

Abstract
Tumor necrosis factor-like cytokine 1A (TL1A, TNFSF15) is implicated in inflammatory bowel disease (IBD), modulating the location and severity of intestinal inflammation and fibrosis. TL1A expression is increased in inflamed gut mucosa and associated with fibrostenosing Crohn’s disease. Tl1a-overexpression in mice leads to spontaneous ileitis, and exacerbated induced proximal colitis and fibrosis. IBD is associated with shifts in the gut microbiome, but the effect of differing microbial populations and their interaction with TL1A on fibrosis has not been investigated. We demonstrate that the profibrotic and inflammatory phenotype resulting from Tl1a-overexpression is abrogated in the absence of resident microbiota. To evaluate if this is due to the absence of a unique bacterial population, as opposed to any bacteria per se, we gavaged germ-free (GF) wild-type and Tl1a-transgenic (Tl1a-Tg) mice with stool from specific pathogen-free (SPF) mice and a healthy human donor (Hu). Reconstitution with SPF, but not Hu microbiota, resulted in increased intestinal collagen deposition and fibroblast activation in Tl1a-Tg mice. Notably, there was reduced fibroblast migration and activation under GF conditions compared to native conditions. We then identified several candidate organisms that correlated directly with increased fibrosis in reconstituted mice and showed that these organisms directly impact fibroblast function in vitro. Thus, Tl1a-mediated intestinal fibrosis and fibroblast activation are dependent on specific microbial populations.
Introduction
Tl1a (a protein encoded by TNFSF15) is a member of the tumor necrosis factor (TNF) superfamily that binds to death domain receptor 3 (DR3), expressed on a variety of cell types including immune cells, epithelial cells, and fibroblasts (1–5). Modulating an array of immune responses, Tl1a can be produced by endothelial cells in response to IL-1β and TNFα, by macrophages and dendritic cells in response to Toll-like receptor stimulation, as well as in some lymphoid lineage cells (2,3,6–9). A TNFSF15 haplotype is associated with higher Tl1a production, increased risk of CD, intestinal fibrostenosis, and greater need for surgery (10–13). In mice, constitutive Tl1a expression induced increased collagen deposition in the colon without detectable histologic colitis as well as increased collagen deposition in the ileum with spontaneous ileitis (14–17). Under colitogenic conditions induced by chronic DSS treatment or adoptive T-cell transfer, there was increased collagen deposition with fibrostenotic lesions in the gut that caused intestinal obstruction in the Tl1a-Tg mice (12). These results support the role of Tl1a in fibrogenesis that can lead to fibrostenosis in the setting of chronic inflammation, which is a common complication of CD leading to resection.
The intestinal microbiome has been linked with many inflammatory diseases including IBD (18–21). Although previous studies have found alterations in various bacterial taxa in IBD patients, and a recent study found an association with a fibrostenotic disease cohort, none have correlated specific microbes with degree of fibrosis and fibroblast phenotype (22). Most of these studies have been largely associative without the ability (by design) to prove causality. The question remains, therefore, whether altered microbiota associated with IBD contribute to the disease phenotype or are its consequence. In rodent models, comparison of experimental IBD models under GF conditions have yielded disparate results with development of colitis in most spontaneous genetically engineered models dependent on resident microbiota, or uniquely, potentiated DSS-induced colitis in GF mice (23–25). No animal experiments have evaluated the contribution of the microbiome to fibrosis in the context of IBD. In this study, we show that the native murine fecal microbiota is required for optimal Tl1a-dependent fibroblast activation and transformation into myofibroblasts. Moreover, we provide evidence that the intestinal fibrotic phenotype requires specific microbial cues provided by mouse microbiota from an SPF facility but absent in human feces from a healthy donor. Our analysis further identified several candidate organisms that correlate directly with degree of fibrosis in reconstituted hosts and impact fibroblasts in vitro. To our knowledge, this is the first study to establish a potential causal role for the microbiome in intestinal fibrosis, fibroblast activation and function.
Results
The intestinal microbiome is required for Tl1a-enhanced intestinal inflammation, collagen deposition and fibroblast migration
In agreement with our prior results, Tl1a-Tg mice raised under SPF microbial conditions display significant spontaneous ileitis, as evident by increased histopathological scoring under H&E, with over a 2-fold increase in average histopathology compared with wild type mice (Figure 1A) (16). No histologically apparent cecal inflammation was observed (Figure 1B). Despite this, Tl1a-Tg mice demonstrate increased cecal collagen deposition (Figure 1D), underscoring the importance of TL1A as a mediator of fibrosis that can act independently of its pro-inflammatory effects.
Figure 1. The intestinal microbiome is required for Tl1amediated location and degree of intestinal inflammation and collagen deposition.
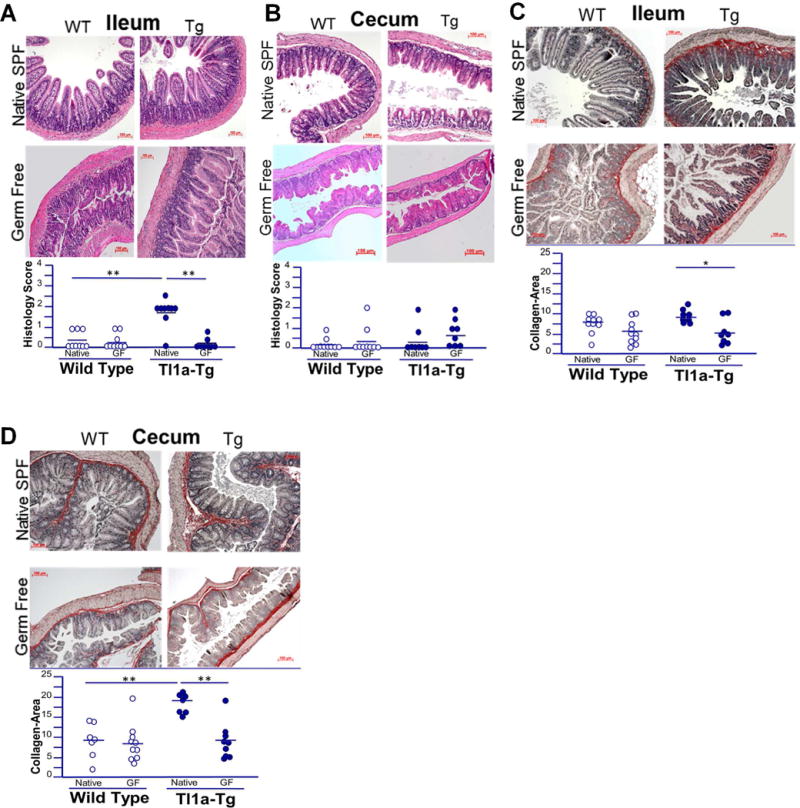
A-B) Representative H&E stains of ileal and cecal sections of 5-month-old WT and Tl1a-Tg mice, raised in germ free and native SPF conditions. C-D) Representative Sirius red staining of collagen deposition in ileal and cecal sections of 5-month-old WT and Tl1a-Tg mice, raised in GF and native SPF conditions. Dot plots with means displayed; n = 7-10 mice per group. * indicates p<0.05; ** p<0.01.
As the microbiome is relevant to inflammation in several diseases, we evaluated the effects of GF conditions on Tl1a-mediated intestinal inflammation. The absence of a microbiome abrogated the spontaneous ileitis induced by Tl1a-overexpression, as there were no significant differences in ileal histopathology between GF Tl1a-Tg and GF WT mice (Figure 1A).
We next evaluated if the fibrosis observed in Tl1a-Tg mice was dependent upon the microbiome. The absence of microbes significantly reduced ileal and cecal collagen deposition in GF Tl1a-Tg mice compared with microbiome intact Tl1a-Tg mice (Figure 1C-D). No difference was observed in WT mice.
Since we observed notable changes in fibrosis between Tl1a-Tg and WT mice under native SPF conditions that were abrogated under GF conditions, we sought to determine the impact of Tl1a-overexpression and resident microbiota on fibroblast phenotype. Colonic fibroblasts isolated from Tl1a-Tg mice raised under native microbial conditions displayed significantly increased migratory capacity after simulated wounding compared with those from WT mice (Figure 2A-B). Tl1a-overexpression also increased fibroblast adhesion, which was unchanged in GF mice (Supplementary Figure 1). The enhanced rate of fibroblast gap-closure observed with Tl1a-overexpression under native conditions was eliminated under GF conditions, consistent with the observed reduction in histological fibrosis (Figure 2A-B). Interestingly, the absence of microbiome reduced fibroblast migratory capacity even in WT mice, but to a lesser extent than in Tl1a-Tg mice. These results indicate that the intestinal microbiome is required for Tl1a-mediated intestinal fibrosis and influences fibroblast migratory function.
Figure 2. The intestinal microbiome is required for Tl1a-enhanced fibroblast migration.
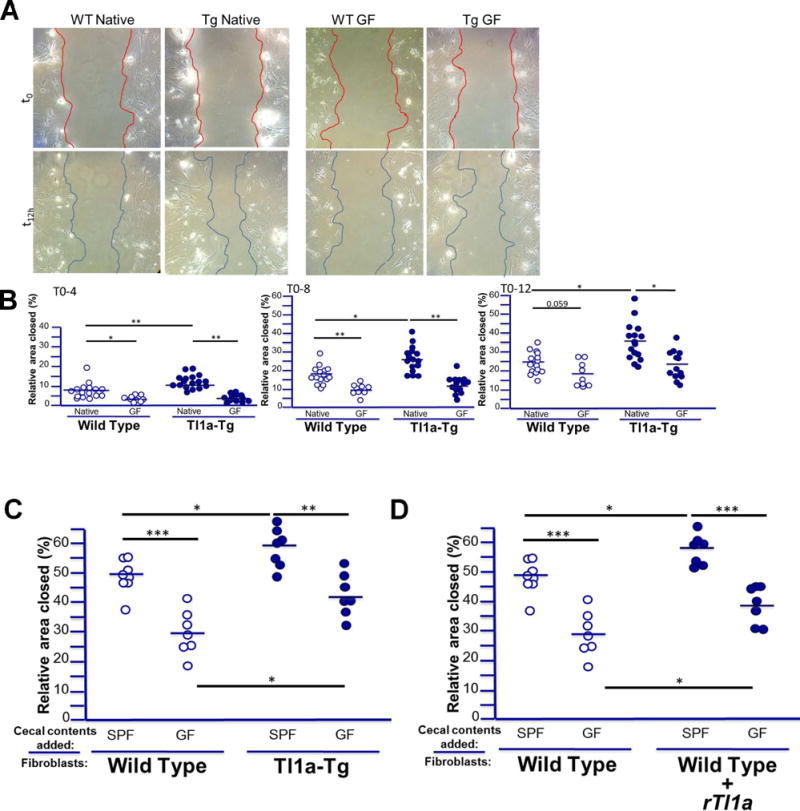
A) Representative images of gap-closure assay after simulated wound shown for WT and Tl1a-Tg mice under native (left) and GF conditions (right) at initial wound (upper panels) and after migration (lower panel). B) Relative % area of gap closed indicated for fibroblasts isolated from WT and Tl1a-Tg mice raised under native SPF or GF conditions with 4, 8, and 12 hours of migration after initial wound. C) Relative % area of gap closed indicated for fibroblasts isolated from WT and Tl1a-Tg mice raised under native SPF conditions and supplemented with cecal washings from either native SPF or GF WT mice during 16 hours of migration after initial wound. D) Relative % area of gap closed indicated for fibroblasts isolated from native SPF WT mice and supplemented with cecal washings as in (C), or with either of the cecal washings and recombinant mouse Tl1a (100ng/ml). Data points represent fibroblast isolation from individual mice and bars represent means. * indicates p<0.05, ** p<0.01, *** p<0.005.
An important question arising from these results is whether the observed findings are due to direct effects of SPF microbiota and Tl1a (alone or in concert) on fibroblasts themselves, or if the microbiome and Tl1a in Tl1a-Tg mice affect other non-fibroblast cell types, which then promote fibroblast activation and profibrotic phenotype. Bacterial components and products can induce fibroblast activation (26), but it is unclear if they can promote fibroblast migration directly. Moreover, the direct effect of bacterial stimulus on fibroblasts in the context of host Tl1a-overexpression has not been evaluated. We therefore assessed if bacterial products isolated from the cecal luminal washings of native WT SPF mice could promote fibroblast migration directly, and if this effect was enhanced by host Tl1a-overexpression. WT fibroblasts exposed to native SPF cecal washings demonstrate significantly increased migration compared to those exposed to cecal washings from GF mice (Figure 2C). This direct effect of cecal bacterial components was enhanced by host Tl1a-overexpression in Tl1a-Tg mice. Thus, a significant part of the commensal microbiome’s effect on fibroblast migration seen in Figure 2A-B may be due to direct effects mediated by bacterial components (or products) on fibroblasts themselves.
We next asked if this enhanced migratory phenotype in fibroblasts from Tl1a-Tg mice was partly due to direct Tl1a-mediated effects on fibroblasts; namely, can Tl1a promote fibroblast migration directly and do so in concert with the direct effect of SPF microbiota on fibroblasts seen in Figure 2C? We have previously demonstrated that fibroblasts express the Tl1a receptor DR3, and upon treatment with Tl1a in vitro, demonstrate expression of alpha-smooth-muscle actin (indicative of activation of myofibroblasts) and collagen (27). Consequently, fibroblasts treated with Tl1a in vitro demonstrate expression of alph-smooth-muscle actin (activation to myofibroblasts) and collagen (27). We therefore hypothesized that direct stimulation of fibroblasts with Tl1a would increase migration. WT fibroblasts treated with Tl1a (and without bacterial components) in vitro displayed significantly increased migration compared with untreated cells, suggesting a direct effect of Tl1a on fibroblast migration (Figure 2D). To determine if Tl1a can enhance the fibroblast migratory response to bacterial components, we conducted the same experiments as in Figure 2C in the context of exogenous Tl1a stimulation. WT fibroblasts treated with both Tl1a and SPF cecal bacterial products together demonstrated enhanced migration compared with those treated solely with the bacterial products (Figure 2D). These data demonstrate that resident bacteria and Tl1a can both stimulate fibroblasts, and the direct effect of resident bacteria on fibroblasts is enhanced directly by Tl1a.
Gavage with murine but not human fecal microbiota promotes intestinal inflammation and collagen deposition in Tl1a transgenic mice
As these data demonstrate that the microbiome is required for Tl1a-mediated fibrosis in intestinal regions with and without underlying inflammation, we sought to evaluate if this phenotype is due to the absence of a unique bacterial population, as opposed to any bacterial colonization per se. We used two distinct microbiota to test the hypothesis that the pro-fibrotic phenotype observed in Tl1a-Tg mice under native microbial conditions was due to a specific bacterial population adapted to the mouse intestine rather than the presence of any gut bacteria. GF mice were gavaged with stool collected from wild-type mice housed in SPF or with stool from a healthy human (Hu) donor and evaluated 2 months later.
WT mice displayed no increase in intestinal inflammation or collagen deposition when colonized with either SPF or Hu flora, indicating that in the absence of Tl1a-overexpression the species-specific microbiome does not induce intestinal inflammation or fibrosis (Figure 3A-D). Tl1a-transgenic mice colonized with SPF microbiota demonstrated increased collagen deposition in both inflamed ileum and non-inflamed cecum, consistent with findings in mice under native conditions (Figure 3A-D, Figure 1). In contrast, Tl1a-transgenic recipients of Hu microbiota showed no increase in ileal or cecal collagen deposition or inflammation. Together, these data indicate that Tl1a-mediated intestinal fibrosis is modulated by the composition of the intestinal microbiome and suggest that this phenotype is induced by microbes selectively contained in the SPF mouse microbiota but missing from the human microbiota.
Figure 3. Tl1a-Tg mice have increased intestinal inflammation and collagen deposition in the presence of mouse microbiota, but not human intestinal microbiota.
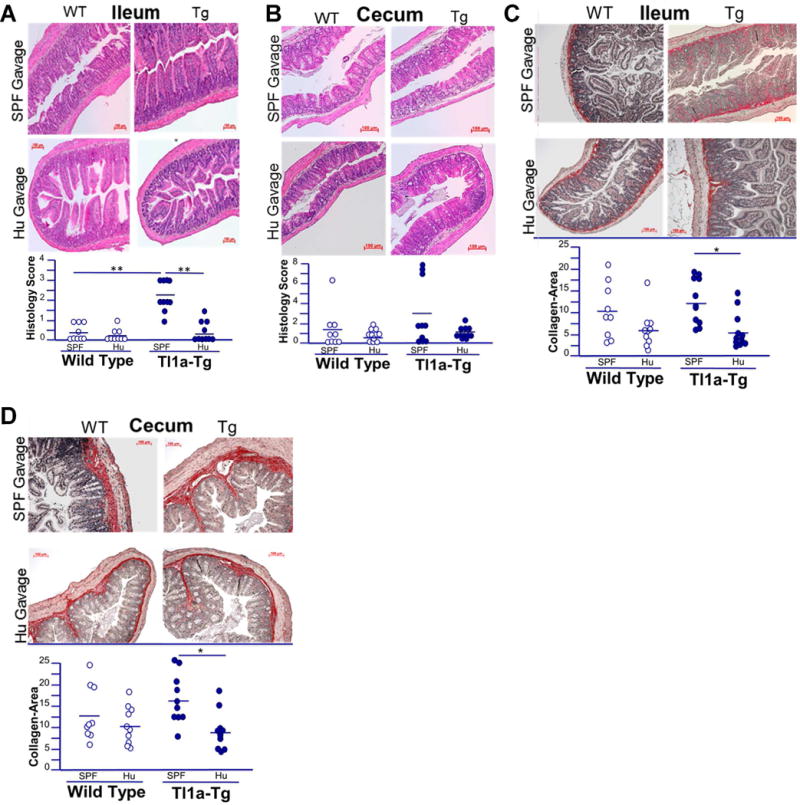
A-B) Representative H&E stains of ileal and cecal sections of WT and Tl1a-Tg mice, raised under germ free conditions and then gavaged with either human (Hu) or specific pathogen free (SPF) microbiota. C-D) Representative Sirius red staining of collagen deposition in ileal and cecal sections of WT and Tl1a-Tg mice, raised under germ free conditions and then gavaged with either Hu or SPF microbiota. Dot plots with means displayed; n = 7-10 mice per group. * indicates p<0.05; ** p<0.01.
Murine microbiota potentiate Tl1a-mediated intestinal fibroblast differentiation to myofibroblasts
Fibroblast activation has been shown to occur after bacterial stimulation e.g. with lipopolysaccharide (26). Previously, we showed that there is an increase in the proportion of intestinal myofibroblasts in Tl1a-Tg mice raised under conventional SPF conditions (27). We investigated whether absence of microbial stimulation (i.e. under GF conditions) impairs fibroblast differentiation to myofibroblasts. GF Tl1a-Tg mice did not display an increased number or proportion of activated fibroblasts in the cecum compared to GF WT mice (Figure 4A-B). Colonization with SPF microbiota induced intestinal myofibroblasts in both WT and Tl1a-Tg mice relative to GF conditions or colonization with Hu microbiota (Figure 4A-B). SPF microbiota, but not Hu microbiota also restored the increased proportion of myofibroblasts in Tl1a-Tg mice compared to WT controls (54.7% vs. 36.7%). Interestingly, GF Tl1a-Tg mice had reduced myofibroblast proportion compared to GF WT mice, which was not seen in the presence of Hu microbiota (Figure 4A-B). These results show that fibroblast activation in the cecum induced by Tl1a overexpression is microbiota-dependent and that microbial composition affects fibroblast differentiation into myofibroblasts.
Figure 4. Tl1a-Tg mice have increased fibroblast activation in the presence of mouse microbiota, but not human intestinal microbiota.
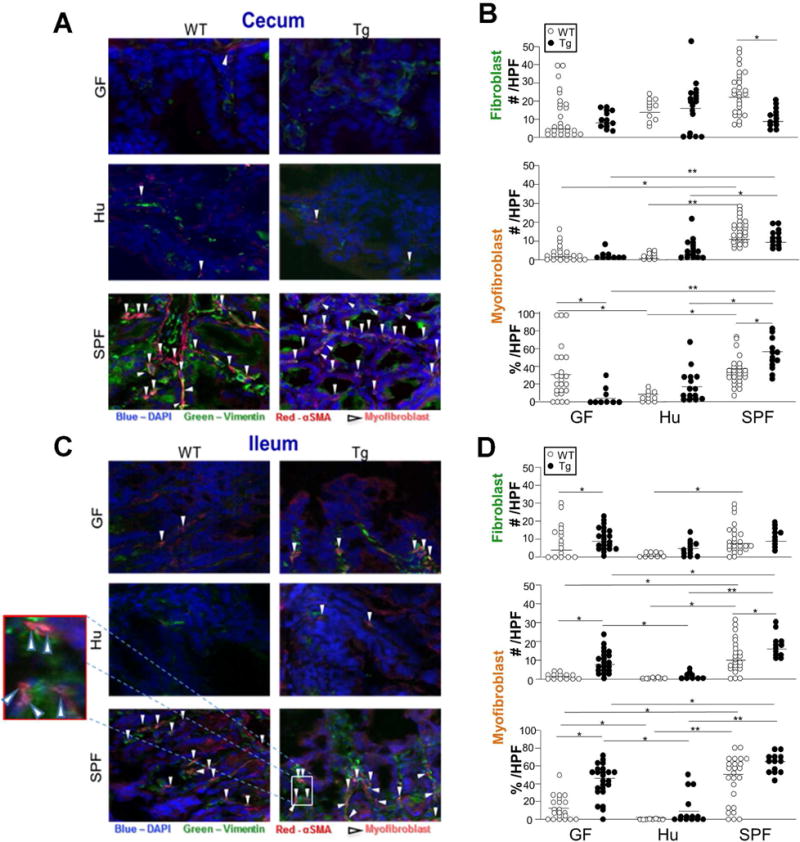
Immunofluorescent staining of vimentin (green) and αSMA (red) from cecal (A) and ileal (C) sections of 5-month-old WT and Tl1a-Tg mice raised in germ free conditions and then gavaged with either Hu or SPF microbiota. White arrows denote myofibroblasts that co-localize vimentin and αSMA at 400× magnification. Total numbers and percentage of myofibroblasts over total vimentinpositive cells per individual HPF in cecum (B) or ileum (D) were quantitated and displayed as dot plots with means. Pooled data of total individual HPFs, representative of 4-6 mice per group. * indicates p<0.05; ** p<0.01.
To assess whether Tl1a mediated fibroblast activation in the ileum also requires the microbiome, we quantitated myofibroblasts in Tl1a-Tg mice under GF conditions. In contrast to the cecum, there was an increased proportion of activated myofibroblasts in the ileum of GF Tl1a-Tg mice compared with GF WT mice (Figure 4C-D). We next tested whether the degree of Tl1a-mediated fibroblast activation is affected by the specific microbiome. SPF gavage increased myofibroblast numbers and proportion in both WT and Tl1a-Tg mice while preserving the relative increase in myofibroblasts in Tl1a-Tg mice (Figure 4C-D). In contrast, mice colonized with Hu microbiota had reduced proportion of fibroblast activation compared with GF conditions, suggesting that members of the Hu microbiota may have inhibited fibroblast activation. This did not result in significant histopathological differences in collagen deposition, however. Taken together, these results suggest greater modulation of Tl1a-mediated fibroblast activation by the microbiome in the cecum (without concomitant changes in inflammation), for which Tl1a can potentially compensate in the ileum, (and in which there is significant increases in inflammation). This may reflect distinct microbial communities and mucosal immunity in the ileum, reflected in the significant increase in ileal inflammation in Tl1a-Tg mice. Indeed, differing microbial composition between the ileum and the cecum has been well-documented in mice and humans, including patients with IBD who show distinct microbiome profiles between subsets with ileal vs. colonic disease (48). Consistent with this, sequencing of mucosal and luminal microbial communities in the ileum and cecum demonstrated distinct microbial populations in the ileum vs. the cecum in both Tl1a-Tg and WT mice colonized with SPF microbiota (Supplemental Figure 2). These data illustrate biogeographic differences in the host-microbe interactions underlying intestinal collagen deposition. Additionally, our data demonstrated that microbial composition modulates the degree of Tl1a-mediated myofibroblast activation independent of the intestinal location.
Fibrosis severity is associated with the abundance of specific microbes found in the mouse SPF microbiome
Since SPF and Hu microbiota had differential effects on collagen deposition and fibroblast activation, we hypothesized that the abundance of specific bacteria would be associated with the degree of fibrosis seen in recipient mice. To evaluate this, we performed 16S rRNA sequencing to characterize the ileal and cecal microbiome of colonized mice and then employed multivariate models to identify microbes with a statistically significant positive or negative association with fibrosis score. These microbes were then used to construct co-occurrence/co-exclusion networks with fibrosis severity to identify the microbes that were directly associated with increased or decreased fibrosis rather than merely having a co-occurrence or co-exclusion relationship with fibrosis-associated microbes. Separate analyses were performed of the cecum and ileum of humanized and SPF-colonized ex- GF mice (Figure 5). In the cecum of SPF-colonized mice, which demonstrated significant collagen deposition and fibroblast activation, we identified several microbes not present in Hu-gavaged mice that clustered tightly with fibrosis (Figure 5A). This included groups of mucolytic bacteria such as Mucispirillum schaedleri and Ruminococcus. Additionally, Anaeroplasma were also significantly associated with fibrosis in the cecum of SPF-colonized mice. Members of Oscillospira and Coprococcus were negatively correlated with fibrosis in the cecum (Figure 5A).
Figure 5. Microbial networks associated with cecal and ileal fibrosis in germ-free mice colonized with SPF or Hu microbiota.
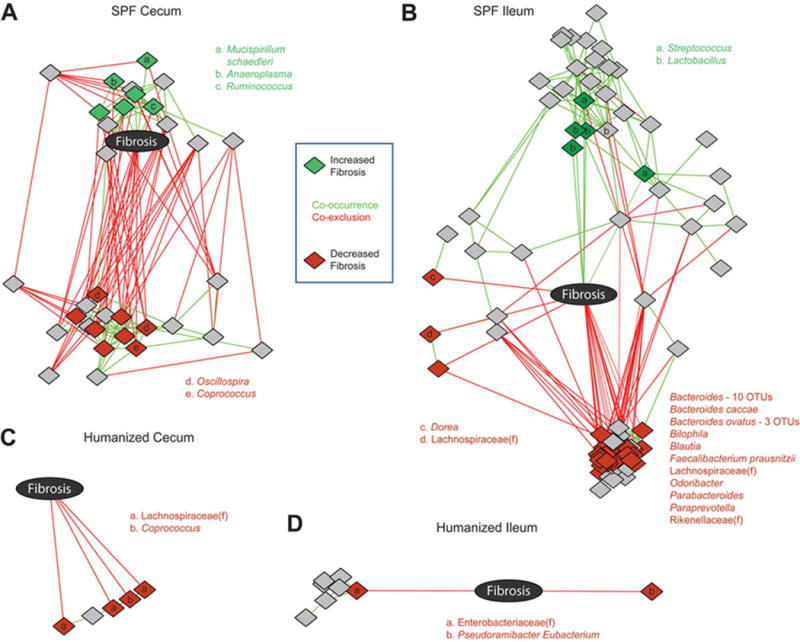
A) Microbes associated with fibrosis in DESeq2 models were used to construct cooccurrence/ co-exclusion networks with fibrosis score. Networks were constructed in Cytoscape with an edge-weighted, spring-embedded layout, which groups nodes with highly interconnected nodes. Diamonds represent OTUs, colored by association with fibrosis: green (associated with increased fibrosis), red (associated with reduced fibrosis), gray (indirect association only). Letters indicate the genus of fibrosis-associated OTUs; unmarked OTUs are unclassified members of the Clostridiales order. Similar network plots were constructed for fibrosis in the ileum of SPF colonized mice (B), cecum of Hu mice (C), and ileum of Hu mice (D). In (B), the names and numbers of OTUs associated with reduced fibrosis are shown next to a dense cluster in which it is difficult to discern individual OTUs. OTUs that are unclassified members of microbial families are indicated with (f).
In the ileum of SPF-colonized mice, we observed that there were competing sets of microbes associated with enhanced or reduced fibrosis severity (Figure 5B). For example, members of the Streptococcus and Lactobacillus genera were found to be positively associated with fibrosis, whereas Faecalibacterium prausnitzii and members of Bacteroides were negatively associated with fibrosis. Consistent with the absence of histological fibrosis, we observed only negative correlations between microbial species and fibrosis in both the cecum and ileum of Hu-colonized mice (Figure 5C-D).
Differential effects of bacteria positively or negatively correlated with fibrosis on in vitro fibroblast function
Next, we determined whether bacterial strains that positively or negatively correlated with cecal fibrosis severity in vivo could alter fibroblast function directly in in vitro. Cell lysates of Ruminoccocus and M. schaedleri, two bacterial strains that were positively correlated with the degree of fibrosis, promoted fibroblast migration and collagen expression compared with negatively-correlated Oscillospira, which had comparatively less pronounced effects (Figure 6). These results show that microbes that positively or negatively correlate with fibrosis in vivo can directly and disparately impact fibroblasts in vitro. Furthermore, one potential causal mechanism is now suggested by which specific organisms in the gut microbiome mediate fibrosis.
Figure 6. Differential effects of bacteria positively or negatively correlated with fibrosis on fibroblast migration and collagen expression.
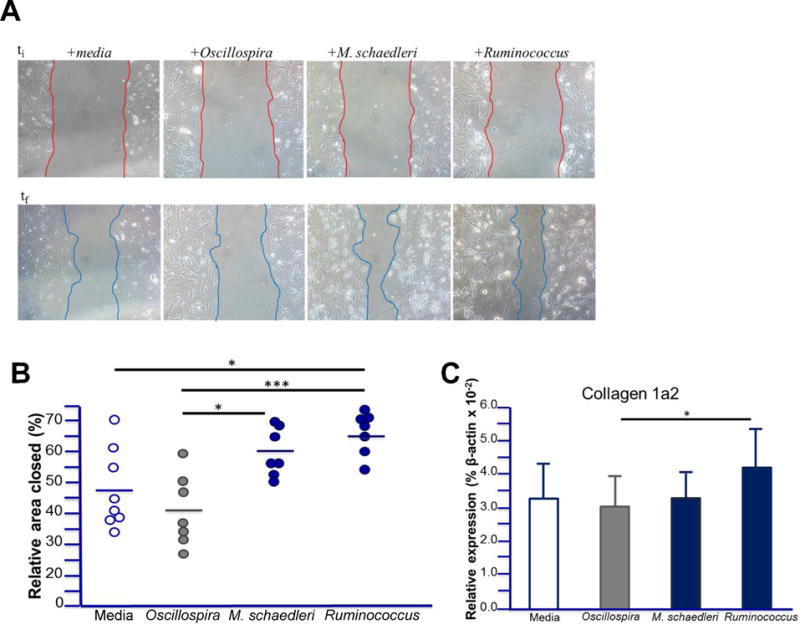
A) Representative images of gap-closure assay after simulated wound on fibroblasts isolated from native WT mice at initial wound (upper panels) and after migration (lower panel) in the presence of bacterial lysates. B) Relative % area of gap closed for fibroblasts isolated from native WT mice and supplemented with indicated bacterial lysates (25 μg/ml) during migration. Data points represent fibroblast isolation from individual mice and bars represent means. C) Relative expression of collagen from fibroblasts in (B) after migration in the presence of the specified bacterial lysates. Data are represented as means ± SD; * indicates p<0.05, ** p<0.01, *** p<0.005.
Discussion
To our knowledge, this is the first study that causally implicates the intestinal microbiome in intestinal fibrosis, demonstrating that fibrosis requires the presence of resident microbiota and that Tl1a-mediated fibrosis is dependent upon specific bacteria or bacterial consortia. Furthermore, we show that microbes that positively or negatively correlate with intestinal fibrosis in vivo have direct (and opposing) effects on fibroblast function in vitro. These results also suggest that microbiome-TL1A interactions may influence the degree and location of intestinal fibrosis in IBD, which has up to now been attributed to the severity of inflammation. Accordingly, no histologically significant cecal inflammation was observed under SPF microbial conditions but despite this, Tl1a-Tg mice demonstrate increased cecal collagen deposition, underscoring the importance of TL1A as a modulator of the location and severity of mucosal inflammation, as well as a pro-fibrotic mediator that can act independently of its pro-inflammatory effects. Indeed, this disjunction between inflammation and fibrosis is clinically significant. While inflammatory disease may be associated with significant fibrotic change, as increased inflammation perpetuates the cascade of mucosal repair, the frequency of fibrostenosing complications remains significant despite immunosuppressive therapy in CD patients in the form of steroids or immunomodulators (45). Findings that provide insight into unique pro-fibrotic mediators—whether cytokine- or microbiome-driven (or both)—are highly relevant for clinical disease.
This theme apparent in the histopathological results was mirrored in our results for fibroblast activation, which further underscore the relevance of a pro-fibrotic SPF consortia and Tl1a. Given their expression of Toll-like-receptors, fibroblasts have the capacity to become activated by bacterial products (26). Consistent with this, Tl1a-mediated fibroblast activation required the microbiome in the cecum (despite no concomitant changes in inflammation). However, in the ileum, (in which there is significant increase in inflammation), Tl1a could partially compensate for the lack of the microbiome and promote some fibroblast activation. This might point to unique effects of Tl1a on fibroblast activation that are tissuespecific, but may still ultimately require a specific consortium of organisms in SPF to yield histopathologically evident fibrosis, as Tl1a-Tg mice still had reduced ileal fibrosis under GF conditions and with Humanized microbiome compared with conventional SPF microbiota (Figure 1 and 3). The effect of SPF microbiota on ileal fibrosis was not as dramatic as in the cecum. One possible explanation for this effect is the differing microbial communities that colonize the ileum vs. the cecum. We show that fibrosis correlated much more closely with several organisms in the cecum compared with the ileum where there were equal positive and negative “pulls” at fibrosis, with a few positively correlated organisms and numerous negatively correlated organisms. The organisms that are tightly correlated with increased fibrosis in the cecum are less abundant in the ileum. Similarly, the ileum harbors numerous organisms that are negatively correlated with fibrosis compared with the cecum where there are fewer such organisms. A related potential explanation, is the quantity, structure, and type of mucous seen in the small intestine vs. the large intestine. The differing mucin composition may account for observed differences in bacterial composition in different regions of the gut (49). The mucin-rich large intestine can harbor anaerobic organisms with a repertoire of glycosidic enzymes that disassemble complex mucus glycans to be used as a carbon source (50). We identified mucin-degrading bacteria (M. schaedleri) to correlate with fibrosis in the cecum. The effect of differing microbial compositions in the cecum vs. ileum on fibrosis may be mediated through bacterial modulation of fibroblast phenotype, since specific bacteria may have direct and opposing effects on fibroblast, as we demonstrated in vitro.
Cecal and ileal colonization with SPF microbiota induced intestinal myofibroblasts in both WT and Tl1a-Tg mice relative to GF conditions or colonization with Hu microbiota. Notably, however, colonization with SPF microbiota (but not Hu microbiota), in addition to Tl1a-overexpression, resulted in overall increased proportion of myofibroblasts. Interestingly, GF Tl1a-Tg mice had reduced myofibroblast proportion in the cecum compared to GF WT mice, which was not seen in the presence of Hu microbiota. This raises the question as to the specific contribution of Tl1a-overexpression vs. microbial changes in previously referenced colitigenic models conducted in native microbiome-intact mice (12, 27). It would, therefore, be important to determine what effects Tl1a-overexpression, independent of any bacterial stimulation, but in the presence of other mucosal stimulation, such as inflammatory insults due to DSS for example, may have on intestinal fibrosis and inflammation; or the effects that differing microbial populations may impart on experimental colitis.
These results also have novel implications for a microbiome effect on fibroblast function in concert with Tl1a. Consistent with the above noted points regarding the disconnect between pro-inflammatory and profibrotic stimuli, colonic fibroblasts do not migrate in response to classic pro-inflammatory cytokines such as TNFα or IL-1, but rather require traditionally “pro-fibrotic” cytokines such as TGFβ to induce migration (46). It is notable that in our migration assay Tl1a significantly and directly increased fibroblast migration compared with controls, again suggesting a direct contribution of Tl1a to the pro-fibrotic pathway, which may act independently of inflammation. Importantly, our data demonstrate that the gut microbiota can promote this effect directly in concert with Tl1a. These findings propose novel roles for both Tl1a and SPF microbiota in fibroblast function.
In this study, we utilized a novel correlation of direct changes in fibrosis with specific bacterial abundance in a region-specific manner. In the cecum of SPF-colonized mice, which demonstrated significant collagen deposition and fibroblast activation, we identified several microbes that clustered tightly with fibrosis. This included Mucispirillum schaedleri, a mucous degrading organism that has been reported to discriminate between colitis and remission (28) in a mouse model but has not been linked to fibrosis. Ruminococcus are another group of mucolytic bacteria that have been observed to be increased in CD in some studies, associated with the stricturing phenotype in a recent pediatric CD study, and contribute to experimental colitis (22, 29, 30, 44). Ruminococcus and M. schaedleri were capable of directly modulating fibroblast function in vitro. Therefore, further studies demonstrating potential causal efficacy of these correlated organisms in vivo are warranted. It would be interesting to assess in future studies whether M. schaedleri, or previously identified species of Ruminococcus, are present in Tl1a-Tg mice with fibrostenosis under colitic conditions and in CD patients with the high risk TNFSF15 haplotype and stricturing disease. Furthermore, it would be informative to assess mucus structure, mucosal barrier function, and fibroblast activation in mice and humans with and without these mucous degrading organisms. Lastly, Anaeroplasma, a genus which has been previously associated with experimental colitis (31), was also significantly associated with fibrosis in the cecum of SPF-colonized mice. In terms of organisms that were associated with reduced fibrosis in the cecum, Coprococcus, a genus that has been reported to be depleted in patients with CD (34) was associated with reduced fibrosis in both, mice reconstituted with SPF microbiota and human microbiota. Oscillospira have been associated with gut health, and their reduced abundance has been implicated in a variety of diseases including CD (47). Notably, compared with positively correlated organisms, Oscillopsira mitigated fibroblast function in vitro.
In the ileum of SPF-colonized mice, we observed that there were competing sets of microbes associated with either enhanced or reduced fibrosis severity. It is worth noting that ileal inflammation may have impacted the microbial variations and associations with fibrosis seen, compared with the cecum which had very tight microbial associations with fibrosis in the context of no significant inflammation. Members of the Streptococcus and Lactobacillus genera were found to be positively associated with fibrosis in the ileum. This is concordant with recent human data indicating that fecal abundance of these microbes is associated with another fibrotic complication of IBD, primary sclerosing cholangitis (32). Many organisms that were associated with reduced fibrosis in SPF-colonized mice have previously been observed to be depleted in patients with CD, including Faecalibacterium prausnitzii, which has been well-described to have anti-inflammatory properties (29). Additionally, members of the Lachnospiraceae family, which contains many butyrate producers that are decreased in CD patients (33), were associated with reduced fibrosis in the ileum of SPF-colonized mice. To our knowledge, this is the first study to link these microbes not just to protection from inflammation, but also fibrotic disease. Thus, it would be important to determine mechanistically how these short-chain fatty acid producers affect intestinal fibrosis in addition to inflammation. One possibility is a direct effect of these microbes (or their products) on fibroblast function, as our data suggest.
Interestingly, our microbiome-fibrosis correlation studies also underscore the disjunction between inflammation and fibrosis noted above. Sulfite-reducing bacteria such as Bilophila have been associated with a pro-inflammatory T helper type 1 immune response and an ability to induce experimental colitis (35). Despite the potential pro-inflammatory effects of such bacteria, we found that Bilophila correlated with a reduction in fibrosis in the ileum of SPF-colonized mice. Thus, hydrogen sulfide, one of the metabolic products of these bacteria, may have opposing effects on intestinal inflammation compared with fibrosis (36, 37).
To our knowledge, this is the first study that causally implicates the intestinal microbiome in intestinal fibrosis, demonstrating that Tl1a-mediated fibrosis is dependent upon specific bacteria or bacterial consortia and that those bacteria can directly affect fibroblast function. Thus, a focus on TL1A pathways acting in concert with the microbiome may identify future therapeutic targets for fibrostenosing Crohn’s disease.
Materials and Methods
Gnotobiotic experiments
Tl1a-Tg (which have sustained TL1A expression) and WT mice, both on C57Bl/6 background were re-derived into germ-free status and bred under sterile conditions at the National Gnotobiotic Rodent Resource Center, Chapel Hill, NC. Tl1a-Tg mice and WT littermates at 2-4 months of age were orally gavaged with 200 uL of a 1:10 suspension of stool from either Cedars-Sinai specific pathogen free (SPF) mice or a healthy human donor diluted in pre-reduced phosphate-buffered saline. Mice were euthanized after 2 months of colonization for assessment of intestinal fibrosis and histopathology. Mucosal areas of collagen deposition identified by Picrosirius red-stained gut sections were quantitated for the relative degree of fibrosis using ImageJ software, as previously described (27). Two animal pathologists scored H&E stained sections in a blinded manner using previously described histopathological scoring system used in GF experiments (38).
Fibroblast gap-closure assays
Mouse primary colonic fibroblasts were isolated as previously described (27). Equal numbers of fibroblasts per group (1×105 cells) were seeded in 8 chamber slides and cultured for 24-48h until a monolayer was formed. A scratch was created with a P200 pipette tip. Cell debris was removed by washing cells with PBS and then cell-culture medium was replaced with time-lapse images taken every 4 hours under an Olympus CK2 microscope at 100× magnification. The area of the gap between the two migrating fronts of the cells was quantified using ImageJ software and relative percent area of gap closed at the indicated time points was calculated as (area t0 – area tx)/area t0. For assays involving supplementation with cecal washings, cecal contents from native SPF WT and GF mice were released by flushing with 1 ml of distilled deionized water, as previously described (38). The washings were then homogenized by vortexing, and pelleted by centrifugation. Supernatant were collected, filtered through a 0.22 μm filter, and was added directly to the cells after simulated wound, at a 1:20 dilution (5% volume). In the indicated assays, mouse recombinant Tl1a (R&D Systems, Minneapolis, MN) was added at a concentration 100 ng/ml for 4 hours prior to simulated wound and then maintained during the indicated migration period. For assays involving the addition of bacterial lysates, Oscillospira sp., Mucispirillum schaedleri and Ruminococcus gnavus were cultured anaerobically on chocolate blood agar. Fresh bacterial colonies were resuspended in sterile PBS and lysed. After simulated wound, 25 μg/ml lysate was added to the culture chamber, as describe previously (35). Cell migration was assessed after 16 h of incubation.
For fibroblast adhesion assays, an equal number of cells were seeded into 24-well plates and allowed to settle for either 20 or 80 minutes, after which the wells were washed twice with PBS to remove nonadherent cells. Adherent cells were counted for 5 visual fields/well (representing four quadrants and the center of the well) at 200× magnification, then averaged. The average number of adherent cells per visual field is then displayed for each well.
Histological myofibroblast quantification
Fibroblast and myofibroblasts were quantified by anti-vimentin and anti-α-Smooth Muscle Actin immunofluorescence-stained OCT tissue sections. 4 μm frozen sections were fixed with 10% formalin, blocked in 10% BSA, 0.1% Triton X-100 TBST, and stained overnight at 4 degree C with primary antibodies: rabbit polyclonal anti-αSMA Ab (Abcam, Cambridge, MA) at 1:100 dilution and chicken polyclonal anti-Vimentin Ab (Abcam, Cambridge, MA) at 1:2000 dilution. Secondary antibody at 1:500 dilution was added for 2 hours at room temperature with donkey anti-rabbit IgG-Alexa-fluor-647 and goat anti-chicken IgY- DyLight 488 (Abcam, Cambridge, MA). Images were captured with Leica TCS spectral microscope. Total numbers and percentage of myofibroblasts (that colocalize fluorescence) over total vimentin-positive cells per HPF in ileum or cecum were quantitated by two independent investigators.
Quantitative real-time PCR analysis
Total RNA was isolated from cultured fibroblasts using Qiagen RNeasy Micro Kit according to the manufacturer’s protocol. Two-hundred and fifty nanograms of total RNA was used in each RT reaction, with oligo(dT) as primer, using the Omniscript kit and protocol (Qiagen). Collagen 1a2 and β-actin transcripts were amplified by quantitative real-time RT-PCR with TaqMan probes and primers (ThermoFisher Scientific, Waltham MA, USA). PCR was done on 1/4 the RT reaction in duplicate as follows: 50°C for 2 min, 95°C for 2 min, then 45 cycles at 95°C for 15 s, and 60°C for 1 min. Assays were performed following the predeveloped TaqMan assay reagents protocol for Platinum qPCR mix (Invitrogen Life Technologies) in a Mastercycler Ep realplex2 (Eppendorf). The Mastercycler System Interface was used to analyze samples. Duplicates differing by less than one cycle were averaged and amount of transcript was analyzed. Replicate Ct values were normalized to replicate reference gene (β- actin) Ct values (ΔCt), and relative expression was calculated with respect to the indicated reference sample (ΔΔCt), expressed as percentage of β-actin.
Microbial correlation with fibrosis
Cecal and ileal luminal content was released by flushing with distilled deionized water then the mucosa-associated bacteria were released by DTT treatment according to our published protocol (38). DNA extraction and sequencing of the 16S ribosomal RNA gene was then performed for luminal and mucosal samples as previously described (39). In brief, bacterial DNA was extracted using the MO BIO Powersoil kit with bead beating. The V4 region of the 16S gene was amplified and barcoded using 515f/806r primers then 150×2 bp sequencing was performed on an Illumina HiSeq 2500. Raw data was processed in QIIME 1.9.1 and 97% operational taxonomic units (OTUs) were identified by closed reference OTU picking against the Greengenes database (40). OTUs associated with fibrosis were identified using DESeq2, an algorithm that employs multivariate negative binomial models to identify differentially abundant features (41). Separate analyses were performed of the cecum and ileum of humanized and SPF colonized ex-GF mice. The models included fibrosis score by Sirius Red staining and sample type, sex, and TL1a genotype as covariates. Results were adjusted for multiple hypothesis testing (42). OTUs with adjusted p-values <0.05 were inputted into CoNet along with fibrosis scores to generate co-occurrence networks (43). This analysis involved four metrics (Spearman correlation, Bray Curtis dissimilarity, Jensen Shannon dissimilarity, scaled variance of log ratios) from which a merged p-value was obtained by Fisher’s method and corrected with the Benjamini-Hochberg method. The resulting network was visualized in Cytoscape 3.2.1 (http://cytoscape.org) with an edge-weighted, spring-embedded layout.
Statistics
Data are presented as dot plots and means with group differences tested using standard methods depending on variables measured: Student’s t-test for comparisons between two groups or Mann Whitney test for comparisons between two groups requiring non-parametric testing. When indicated, 1-way Analysis of Variance (ANOVA) with Tukey’s honestly significant difference (HSD) test for multiple comparisons was used. In all settings, a P value of less than 0.05 indicated a statistically significant difference in the parameter being compared. Additional statistical methods were used for microbial correlations with fibrosis, as described.
Study approval
This study was carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animal studies were approved by the CSMC Animal Care and Use Committee, under IACUC protocol 4942.
Supplementary Material
Acknowledgments
This work is supported by the National Institutes of Health (NIH) NIH R01 DK056328-16 (NJ, SRT and DQS), NIH K08 Career Development Award DK093578 (DQS), NIH T32 DK07180-40 (NJ), Specialty Training and Advanced Research (STAR) Program at UCLA (NJ), The Crohn’s and Colitis Foundation and the F. Widjaja Foundation Inflammatory Bowel & Immunobiology Research Institute (NJ, SRT and DQS), 5-P30-DK034987 and 5-P40-OD010995 (RBS).
Footnotes
Author contributions
NJ, JJ, CWYH, SD, JB, KSM, RBS, SRT, and DQS designed experiments; NJ, JJ, KK, VL, YK, KA, AMH, AVA, CWYH, RBS performed experiments and analyzed data; and NJ, JJ, RBS, KSM, SRT, and DQS wrote the manuscript.
Disclosure
The authors have declared that no conflicts of interests exist.
References
- 1.Kitson J, et al. A death-domain-containing receptor that mediates apoptosis. Nature. 1996;384:372–5. doi: 10.1038/384372a0. [DOI] [PubMed] [Google Scholar]
- 2.Chinnaiyan AM, et al. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 1996;274:990–2. doi: 10.1126/science.274.5289.990. [DOI] [PubMed] [Google Scholar]
- 3.Bodmer JL, et al. TRAMP, a novel apoptosis-mediating receptor with sequence homology to tumor necrosis factor receptor 1 and Fas(Apo-1/CD95) Immunity. 1997;6:79–88. doi: 10.1016/s1074-7613(00)80244-7. [DOI] [PubMed] [Google Scholar]
- 4.Screaton GR, et al. LARD: a new lymphoid-specific death domain containing receptor regulated by alternative pre-mRNA splicing. Proc Natl Acad Sci USA. 1997;94:4615–9. doi: 10.1073/pnas.94.9.4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan KB, et al. Characterization of a novel TNF-like ligand and recently described TNF ligand and TNF receptor superfamily genes and their constitutive and inducible expression in hema-topoietic and non-hematopoietic cells. Gene. 1997;204:35–46. doi: 10.1016/s0378-1119(97)00509-x. [DOI] [PubMed] [Google Scholar]
- 6.Shih DQ, et al. Microbial induction of inflammatory bowel disease associated gene TL1A (TNFSF15) in antigen presenting cells. Eur J Immunol. 2009;39:3239–50. doi: 10.1002/eji.200839087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Lamki RS, et al. TL1A both promotes and protects from renal inflammation and injury. J Am Soc Nephrol. 2008;19:953–60. doi: 10.1681/ASN.2007060706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bamias G, et al. Role of TL1A and its receptor DR3 in two models of chronic murine ileitis. Proc Natl Acad Sci USA. 2006;103:8441–6. doi: 10.1073/pnas.0510903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prehn JL, et al. The T cell costimulator TL1A is induced by FcgammaR signaling in human monocytes and dendritic cells. J Immunol. 2007;178:4033–8. doi: 10.4049/jimmunol.178.7.4033. [DOI] [PubMed] [Google Scholar]
- 10.Picornell Y, et al. TNFSF15 is an ethnic-specific IBD gene. Inflamm Bowel Dis. 2007;13:1333–8. doi: 10.1002/ibd.20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michelsen KS, et al. IBD-associated TL1A gene(TNFSF15) haplotypes determine increased expression of TL1A protein. PLoS One. 2009;4:e4719. doi: 10.1371/journal.pone.0004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrett R, et al. Constitutive TL1A expression under colitogenic conditions modulates the severity and location of gut mucosal inflammation and induces fibrostenosis. Am J Pathol. 2012;180:636–49. doi: 10.1016/j.ajpath.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirano A, et al. Association study of 71 European Crohn’s disease susceptibility loci in a Japanese population. Inflam Bowel Dis. 2013;19:526–33. doi: 10.1097/MIB.0b013e31828075e7. [DOI] [PubMed] [Google Scholar]
- 14.Meylan F, et al. The TNF-family cytokine TL1A drives IL-13- dependent small intestinal inflammation. Mucosal Immunol. 2011;4:172–85. doi: 10.1038/mi.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taraban VY, et al. Sustained TL1A expression modulates effector and regulatory T-cell responses and drives intestinal goblet cell hyperplasia. Mucosal Immunol. 2011;4:186–96. doi: 10.1038/mi.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shih DQ, et al. Constitutive TL1A (TNFSF15) expression on lymphoid or myeloid cells leads to mild intestinal inflammation and fibrosis. PLoS One. 2011;6:e16090. doi: 10.1371/journal.pone.0016090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng L, et al. Sustained Tl1a Expression on Both Lymphoid and Myeloid Cells Leads to Mild Spontaneous Intestinal Inflammation and Fibrosis. Eur J Micro & Immunol. 2013;3:11–20. doi: 10.1556/EuJMI.3.2013.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bibiloni R, Mangold M, Madsen KL, Fedorak RN, Tannock GW. The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn’s disease and ulcerative colitis patients. J Med Microbiol. 2006;55:1141–9. doi: 10.1099/jmm.0.46498-0. [DOI] [PubMed] [Google Scholar]
- 19.Gevers D, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–92. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–99. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 22.Kugathasan S, et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet. 2017;389:1710–1718. doi: 10.1016/S0140-6736(17)30317-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitajima S, Morimoto M, Sagara E, Shimizu C, Ikeda Y. Dextran sodium sulfate-induced colitis in germ-free IQI/Jic mice. Exp Anim. 2001;50:387–95. doi: 10.1538/expanim.50.387. [DOI] [PubMed] [Google Scholar]
- 24.Sartor RB, Wu GD. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology. 2017;152:327–339. doi: 10.1053/j.gastro.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rath HC, et al. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J Clin Invest. 1996;98:945–53. doi: 10.1172/JCI118878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. 2003;124:1866–78. doi: 10.1016/s0016-5085(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 27.Shih DQ, et al. Inhibition of a novel fibrogenic factor Tl1a reverses established colonic fibrosis. Mucosal Immunol. 2014;7:1492–503. doi: 10.1038/mi.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rooks MG, et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J. 2014;8:1403–17. doi: 10.1038/ismej.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joossens M, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60:631–7. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- 30.Png CW, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105:2420–8. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- 31.Lupp C, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:119–29. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 32.Sabino J, et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut. 2016;65:1681–9. doi: 10.1136/gutjnl-2015-311004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frank DN, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–5. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaakoush NO, et al. Microbial dysbiosis in pediatric patients with Crohn’s disease. J Clin Microbiol. 2012;50:3258–66. doi: 10.1128/JCM.01396-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devkota S, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–8. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng D, et al. Exogenous Hydrogen Sulfide Attenuates Cardiac Fibrosis Through Reactive Oxygen Species Signal Pathways in Experimental Diabetes Mellitus Models. Cell Physiol Biochem. 2015;36:917–29. doi: 10.1159/000430266. [DOI] [PubMed] [Google Scholar]
- 37.Zeng O, et al. Effect of Novel Gasotransmitter hydrogen sulfide on renal fibrosis and connexins expression in diabetic rats. Bioengineered. 2012;7:314–320. doi: 10.1080/21655979.2016.1197743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaubeck M, et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut. 2016;65:225–37. doi: 10.1136/gutjnl-2015-309333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacobs JP, et al. Microbial, metabolomic, and immunologic dynamics in a relapsing genetic mouse model of colitis induced by T-synthase deficiency. Gut microbes. 2017;8:1–16. doi: 10.1080/19490976.2016.1257469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faust K, Raes J. CoNet app: inference of biological association networks using Cytoscape. F1000Res. 2016;5:1519. doi: 10.12688/f1000research.9050.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eun CS, et al. Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10−/− mice. Infect Immun. 2014;82:2239–46. doi: 10.1128/IAI.01513-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Latella G, Papi C. Crucial steps in the natural history of inflammatory bowel disease. World J Gastroenterol. 2012;18:3790–9. doi: 10.3748/wjg.v18.i29.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drygiannakis I, et al. Proinflammatory cytokines induce crosstalk between colonic epithelial cells and subepithelial myofibroblasts: implication in intestinal fibrosis. J Crohns Colitis. 2013;7:286–300. doi: 10.1016/j.crohns.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 47.Walters WA, et al. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 2014;588:4223–33. doi: 10.1016/j.febslet.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Willing BP, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 49.Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A. 2011;108:4659–65. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, et al. The outer mucus layer hosts a distinct intestinal microbial niche. Nat Commun. 2015;6:8292. doi: 10.1038/ncomms9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.