d (A & B) and lavage samples taken for C3a ELISA. Note that Af induced membrane complement deposition as marked by the long-lived membrane associated C3 activation fragment, C3d. C3d deposition was increased in Af-CRS mice (B, Arrows) as compared to control (A). Confirmation of local C3 activation was seen in nasal lavage fluids were increased C3a anaphylatoxin concentrations were seen 72 hours post Af inoculation, that was significantly reduced by pretreatment with C3aRA (C). (#p<0.05)(n=4-6). ELISA measurement of inflammatory chemokines/cytokines CCL20, GM-CSF, and KC, and type 2 cytokines IL-4, IL-5 and IL-13 post inoculation with either Af or C3a, C3aRA pre-treated Af inoculated mice. Note that all challenges induce cytokine production, with the exception of IL-13 (data not shown), and that C3aR antagonism pre-treatment significantly reduces all measured cytokine/chemokines as compared to Af alone. (#p<0.05, control vs treatment groups, **p<0.01 GM-CSF, CCL20, KC, IL-4, IL-5 treatment group vs C3aRA treatment) (n=4-5).
In concordance with immunohistochemical studies, nasal lavage fluids had significantly elevated C3 activation product, C3a, following Af stimulation (Fig. 5C). It is thought that C3 release and synthesis is in part mediated by an autocrine feedback loop, whereby C3a interacts with C3aR to promote C3 and C3a synthesis and storage.24 To examine this in-vivo, we pretreated mice with intranasal C3aRA intranasally challenged mice with Af 24 hours later. After an additional 72 hours, we measured nasal lavage levels of C3a. Pre-challenge C3aRA treatment led to a significant reduction in C3a levels, suggesting that local sinonasal C3a release is in part mediated via an autocrine C3a/C3aR axis (Fig. 5C). Of note systemic administration of C3aRA had no impact on local C3a levels following acute Af challenge (data not shown).
Af and C3a sinonasal inoculation promotes inflammation and type 2 cytokines expression and are inhibited by C3aR antagonism
To further explore the interplay between Af and C3a in terms of sinonasal inflammation, we acutely challenged mice that had been exposed to 4 weeks of Af challenge and that had developed CRS, with either Af or C3a (50 ng i.n), and 3 days later analyzed lavage samples for inflammatory chemokine/cytokines and type 2 cytokines levels. CCL20, KC (human IL-8 equivalent), GM-CSF, IL-4, IL-5 and IL-13 were selected because are upregulated in CRSwNP patients and have been shown to be associated with more severe disease.41, 59–63 Both Af and C3a exposure promoted robust GM-CSF, CCL20, KC, IL-4, and IL5 expression as determined by ELISA (Fig. 5D). While IL-13 was measured, we could determine no detectable levels in our assay system. Interestingly, pretreatment of mice with C3aR antagonist, prior to Af and C3a challenge, significantly reduced cytokine levels to that seen in control mice for all measured analytes (Fig. 5D).
C3aR deficiency or antagonism inhibits CRS development
To explore the therapeutic potential of our findings we employed a commercially available C3a receptor antagonist (C3aRA), which we delivered systemically by intraperitoneal injection, or locally by intranasal route. Both C3aR deficiency and C3aRA, irrespective of route of delivery, reduced histologic evidence of sinonasal respiratory epithelial injury and inflammation as compared to untreated controls (Fig. 6). We also observed decreased inflammatory cell infiltrates, eosinophil numbers, goblet cell hyperplasia, and epithelial thickness in C3aR deficient or C3aR inhibited mouse (Fig. 7A-D). Interestingly, localized C3aRA deliver to the sinus cavity by intranasal route was similarly protective as both systemic antagonism and complete C3aR deficiency.
Figure 6. Complement C3a Receptor deficiency or therapeutic antagonism reduce sinonasal inflammation and epithelial remodeling.
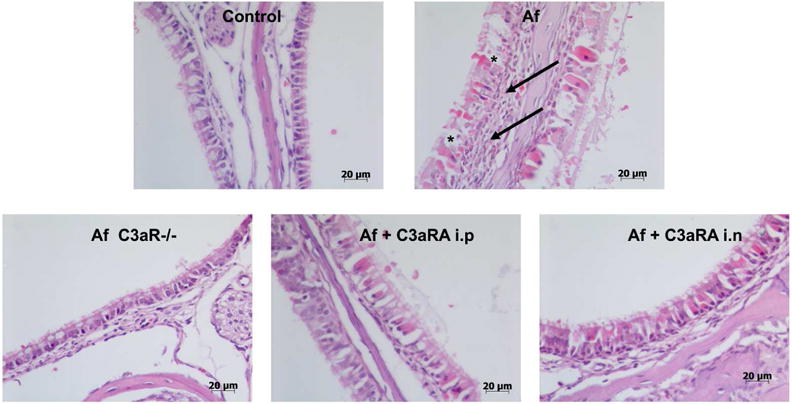
Hematoxylin and eosin stained sections of sinus mucosa from animals in each treatment group. Note the extensive sub-epithelial inflammation characterized by eosinophil, macrophage and neutrophil infiltrates in Af challenged mice as compared to control, C3aR−/− and treatment groups (arrows). Further note the more pronounced mucus producing cells lining the sinus lumen (*). Images representative of n=6 in each group, scale as shown.
Figure 7. Quantification of sinonasal inflammation, epithelial injury and goblet cell hyperplasia demonstrates the protective effect of Complement C3a Receptor deficiency and therapeutic antagonism.
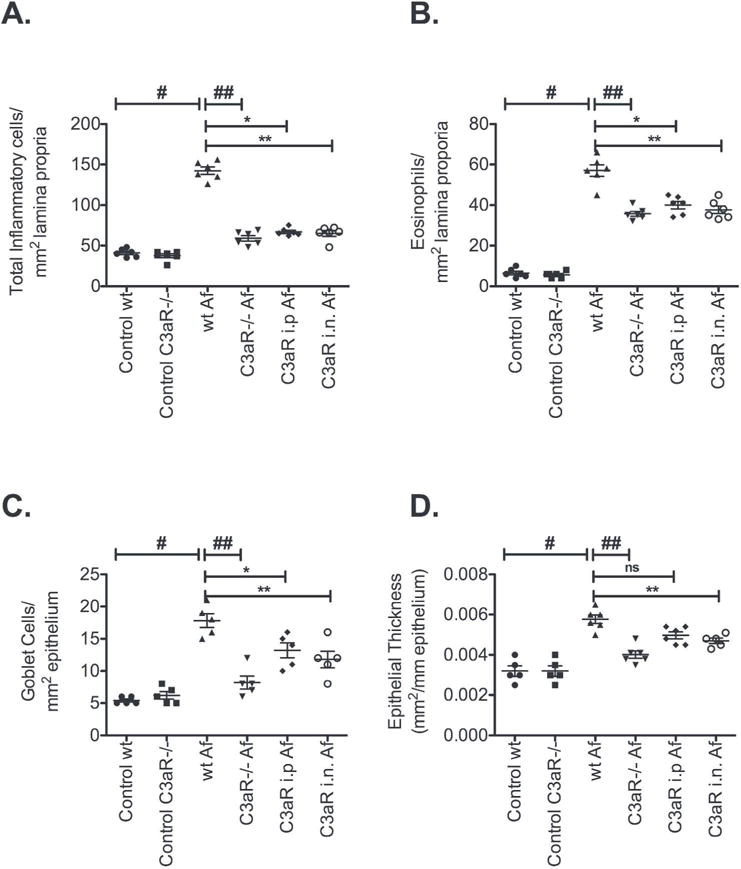
(A). Inflammatory cell infiltrate, (B). Eosinophil accumulation, (C). Goblet cell hyperplasia, and (D). Epithelial injury/thickness. Data are expressed as mean ± SE; All statistical analysis are pairwise comparisons Control wildtype vs wildtype Af (#p<0.001), wildtype Af vs C3aR−/− Af (##p<0.01), wildtype Af vs C3aR i.n (*p<0.01), wildtype Af vs C3aR i.p. (**p< 0.01).
Discussion
CRSwNP is a disease characterized by uncontrolled inflammation that is often refractory to current standard treatments. While many immune mechanisms have been studied in the development and treatment of CRS, no single pathway has been shown to be disease causing or modifying. Innate and adaptive immune factors undoubtedly synergize to promote disease, and as such, targeting pathways that can modify both systems is an approach that may well prove efficacious. One such pathway that we investigated here is the complement system. The complement cascade can induce cell death, promote inflammation, and program adaptive immunity. Given these broad ranging functions, we analyzed plasma and local concentrations of C3 in CRSwNP patients to determine whether an association existed between C3 levels and subjective disease severity. Plasma C3 levels were not associated with CRSwNP subjective disease severity, an observation in line with previous findings.33, 34 While some studies have demonstrated increased plasma C3 concentrations, as compared to controls, these increases were not associated with disease severity.33, 34 C3 is an acute phase reactant, and consequently while measurements of plasma concentrations are often elevated in chronic inflammatory conditions, they may also be depressed by increased catabolism that synthesis of new C3 is temporally not able to restore. Therefore, differences in sample preparation, time of sample procurement, and stage of disease in the patient population may explain why we saw no difference in plasma concentrations in CRSwNP patients as compared to controls. Changes in other complement components that correlate with disease severity have been described, with deficiency in C4A and MBL associated with increased disease, likely due to increased susceptibility to infection.33, 35
While 90% of all complement proteins are produced within the liver, there is a growing appreciation that complement synthesized in the microenvironment is instrumental in orchestrating local immune responses. Novel studies by Liszewski et al (2013).,24 highlighted that lymphoid, myeloid and non-myeloid cells can synthesize, store and release the active C3 components C3a and C3b. Given these findings we hypothesized that local complement production may be increased in patients with CRSwNP and may exacerbate disease. Our mucus proteomic pathway enrichment analysis identified two significant pathways, the Initial Triggering of Complement and Complement Cascade pathways, both of which included C3, C4a, and factor B. The approximate 2-fold changes we noted in C3 were in keeping with a previous proteomic study that utilized nasal mucus samples from 29 allergic rhinitis patients.64 When analysis of local sinonasal mucus sample C3 levels were correlated to SNOT22 scores, unlike plasma concentrations we demonstrated a positive association between C3 levels and disease severity, suggesting that locally elevated C3 plays a role in CRSwNP.
We analyzed sinonasal tissues and observed that the predominant cell type positive for C3 and C3a were HSNEC. The epithelium plays a central role in controlling inflammation in the sinonasal cavity, and is important in modulating immune cell functions, and therefore the production of complement by HSNEC may be important in modulating immunity. Furthermore, previous studies in the lower respiratory tract have shown that bronchial epithelial cells can produce and release C3 in response to pro-inflammatory cytokine stimulation.65 Analysis of HSNECs revealed intracellular stores of C3 that were present at supranormal levels in CRSwNP, as compared to controls. The higher concentrations of C3/C3a that we found in HSNEC with CRSwNP, is in part corroborated by our previous PCR studies where we demonstrated increase C3 mRNA transcripts in CRSwNP,9 and could account for the intrinsically pro-inflammatory microenvironment seen in CRSwNP patients. To provide further support of our flow cytometry analysis we performed complement mRNA analysis and demonstrate that C3, as well as C4, C5 and fB are upregulated in CRSwNP as compared to controls. Interestingly, we showed that early pathway activation factor C1q, and terminal pathway C9 were not expressed at the gene level in HSNEC epithelial cells at baseline. When stimulated with the ubiquitous fungal antigen Af, C3 and C3a was released in greater quantities by CRSwNP HSNEC. Taken together, these data show for the first time the presence of intracellular stores of C3 and C3a in HSNECs, which may contribute to the intrinsically pro-inflammatory nature of HSNECs from patients with CRSwNP. The presence and role of epithelial intracellular complement in injury modulation is an evolving concept, and while it is difficult to delineate the relative contribution of local and systemically derived complement sources in disease progression, the presence of C3 at supranormal levels in the local microenvironment could well orchestrate a heightened pro-inflammatory milieu capable of exacerbating inflammation, and thus disease.
While our data and that of others,24, 66, 67 supports the hypothesis that activated cellular stores of complement can reside within discrete cellular compartments within cells, a recent study has suggested that these stores reflect an active transportation of complement through the cell from serum sources as opposed to local cell production.68 Elvington et al., 2017 eloquently demonstrated that intracellular stores of complement are the result of uptake from serum,68 and that cells, particularly those exposed to fluid phase complement components, actively cycle complement proteins between the extra and intracellular space. In our studies, HSNEC were cultured and maintained in a serum free system devoid of exogenous complement,, and are cultured for up to two weeks prior to performance of presented experiments, thus making it unlikely that the intracellular stores seen in these studies are derived from passive uptake of serum complement, but rather reflect a de novo local production of complement by barrier epithelial cells. Taken together, these studies highlight the important role of locally generated complement and its activation in injury, cytokine production and disease pathogenesis.
Interestingly, while baseline levels of C3/C3a were significantly different between CRSwNP and controls, only CRSwNP responded to escalating doses of Af stimulation by releasing C3, C3a, C4, C5 and fB. The mechanism/s by which Af results in C3 release has not been determined, but it has been postulated that responses to allergens may be mediated by allergen receptors present on diseased epithelial.69, 70 Whether these receptors are differentially expressed on HSNEC from CRSwNP and controls has not been investigated, but given our data it is an area for future investigations. An alternative explanation for these differences could be directly related to differences in basal complement activity between controls and CRSwNP. Af can activate complement independently of canonical pathways of activation. For example, although Af antibodies have been identified, there are no antibodies in our in-vitro system, and thus it’s likely that components of the mycelial Af wall activate C3,71, 72 generating C3a. Given the increased presence of both C3 and C3a in CRSwNP, it is plausible that cleavage of C3 leads to increased C3a signaling and stimulation of C3 production. C3a is a potent chemotactic peptide, can induce pro-inflammatory cytokine release via engagement with its receptor C3aR, and can further promote T effector cell proliferation and T regulatory cell inhibition; all factors that would propagate CRSwNP. In particular, complement anaphylatoxins such as C3a have the ability to skew T-cell responses and suppress T regulatory cell functions.22, 73–78 In human T-cell studies in which C3a intracellular stores were first described, it was postulated that C3a acts in an autocrine fashion to promote C3/C3a production via increased C3aR expression.24 Blockade of C3a signaling in T-cells led to reduced intracellular and extracellular release of C3/C3a.24 These experiments then lead us to test this hypothesis in-vivo, were we observed that C3aRA could in fact diminish Af-induced release of C3 in-vitro. Pretreatment of mice with C3aRA effectively reduced sinonasal levels of C3a upon Af stimulation in sensitized mice. These data support a mechanism by which C3a activation stimulates C3a production through a C3a/C3aR axis. We show that C3aR expression is elevated in HSNEC from CRSwNP patients in-vitro, perhaps suggesting that increased intracellular stores are in part modulated by increased C3aR expression and signaling.
Complement inhibition has become a clinical reality with the introduction of eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria (PHN).79 Eculizumab functions by inhibiting C5 activation and is therefore consider safe for systemic administration as it leaves intact complement detection pathways, activation and opsonin functions.80, 81 While C3 inhibitors exist, such as compstatin, their utilization systemically is not without risk due to systemic complement functions.82 Fortunately, the sinus cavity, lends itself anatomically to topical application. Here we demonstrated that C3a receptor signaling through genetic deficiency, systemic antagonist inhibition, or local antagonist delivery provided protection from the development of CRS like inflammation and epithelial injury. Complement anaphylatoxin deficiency and inhibition has been extensively studied, with blockade of C3a and C5a receptors shown to provide protection in rodent asthma models, such as house dust mite and ovalbumin and Af allergen challenge, and with C3a and C5a shown to play differing roles in allergy induction and hypersensitivity.75, 83, 84 While these model systems are very similar in their approach, none of these studies have previously investigated the upper respiratory tract, and none have accessed the impact of locally delivered receptor antagonist as therapeutic interventions. In these models, C3aR antagonism or deficiency is associated with reduced mucus hyperplasia, epithelial injury and decreased small airway remodeling.84 Similarly, in our investigations in the upper airway, C3aR antagonism reduces hyperplasia, epithelial injury, and immune cell infiltration, all of which reduces CRS-like inflammation as determined by histological evaluation.
In conclusion, here we demonstrate that local sinonasal C3 concentrations are associated with disease severity, that HSNEC have intracellular stores of C3 that are present at supranormal levels in CRSwNP, and that these stores are quickly mobilized into the extracellular space upon stimulation. Building upon these findings we demonstrated that in-vivo local sinonasal inhibition of C3a/C3aR axis in a mouse model of CRS-like inflammation lead to reduction in proinflammatory cytokine and type 2 cytokine release. Finally, we demonstrate the therapeutic potential of local sinonasal complement inhibition as a means to modulate local inflammation and the development of CRS, a strategy that will likely protect systemic and local complement immune functions.
Supplementary Material
Key Findings.
Heightened sinonasal complement activation is present in CRSwNP, and local sinonasal complement C3 levels correlate with subjective patient disease severity.
Complement anaphylatoxin antagonism decreases inflammatory infiltrate and local cytokine production associated with murine fungal extract-induced CRS.
Sinonasal delivery of C3aR antagonist decreases inflammatory infiltrate associated with murine fungal-induced CRS.
Capsule summary.
C3a receptor inhibition ameliorates chronic rhinosinusitis.
Acknowledgments
These studies were funded by grants from the NIH to CA (R01HL091944), Flight Attendants Medical Research Institute Clinical Innovator award 092079 (CA) and 92401 (JKM). JKM is supported by the South Carolina Clinical & Translational Research (SCTR) Institute, with an academic home at the Medical University of South Carolina, NIH/NCATS Grant Numbers KL2 TR001452 & UL1 TR001450. Supported in part by core funds from the Hollings Cancer Center, Medical University of South Carolina (P30 CA138313), and the MUSC Proteomics Center, South Carolina Centers for Economic Excellence SmartState program. BN was supported in part by an award to the South Carolina SmartState Center of Economic Excellence in Proteomics, the Medical University of South Carolina Proteomics Center, and P20GM103542 from the NIGMS.
Identification of certain commercial equipment, instruments, software or materials does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the products identified are necessarily the best available for the purpose.
The Medical University of South Carolina Institutional Review Board granted approval prior to initiation of the study and informed written consent was obtained from all participants. All procedures were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Abbreviations
- Af
Aspergillus fumigatus Extract
- BEGM
Basal Epithelial Cell Growth Medium
- C3
Complement 3
- C3a
Complement anaphylatoxin 3a
- C4a
Complement 4a
- C3aR
Complement 3a Receptor
- C3aR−/−
Complement 3a Receptor Deficient
- C3aRA
Complement 3a Receptor Antagonist
- C5
Complement 5
- C5a
Complement anaphylatoxin 5a
- C5aR
Complement 5a Receptor Antagonist
- CCL20
Chemokine (C-C motif) ligand 20
- CRS
Chronic Rhinosinusitis
- CRSsNP
Chronic Rhinosinusitis without Nasal Polyps
- CRSwNP
Chronic Rhinosinusitis with Nasal Polyps
- ELISA
Enzyme-linked Immuno Sorbent Assay
- EPOS
European Position Paper on Rhinosinusitis and Nasal Polyps
- EpCAM
Epithelial cell adhesion molecule
- GM-CSF
Granulocyte macrophage - colony stimulating factor
- H&E
Hematoxylin and eosin
- i.p.
Intraperitoneal
- KC
Keratinocyte chemoattractant
- MBL
Mannose Binding Lectin
- HSNEC
Human Sinonasal Epithelial Cells
- SNOT22
Sinonasal outcomes test-22
Footnotes
None of the listed authors have any potential conflicts to disclose related to the research presented herein.
References
- 1.Benninger MS, Ferguson BJ, Hadley JA, Hamilos DL, Jacobs M, Kennedy DW, et al. Adult chronic rhinosinusitis: Definitions, diagnosis, epidemiology, and pathophysiology. Otolaryngology - Head and Neck Surgery. 2003;129(3, Supplement 1):S1–S32. doi: 10.1016/s0194-5998(03)01397-4. [DOI] [PubMed] [Google Scholar]
- 2.Chan Y, Kuhn FA. An update on the classifications, diagnosis, and treatment of rhinosinusitis. Curr Opin Otolaryngol Head Neck Surg. 2009;17(3):204–208. doi: 10.1097/MOO.0b013e32832ac393. [DOI] [PubMed] [Google Scholar]
- 3.Mulligan JK, Nagel W, O’Connell BP, Wentzel J, Atkinson C, Schlosser RJ. Cigarette smoke exposure is associated with vitamin D3 deficiencies in patients with chronic rhinosinusitis. The Journal of allergy and clinical immunology. 2014;134(2):342–349. doi: 10.1016/j.jaci.2014.01.039. [DOI] [PubMed] [Google Scholar]
- 4.Bachert C, Wagenmann M, Rudack C, Hopken K, Hillebrandt M, Wang D, et al. The role of cytokines in infectious sinusitis and nasal polyposis. Allergy. 1998;53(1):2013. doi: 10.1111/j.1398-9995.1998.tb03767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schleimer RP, Kato A, Peters A, Conley D, Kim J, Liu MC, et al. Epithelium, Inflammation, and Immunity in the Upper Airways of Humans: Studies in Chronic Rhinosinusitis. Proc Am Thorac Soc. 2009;6(3):288–294. doi: 10.1513/pats.200808-088RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tieu DD, Kern RC, Schleimer RP. Alterations in epithelial barrier function and host defense responses in chronic rhinosinusitis. The Journal of allergy and clinical immunology. 2009;124(1):37–42. doi: 10.1016/j.jaci.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basinski TM, Holzmann D, Eiwegger T, Zimmermann M, Klunker S, Meyer N, et al. Dual nature of T cell-epithelium interaction in chronic rhinosinusitis. Journal of Allergy and Clinical Immunology. 2009;124(1):74–80.e78. doi: 10.1016/j.jaci.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 8.Davis KS, Casey SE, Mulligan JK, Mulligan RM, Schlosser RJ, Atkinson C. Murine complement deficiency ameliorates acute cigarette smoke-induced nasal damage. Otolaryngology–head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery. 2010;143(1):152–158. doi: 10.1016/j.otohns.2010.02.022. [DOI] [PubMed] [Google Scholar]
- 9.Schlosser RJ, Mulligan RM, Casey SE, Varela JC, Harvey RJ, Atkinson C. Alterations in gene expression of complement components in chronic rhinosinusitis. American journal of rhinology & allergy. 2010;24(1):21–25. doi: 10.2500/ajra.2010.24.3399. [DOI] [PubMed] [Google Scholar]
- 10.Lane AP, Truong-Tran QA, Schleimer RP. Altered expression of genes associated with innate immunity and inflammation in recalcitrant rhinosinusitis with polyps. American journal of rhinology. 2006;20(2):138–144. [PMC free article] [PubMed] [Google Scholar]
- 11.Van Roey GA, Vanison CC, Wu J, Huang JH, Suh LA, Carter RG, et al. Classical complement pathway activation in the nasal tissue of patients with chronic rhinosinusitis. The Journal of allergy and clinical immunology. 2016 doi: 10.1016/j.jaci.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyaguchi M, Uda H, Sakai S, Kubo T, Matsunaga T. Immunohistochemical studies of complement receptor (CR1) in cases with normal sinus mucosa and chronic sinusitis. Archives of oto-rhino-laryngology. 1988;244(6):350–354. doi: 10.1007/BF00497463. [DOI] [PubMed] [Google Scholar]
- 13.Ogunleye AO, Arinola OG. Immunoglobulin classes, complement factors and circulating immune complexes in chronic sinusitis patients. Afr J Med Med Sci. 2001;30(4):309–312. [PubMed] [Google Scholar]
- 14.Schlosser RJ, Mulligan RM, Casey SE, Varela JC, Harvey RJ, Atkinson C. Alterations in gene expression of complement components in chronic rhinosinusitis. American journal of rhinology & allergy. 2010;24(1):21–25. doi: 10.2500/ajra.2010.24.3399. [DOI] [PubMed] [Google Scholar]
- 15.Van Roey GA, Vanison CC, Wu J, Huang JH, Suh LA, Carter RG, et al. Classical complement pathway activation in the nasal tissue of patients with chronic rhinosinusitis. The Journal of allergy and clinical immunology. 2017;140(1):89–100.e102. doi: 10.1016/j.jaci.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nature reviews Nephrology. 2018;14(1):26–47. doi: 10.1038/nrneph.2017.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujita T, Endo Y, Nonaka M. Primitive complement system–recognition and activation. Mol Immunol. 2004;41(203):103–111. doi: 10.1016/j.molimm.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 18.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 19.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344(15):1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 20.Kolev M, Le Friec G, Kemper C. The role of complement in CD4(+) T cell homeostasis and effector functions. Seminars in immunology. 2013;25(1):12–19. doi: 10.1016/j.smim.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 21.Heeger PS, Kemper C. Novel roles of complement in T effector cell regulation. Immunobiology. 2012;217(2):216–224. doi: 10.1016/j.imbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N, et al. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med. 2005;201(10):1523–1530. doi: 10.1084/jem.20041967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, et al. Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity. 2015;42(6):1033–1047. doi: 10.1016/j.immuni.2015.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39(6):1143–1157. doi: 10.1016/j.immuni.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pratt JR, Harmer AW, Levin J, Sacks SH. Influence of complement on the allospecific antibody response to a primary vascularized organ graft. Eur J Immunol. 1997;27(11):2848–2853. doi: 10.1002/eji.1830271116. [DOI] [PubMed] [Google Scholar]
- 26.Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med. 2002;8(6):582–587. doi: 10.1038/nm0602-582. [DOI] [PubMed] [Google Scholar]
- 27.Reh DD, Lin SY, Clipp SL, Irani L, Alberg AJ, Navas-Acien A. Secondhand tobacco smoke exposure and chronic rhinosinusitis: a population-based case-control study. Am J Rhinol Allergy. 2009;23(6):562–567. doi: 10.2500/ajra.2009.23.3377. [DOI] [PubMed] [Google Scholar]
- 28.Tammemagi CM, Davis RM, Benninger MS, Holm AL, Krajenta R. Secondhand smoke as a potential cause of chronic rhinosinusitis: a case-control study. Archives of otolaryngology–head & neck surgery. 136(4):327–334. doi: 10.1001/archoto.2010.43. [DOI] [PubMed] [Google Scholar]
- 29.Yamin M, Holbrook EH, Gray ST, Harold R, Busaba N, Sridhar A, et al. Cigarette smoke combined with Toll-like receptor 3 signaling triggers exaggerated epithelial regulated upon activation, normal T-cell expressed and secreted/CCL5 expression in chronic rhinosinusitis. The Journal of allergy and clinical immunology. 2008;122(6):1145–1153.e1143. doi: 10.1016/j.jaci.2008.09.033. [DOI] [PubMed] [Google Scholar]
- 30.Jun SW, Kim TH, Lee HM, Lee SH, Kim WJ, Park SJ, et al. Overexpression of the anaphylatoxin receptors, complement anaphylatoxin 3a receptor and complement anaphylatoxin 5a receptor, in the nasal mucosa of patients with mild and severe persistent allergic rhinitis. The Journal of allergy and clinical immunology. 2008;122(1):119–125. doi: 10.1016/j.jaci.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 31.Vandermeer J, Sha Q, Lane AP, Schleimer RP. Innate immunity of the sinonasal cavity: expression of messenger RNA for complement cascade components and toll-like receptors. Archives of otolaryngology–head & neck surgery. 2004;130(12):1374–1380. doi: 10.1001/archotol.130.12.1374. [DOI] [PubMed] [Google Scholar]
- 32.Xia L, Wang Y, Gao W, Gao H. [Expression of CD55 and CD59 in chronic rhinosinusitis and its significance] Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2014;49(12):1021–1023. [PubMed] [Google Scholar]
- 33.Gaunsbaek MQ, Lange B, Kjeldsen AD, Svane-Knudsen V, Skjoedt K, Henriksen ML, et al. Complement defects in patients with chronic rhinosinusitis. PLoS One. 2012;7(11):e47383. doi: 10.1371/journal.pone.0047383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cui YH, Zhang F, Xiong ZG, You XJ, Gao QX, Liu Z. Increased serum complement component 3 and mannose-binding lectin levels in adult Chinese patients with chronic rhinosinusitis. Rhinology. 2009;47(2):187–191. [PubMed] [Google Scholar]
- 35.Seppanen M, Suvilehto J, Lokki ML, Notkola IL, Jarvinen A, Jarva H, et al. Immunoglobulins and complement factor C4 in adult rhinosinusitis. Clin Exp Immunol. 2006;145(2):219–227. doi: 10.1111/j.1365-2249.2006.03134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fokkens WJ, Lund VJ, Mullol J, Bachert C, Alobid I, Baroody F, et al. EPOS 2012: European position paper on rhinosinusitis and nasal polyps 2012: A summary for otorhinolaryngologists. Rhinology. 2012;50(1):1–12. doi: 10.4193/Rhino12.000. [DOI] [PubMed] [Google Scholar]
- 37.Mulligan JK, Mulligan RM, Atkinson C, Schlosser RJ. Human sinonasal epithelial cells direct dendritic cell function and T-cell T helper 1/T helper 2 skewing following Aspergillus exposure. International Forum of Allergy and Rhinology. 2011;1(4):268–274. doi: 10.1002/alr.20055. [DOI] [PubMed] [Google Scholar]
- 38.Mulligan JK, Nagel W, O’Connell BP, Wentzel J, Atkinson C, Schlosser RJ. Cigarette smoke exposure is associated with vitamin D3 deficiencies in patients with chronic rhinosinusitis. The Journal of allergy and clinical immunology. 2014 doi: 10.1016/j.jaci.2014.01.039. [DOI] [PubMed] [Google Scholar]
- 39.Mulligan JK, Mulligan RM, Atkinson C, Schlosser RJ. Human sinonasal epithelial cells direct dendritic function and T-cell T helper 1/T helper 2 skewing following Aspergillus exposure. International forum of allergy & rhinology. 2011;1(4):268–274. doi: 10.1002/alr.20055. [DOI] [PubMed] [Google Scholar]
- 40.Rehl RM, Balla AA, Cabay RJ, Hearp ML, Pytynia KB, Joe SA. Mucosal remodeling in chronic rhinosinusitis. Am J Rhinol. 2007;21(6):651–657. doi: 10.2500/ajr.2007.21.3096. [DOI] [PubMed] [Google Scholar]
- 41.Schlosser RJ, Mulligan JK, Hyer JM, Karnezis TT, Gudis DA, Soler ZM. Mucous Cytokine Levels in Chronic Rhinosinusitis-Associated Olfactory Loss. JAMA otolaryngology–head & neck surgery. 2016 doi: 10.1001/jamaoto.2016.0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sansoni ER, Sautter NB, Mace JC, Smith TL, Yawn JR, Lawrence LA, et al. Vitamin D3 as a novel regulator of basic fibroblast growth factor in chronic rhinosinusitis with nasal polyposis. International forum of allergy & rhinology. 2015;5(3):191–196. doi: 10.1002/alr.21474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banglawala SM, Mulligan JK, Psaltis AJ, Wang EW, Nguyen SA, Mulligan RM, et al. Impact of intraoperative hydrodebrider treatment on postoperative sinonasal inflammation. American journal of rhinology & allergy. 2014;28(5):438–442. doi: 10.2500/ajra.2014.28.4073. [DOI] [PubMed] [Google Scholar]
- 44.Oyer SL, Mulligan JK, Psaltis AJ, Henriquez OA, Schlosser RJ. Cytokine correlation between sinus tissue and nasal secretions among chronic rhinosinusitis and controls. The Laryngoscope. 2013;123(12):E72–78. doi: 10.1002/lary.24305. [DOI] [PubMed] [Google Scholar]
- 45.Denning DW, Park S, Lass-Florl C, Fraczek MG, Kirwan M, Gore R, et al. High-frequency triazole resistance found In nonculturable Aspergillus fumigatus from lungs of patients with chronic fungal disease. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2011;52(9):1123–1129. doi: 10.1093/cid/cir179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mullins J, Seaton A. Fungal spores in lung and sputum. Clinical allergy. 1978;8(5):525–533. doi: 10.1111/j.1365-2222.1978.tb01506.x. [DOI] [PubMed] [Google Scholar]
- 47.Park SJ, Mehrad B. Innate immunity to Aspergillus species. Clin Microbiol Rev. 2009;22(4):535–551. doi: 10.1128/CMR.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tomee JFC, Wierenga ATJ, Hiemstra PS, Kauffman HF. Proteases from Aspergillus fumigatus Induce Release of Proinflammatory Cytokines and Cell Detachment in Airway Epithelial Cell Lines. Journal of Infectious Diseases. 1997;176(1):300–303. doi: 10.1086/517272. [DOI] [PubMed] [Google Scholar]
- 49.Borger P, Koëter GH, Timmerman JAB, Vellenga E, Tomee JFC, Kauffman HF. Proteases from Aspevgillus fumigatus Induce Interleukin (IL)-6 and IL-8 Production in Airway Epithelial Cell Lines by Transcriptional Mechanisms. Journal of Infectious Diseases. 1999;180(4):1267–1274. doi: 10.1086/315027. [DOI] [PubMed] [Google Scholar]
- 50.Mulligan JK, O’Connell BP, Pasquini W, Mulligan RM, Smith S, Soler ZM, et al. Impact of tobacco smoke on upper airway dendritic cell accumulation and regulation by sinonasal epithelial cells. International forum of allergy & rhinology. 2017 doi: 10.1002/alr.21955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lawrence LA, Mulligan JK, Roach C, Pasquini WN, Soler ZM, Banglawala SM, et al. Superoxide dismutase reduces the inflammatory response to Aspergillus and Alternaria in human sinonasal epithelial cells derived from patients with chronic rhinosinusitis. American journal of rhinology & allergy. 2015;29(2):89–93. doi: 10.2500/ajra.2015.29.4155. [DOI] [PubMed] [Google Scholar]
- 52.Bleier BS, Mulligan RM, Schlosser RJ. Primary human sinonasal epithelial cell culture model for topical drug delivery in patients with chronic rhinosinusitis with nasal polyposis. The Journal of pharmacy and pharmacology. 2012;64(3):449–456. doi: 10.1111/j.2042-7158.2011.01409.x. [DOI] [PubMed] [Google Scholar]
- 53.Khalid AN, Woodworth BA, Prince A, Quraishi SA, Antunes MB, Long FH, et al. Physiologic alterations in the murine model after nasal fungal antigenic exposure. Otolaryngology–head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery. 2008;139(5):695–701. doi: 10.1016/j.otohns.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Lu X, Cao PP, Chu Y, Long XB, Zhang XH, et al. Histological and immunological observations of bacterial and allergic chronic rhinosinusitis in the mouse. Am J Rhinol. 2008;22(4):343–348. doi: 10.2500/ajr.2008.22.3184. [DOI] [PubMed] [Google Scholar]
- 55.Kim JH, Yi JS, Gong CH, Jang YJ. Development of Aspergillus protease with ovalbumin-induced allergic chronic rhinosinusitis model in the mouse. American journal of rhinology & allergy. 2014;28(6):465–470. doi: 10.2500/ajra.2014.28.4100. [DOI] [PubMed] [Google Scholar]
- 56.Kim DW, Khalmuratova R, Hur DG, Jeon SY, Kim SW, Shin HW, et al. Staphylococcus aureus enterotoxin B contributes to induction of nasal polypoid lesions in an allergic rhinosinusitis murine model. American journal of rhinology & allergy. 2011;25(6):e255–261. doi: 10.2500/ajra.2011.25.3727. [DOI] [PubMed] [Google Scholar]
- 57.Lane AP, Truong-Tran QA, Myers A, Bickel C, Schleimer RP. Serum amyloid A, properdin, complement 3, and toll-like receptors are expressed locally in human sinonasal tissue. American journal of rhinology. 2006;20(1):117–123. [PMC free article] [PubMed] [Google Scholar]
- 58.Bolger MS, Ross DS, Jiang H, Frank MM, Ghio AJ, Schwartz DA, et al. Complement levels and activity in the normal and LPS-injured lung. Am J Physiol Lung Cell Mol Physiol. 2007;292(3):L748–759. doi: 10.1152/ajplung.00127.2006. [DOI] [PubMed] [Google Scholar]
- 59.Ayers CM, Schlosser RJ, O’Connell BP, Atkinson C, Mulligan RM, Casey SE, et al. Increased presence of dendritic cells and dendritic cell chemokines in the sinus mucosa of chronic rhinosinusitis with nasal polyps and allergic fungal rhinosinusitis. International forum of allergy & rhinology. 2011;1(4):296–302. doi: 10.1002/alr.20046. [DOI] [PubMed] [Google Scholar]
- 60.Liao B, Liu JX, Li ZY, Zhen Z, Cao PP, Yao Y, et al. Multidimensional endotypes of chronic rhinosinusitis and their association with treatment outcomes. Allergy. 2018 doi: 10.1111/all.13411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alt JA, Sautter NB, Mace JC, Detwiller KY, Smith TL. Antisomnogenic cytokines, quality of life, and chronic rhinosinusitis: a pilot study. The Laryngoscope. 2014;124(4):E107–114. doi: 10.1002/lary.24412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perić A, Vojvodić D, Perić AV, Radulović V, Miljanović O. Correlation between cytokine levels in nasal fluid and scored clinical parameters in patients with nasal polyposis. Indian J Otolaryngol Head Neck Surg. 2013;65(Suppl 2):295–300. doi: 10.1007/s12070-011-0447-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Divekar RD, Samant S, Rank MA, Hagan J, Lal D, O’Brien EK, et al. Immunological profiling in chronic rhinosinusitis with nasal polyps reveals distinct VEGF and GM-CSF signatures during symptomatic exacerbations. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2015;45(4):767–778. doi: 10.1111/cea.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomazic PV, Birner-Gruenberger R, Leitner A, Obrist B, Spoerk S, Lang-Loidolt D. Nasal mucus proteomic changes reflect altered immune responses and epithelial permeability in patients with allergic rhinitis. J Allergy Clin Immunol. 2014;133(3):741–750. doi: 10.1016/j.jaci.2013.09.040. [DOI] [PubMed] [Google Scholar]
- 65.Varsano S, Kaminsky M, Kaiser M, Rashkovsky L. Generation of complement C3 and expression of cell membrane complement inhibitory proteins by human bronchial epithelium cell line. Thorax. 2000;55(5):364–369. doi: 10.1136/thorax.55.5.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sunderhauf A, Skibbe K, Preisker S, Ebbert K, Verschoor A, Karsten CM, et al. Regulation of epithelial cell expressed C3 in the intestine - Relevance for the pathophysiology of inflammatory bowel disease? Mol Immunol. 2017;90:227–238. doi: 10.1016/j.molimm.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 67.Satyam A, Kannan L, Matsumoto N, Geha M, Lapchak PH, Bosse R, et al. Intracellular Activation of Complement 3 Is Responsible for Intestinal Tissue Damage during Mesenteric Ischemia. The Journal of Immunology. 2016 doi: 10.4049/jimmunol.1502287. [DOI] [PubMed] [Google Scholar]
- 68.Elvington M, Liszewski MK, Bertram P, Kulkarni HS, Atkinson JP. A C3(H20) recycling pathway is a component of the intracellular complement system. J Clin Invest. 2017;127(3):970–981. doi: 10.1172/JCI89412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reddy VB, Lerner EA. Activation of mas-related G-protein coupled receptors by the house dust mite cysteine protease Der p1 provides a new mechanism linking allergy and inflammation. J Biol Chem. 2017 doi: 10.1074/jbc.M117.787887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wills-Karp M. Allergen-specific pattern recognition receptor pathways. Curr Opin Immunol. 2010;22(6):777–782. doi: 10.1016/j.coi.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rosbjerg A, Genster N, Pilely K, Skjoedt M-O, Stahl GL, Garred P. Complementary Roles of the Classical and Lectin Complement Pathways in the Defense against Aspergillus fumigatus. Front Immunol. 2016;7:473. doi: 10.3389/fimmu.2016.00473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Speth C, Rambach G. Complement Attack against Aspergillus and Corresponding Evasion Mechanisms. Interdiscip Perspect Infect Dis. 2012;2012:463794. doi: 10.1155/2012/463794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hartmann K, Henz BM, Kruger-Krasagakes S, Kohl J, Burger R, Guhl S, et al. C3a and C5a stimulate chemotaxis of human mast cells. Blood. 1997;89(8):2863–2870. [PubMed] [Google Scholar]
- 74.Kemper C, Kohl J. Novel roles for complement receptors in T cell regulation and beyond. Mol Immunol. 2013;56(3):181–190. doi: 10.1016/j.molimm.2013.05.223. [DOI] [PubMed] [Google Scholar]
- 75.Mizutani N, Nabe T, Yoshino S. Complement C3a regulates late asthmatic response and airway hyperresponsiveness in mice. J Immunol. 2009;183(6):4039–4046. doi: 10.4049/jimmunol.0901468. [DOI] [PubMed] [Google Scholar]
- 76.Raedler H, Yang M, Lalli PN, Medof ME, Heeger PS. Primed CD8(+) T-cell responses to allogeneic endothelial cells are controlled by local complement activation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2009;9(8):1784–1795. doi: 10.1111/j.1600-6143.2009.02723.x. [DOI] [PubMed] [Google Scholar]
- 77.Lalli PN, Strainic MG, Lin F, Medof ME, Heeger PS. Decay accelerating factor can control T cell differentiation into IFN-gamma-producing effector cells via regulating local C5a-induced IL-12 production. J Immunol. 2007;179(9):5793–5802. doi: 10.4049/jimmunol.179.9.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kwan WH, Hashimoto D, Paz-Artal E, Ostrow K, Greter M, Raedler H, et al. Antigen-presenting cell-derived complement modulates graft-versus-host disease. The Journal of clinical investigation. 2012;122(6):2234–2238. doi: 10.1172/JCI61019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Risitano AM, Marotta S. Therapeutic complement inhibition in complement-mediated hemolytic anemias: Past, present and future. Semin Immunol. 2016;28(3):223–240. doi: 10.1016/j.smim.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 80.Subias Hidalgo M, Martin Merinero H, Lopez A, Anter J, Garcia SP, Ataulfo Gonzalez-Fernandez F, et al. Extravascular hemolysis and complement consumption in Paroxysmal Nocturnal Hemoglobinuria patients undergoing eculizumab treatment. Immunobiology. 2017;222(2):363–371. doi: 10.1016/j.imbio.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 81.Risitano A. Paroxysmal Nocturnal Hemoglobinuria: The Future Of Complement-Based Therapies. Current drug targets. 2016 [PubMed] [Google Scholar]
- 82.Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunological reviews. 2016;274(1):33–58. doi: 10.1111/imr.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kohl J, Baelder R, Lewkowich IP, Pandey MK, Hawlisch H, Wang L, et al. A regulatory role for the C5a anaphylatoxin in type 2 immunity in asthma. J Clin Invest. 2006;116(3):783–796. doi: 10.1172/JCI26582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang X, Lewkowich IP, Kohl G, Clark JR, Wills-Karp M, Kohl J. A protective role for C5a in the development of allergic asthma associated with altered levels of B7-H1 and B7-DC on plasmacytoid dendritic cells. J Immunol. 2009;182(8):5123–5130. doi: 10.4049/jimmunol.0804276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.