

Abstract
Objective
Few controlled studies are available to guide treatment decisions in juvenile dermatomyositis (JDM). This study evaluated therapies received, changes of treatment over time, and factors associated with medication choices in JDM.
Methods
We performed a retrospective analysis of the number and type of therapies and duration of treatment for 320 patients with JDM enrolled in a North American registry. Kaplan-Meier and logistic regression analysis were used to assess the association of demographic and clinical features and autoantibodies with medication usage.
Results
High-dose oral prednisone was the primary therapy given to 99% of patients. 1997 was determined to be a threshold year for increasing usage of medications other than prednisone. The median time to half the initial oral prednisone dose was shorter in patients diagnosed after vs. before 1997 (10 vs. 19 months, P<0.01). Patients received intravenous methylprednisolone (IVMP), methotrexate, intravenous immunoglobulin, antimalarial drugs, and combination therapy more frequently when diagnosed after 1997. IVMP was frequently received by patients with severe illness onset, anti-p155/140 (anti-TIF1) and anti-MJ (anti-NXP2) autoantibodies. Treatment with methotrexate was associated with older age at diagnosis and anti-MJ autoantibodies, while antimalarial therapy was associated with anti-p155/140 autoantibodies and mild onset severity.
Conclusion
Oral prednisone has been the mainstay of therapy in JDM, and prednisone was reduced faster in patients diagnosed after 1997 when there was also an increase in other medications. Specific medications received by patients with JDM correlated with year and age at diagnosis, myositis autoantibodies, onset severity, and illness features.
Keywords: juvenile dermatomyositis, treatment, medications, myositis autoantibodies, prednisone, methotrexate
Introduction
Juvenile dermatomyositis (JDM) is a systemic autoimmune disease with characteristic skin rashes, chronic muscle inflammation and associated vasculopathy (1). Myositis autoantibodies (MSAs), including anti-p155/140 (anti-transcription intermediary factor 1; TIF1), anti-MJ (anti-nuclear matrix protein 2; NXP2), anti-melanoma differentiation–associated gene 5 (MDA5) autoantibodies, among others, have been identified in the majority of children with JDM, each defining relatively homogeneous groups of patients with distinct clinical, demographic and laboratory manifestations, and outcomes (2–4).
Immunosuppressive therapy is the mainstay of treatment for JDM. Since high doses of daily oral prednisone of longer duration reportedly have led to better functional outcomes and less frequent calcinosis (5), patients with JDM have been treated primarily with high doses of oral prednisone. Based on open-label and retrospective studies, methotrexate (MTX), intravenous pulse methylprednisolone (IVMP), intravenous immunoglobulin (IVIG), cyclosporine and cyclophosphamide were later introduced to decrease the exposure to daily oral prednisone, as well as the duration of active disease (6–10). In recent years, additional therapeutic options for treatment-refractory patients that were often supported by open-label trials and retrospective case series have included mycophenolate mofetil (MMF), tacrolimus, anti-TNF therapy, and rituximab, among others (3, 11–13).
However, because of the rarity of this disease, until very recently, the treatment of JDM lacked uniformity. In a survey of North American pediatric rheumatologists, JDM patients reportedly received varying prednisone doses and varying combinations of other medications (14). In a large observational cohort from Europe and South America, there were differences in oral prednisone dosage and the usage of other medications, including IVMP and MTX, by geographic region (15). These reports indicate that a variety of medications and combinations of medications have been used to treat JDM to date. However, the factors associated with the various treatment choices have not been clear.
The purpose of the present study was to determine which medications were received by patients with JDM in a North American registry, and evaluate differences in therapies by year of diagnosis, MSAs, illness severity at disease onset, and other clinical and demographic features.
Patients and Methods
Patients
Three hundred twenty patients diagnosed prior to 18 years of age fulfilled the Bohan and Peter criteria for probable or definite JDM (16, 17), and were enrolled in National Institutes of Health or Food and Drug Administration investigational review board-approved natural history protocols from March 1989 through November 2015. These patients also had at least 6 months of treatment data available. Patients were recruited for enrollment through myositis patient support groups, advertisement in medical journals, and by writing pediatricians and pediatric and adult rheumatologists, neurologists, and dermatologists. Patients provided a blood sample for autoantibody and genetic testing after written consent/assent was obtained, and the treating physician completed a questionnaire that included clinical, demographic, and laboratory data, and outcomes (18). The enrolling physician also retrospectively completed a standardized treatment questionnaire about the courses of medications received, their start and end dates, and their associated clinical and laboratory responses (19, 20). Medical records for every clinic or hospital visit in a patient’s illness course were reviewed by study staff for 236 (74%) patients to confirm questionnaire data; the remaining records were not available for review. Patients were diagnosed between February 1986 and September 2014 (median date of diagnosis was September 1994 [Interquartile range (IQR): May 1992-February 2001]). Patients were excluded if they were diagnosed before 1986 (n=14), had a follow-up duration of less than 6 months (n=31) or treatment data were not available or insufficient (where the medications, dose and/or start or end dates of a treatment course were unknown, n=35).
The definitions of severity at disease onset and organ symptom scores, the clinical and demographic variables used, and the methods for testing MSAs were described in previous reports (18, 21–24). Severity of onset was graded on a Likert scale (18), and the number of symptoms at diagnosis were used to calculate organ system scores which was defined as the number of symptoms present related to that system at diagnosis, divided by the number of symptoms assessed; values ranged from 0 to 1 (22). MSAs were evaluated in the sera of 311 patients (97%) as part of the study, and results were not available to the treating physician.
Treatments received and responses
A treatment course was defined as a single episode from beginning of administration of a given medication to the termination of treatment with that medication, or combination of medications, in each patient (19, 20). For prednisone, this was until the dose was reduced to 25% of the original dose, or reached a stable dose that was the objective of the taper. A new course of medication consisted of the addition of a new medication or termination of a medication, an increase or decrease in the dose by ≥ 25% of original dose, or if the route of administration changed (i.e., oral versus parenteral MTX).
Medications were categorized as major and minor therapies. Major medication categories included oral prednisone, IVMP, MTX, IVIG, other disease-modifying antirheumatic drugs (DMARDs), cytotoxic drugs and biologic therapies, per the recent classification of the Childhood Arthritis and Rheumatology Research Alliance (CARRA) (25). Other DMARDs included azathioprine, cyclosporine, MMF, tacrolimus and leflunomide. Cytotoxic drugs included IV or oral cyclophosphamide and chlorambucil. Biologic therapies included etanercept, infliximab, rituximab, and abatacept. Antimalarial drugs, which were not considered major medications, included hydroxychloroquine, chloroquine and quinacrine. Other minor medications, such as topical steroids and tacrolimus, and oral non-steroidal anti-inflammatory drugs, were excluded from analysis, because the number of patients who received these medicines was limited (11–17%). Combination therapy was defined as combination usage of any major medication in the same treatment trial. The maximum combination of major therapies was defined as the maximum number of major medications used simultaneously in each patient. Time to discontinuation of a medication was defined as the number of months from the initiation of a drug therapy until final discontinuation.
Statistical Analysis
Statistical analyses were performed using JMP for Windows, version 11.0.0 (SAS Institute Inc, Cary, NC, USA). Logistic regression analysis and Receiver Operating Characteristic (ROC) curve analysis were used for the selection of threshold year after diagnosis to determine if there was a change over time in the introduction of major medications. The sensitivity and specificity of major drug usage was calculated for each year after diagnosis, and the maximum point of Youden’s index (sensitivity-(1-specificity)) was used to choose an appropriate threshold year for usage of other major medications.
Types and numbers of medications received as well as their durations were evaluated by Chi-square analysis or Fisher’s exact test, and by Wilcoxon or Mann-Whitney U test, respectively. P values were adjusted for multiple testing using Bonferroni correction and were determined to be significant when the adjusted P values were ≤ 0.05. Kaplan-Meier analysis using the Log-Rank test was used to evaluate time to event distributions between groups; subgroups were excluded when limited in power by fewer than 10 patients.
The factors associated with usage of IVMP, MTX, IVIG, antimalarial drugs, and two or more major medications in combination were evaluated by univariate logistic regression, and significant variables were confirmed by multivariable logistic regression analysis. Variables assessed included gender, year of diagnosis, race, MSAs, onset severity, age at first treatment, as well muscle and skin symptom score at diagnosis. The impact of these variables on time to discontinuation of each medication was also analyzed by Cox proportional hazard modeling. Odds ratio (OR) and hazard risk ratio (RR) were presented with 95% confidence intervals (CIs).
Results
The demographic characteristics and treatment summary for the total population of JDM patients and patients divided by year of diagnosis are presented in Table 1. We first examined whether there was a threshold point in time in which patients began to receive major medications other than daily oral corticosteroids. Based on the maximum point of Youden’s index, 1997 was determined to be the threshold year for major drug usage in combination with daily oral prednisone therapy (Figure 1).
Table 1.
Demographics, myositis autoantibodies and therapy for 320 patients with juvenile dermatomyositis in this study
| Year of Diagnosis | |||
|---|---|---|---|
| Total | Before 1997 | After 1997 | |
| N (%) or Median [IQR] (Range) | N (%) or Median [IQR] (Range) | N (%) or Median [IQR] (Range) | |
| Total number of patients | 320 (100) | 220 (68.8) | 100 (31.3) |
| Female | 223 (69.7) | 155 (70.5) | 68 (68.0) |
| Age at Diagnosis (in years) | 7.1 [4.6–10.4] (1.3–18.8) | 6.8 [4.6–10.0] (1.3–18.8) | 7.2 [4.6–11.0] (1.9–16.8) |
| Delay of Diagnosis (in months) | 3.9 [1.7–7.4] (0–81.0) | 3.1 [1.6–7.1] (0–77.0) | 4.2 [2.0–8.0] (0.1–81.0) |
| Race | |||
| Caucasian | 223 (69.7) | 154 (70.0) | 69 (69.0) |
| African American | 33 (10.3) | 23 (10.5) | 10 (10.0) |
| Hispanic | 18 (5.6) | 14 (6.4) | 4 (4.0) |
| Other | 46 (14.4) | 29 (13.2) | 17 (17.0) |
| Onset Severity | |||
| Mild | 36 (11.3) | 21 (9.6) | 15 (15.0) |
| Moderate | 193 (60.3) | 142 (64.6) | 51 (51.0) |
| Severe/Very Severe | 91 (28.4) | 57 (25.9) | 34 (34.0) |
| Myositis Specific Autoantibodies (N=302) | |||
| Anti-p155/140 (TIF1) | 117 (38.7) | 71 (37.6) | 46 (54.1) |
| Anti-MJ (NXP2) | 87 (28.8) | 64 (33.9) | 23 (27.1) |
| Anti-MDA5 | 26 (8.6) | 18 (9.5) | 8 (9.4) |
| MSA negative | 44 (14.6) | 36 (19.1) | 8 (9.4) |
| Follow-up duration (months) | 43.6 [22.1–74.4] (6.4–224.0) | 49.9 [26.4–87.3] (6.4–224.0) | 29.1 [17.0–56.9] (7.1–177.7)* |
| Treatment Courses/year | 1.4 [0.7–2.4] (0.08–7.6) | 1.0 [0.6–1.8] (0.1–7.1) | 2.3 [1.6–3.4] (0.1–6.8)** |
| Steroid Courses/year | 1.1 [0.6–2.2] (0–6.9) | 0.9 [0.5–1.6] (0–6.9) | 2.1 [1.1–3.3] (0.1–6.9)** |
| Duration of treatment (months) | 31.0 [19.3–58.8] (3.5–216.9) | 36.0 [20.4–63.1] (3.5–216.9) | 26.9 [14.5–50.3] (5.0–168.1) |
| Duration of steroid treatment (months) | 26.5 [15.9–45.5] (0–181.9) | 28.3 [17.0–49.0] (0–181.9) | 23.9 [13.1–38.1] (3.9–156.7) |
| Percent follow-up time on treatment | 98.9 [76.3–100] (3.5–100) | 88.1 [70.5–100] (3.5–100) | 100 [99.5–100](6.5–100)** |
Abbreviations: IQR, Interquartile range; MSA, Myositis autoantibodies; TIF1, transcription intermediary factor 1; NXP2, antinuclear matrix protein 2; Anti–MDA5, Anti–melanoma differentiation–associated gene 5.
The number of patients with other myositis specific autoantibodies was insufficient for statistical analysis, and included 9 patients with anti-Mi-2 autoantibodies, 8 with anti-aminoacyl-tRNA synthetase autoantibodies including anti-Jo-1, and 2 with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase autoantibodies.
P <0.01,
P<0.001 for Before vs After 1997
Figure 1.
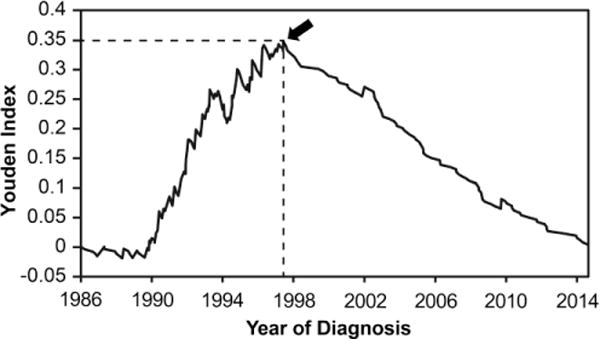
Plot of year of diagnosis versus Youden index. Youden index is defined as (Sensitivity-(1-Specificity)) for major drug usage other than prednisone, and is calculated by Receiver Operating Characteristic (ROC) curve analysis. The sensitivity and specificity of major drug usage beyond oral prednisone was calculated for each year after diagnosis. The arrow indicates the maximum point of the Youden index (0.348). This corresponds to the threshold date for major medication usage beyond oral prednisone, which was July 11, 1997.
There were 320 JDM patients, with 100 patients (31.3%) diagnosed after 1997 who met the study inclusion criteria (Table 1). Patients diagnosed before or after 1997 were similarly distributed by gender, age at diagnosis, diagnosis delay, race, onset severity and MSAs. Patients diagnosed after 1997 had a greater number of drug trials per year and a longer percentage of follow-up time on treatment compared to patients diagnosed before 1997, but they also had a shorter follow-up duration. The patients who were severe at illness onset had a greater number of trials per year than patients who were moderate at onset (median number of drug trials per year of 1.8 [IQR 1.0–3.0] vs. 1.1 [IQR 0.7–2.1], P=0.0009), and this did not differ by year of diagnosis. Patients who had anti-p155/140 (TIF1) autoantibodies had a greater number of treatment trials per year than MSA-negative patients (median number of drug trials per year of 1.6 [IQR 0.8–2.5] vs. 0.8 [IQR 0.5–1.8], P=0.013). There were no significant differences in follow up duration, treatment duration, or time on or off medication by MSAs (anti-p155/140 (TIF1), MJ (NXP2), MDA5, or MSA-negative).
Oral prednisone was the primary therapy prescribed for 99% of JDM patients, which was significantly greater in frequency than the other medications (Table 2). MTX was the second most commonly prescribed medication, and IVMP was the third most frequent medication. IVMP, MTX, IVIG, other DMARDs, antimalarial drugs and cytotoxic/biologic therapies were given more frequently to patients diagnosed after 1997 compared to those diagnosed before 1997 (Table 2). Prednisone or IVMP were most frequently started as the initial treatment, and, in patients diagnosed after 1997, MTX was introduced earlier after the start of initial treatment than in those diagnosed before 1997. Antimalarial drugs were often given within a few months of the start of treatment. Other medications were given later, but there were no significant differences in their timing before and after 1997.
Table 2.
Medications received by 320 patients with juvenile dermatomyositis.
| Number and Percent of Patients | Time to Start Medication* | |||||
|---|---|---|---|---|---|---|
| Ever Used | Before 1997 | After 1997 | All patients | Before 1997 | After 1997 | |
| (N=320) | (N=220) | (N=100) | (N=320) | (N=220) | (N=100) | |
| N (%) | N (%) | N (%) | Months [IQR] | Months [IQR] | Months [IQR] | |
| Oral Prednisone | 318 (99.4) | 218 (99.1) | 100 (100.0) | 0 [0–3] | 0 [0–3] | 0 [0–3] |
| IV Methylprednisolone1 | 182 (56.9)† | 112 (50.9) | 70 (70.0)‡ | 0 [0–251]† | 0 [0–312.8] | 3 [0–183.8] |
| Methotrexate2 | 225 (70.3)†§ | 130 (59.1) | 95 (95.0)** | 2.0 [0.1–13.0]†§ | 6.0 [0.8–22.0] | 0.2 [0–2.0]** |
| IV Immunoglobulin3 | 119 (37.2)†§‖ | 55 (25.0) | 64 (64.0)** | 7.1 [0.6–20.0]†§ | 10.0 [0.5–25.0] | 5.1 [0.7–19.0] |
| Other DMARDs4 | 66 (20.6)†§‖¶ | 25 (11.4) | 41 (41.0)** | 14.5 [3.4–31.2]†‖§ | 23.0 [5.5–51.4] | 13.3 [3.2–24.9] |
| Cytotoxic/Biologics therapies5 | 40 (12.5)†§‖¶ | 12 (5.5) | 28 (28.0)** | 9.0 [3.1–31.5]†‖§ | 5.5 [2.7–13.1] | 9.0 [3.1–42.6] |
| Antimalarial drugs6 | 153 (47.8)†‖⨍# | 83 (37.7) | 70 (70.0)** | 3.3 [0.1–20.7]†‖⨍ | 7.0 [0.1–31.7] | 2.3 [0–12.2] |
| Maximum combination of medications received, Median [IQR] | 2.0 [2.0–3.0] | 2.0 [1.0–3.0] | 4.0 [2.0–4.8]** | |||
| 1 major medication | 76 (23.8) | 71 (32.3) | 5 (5.0)** | 0 [0–0] | 0 [0–0] | 0 [0–0] |
| 2 major medications7 | 107 (33.4) | 83 (37.7) | 24 (24.0) | 2.9 [0.1–13.9]†† | 3.0 [0.1–22.0] | 1.0 [0–2.9] |
| 3 major medications8 | 71 (22.2) | 51 (23.2) | 20 (20.0) | 8.0 [1.0–23.0]†† | 11.5 [2.0–37.2] | 5.7 [1.1–21.2] |
| 4 or more major medications | 64 (20.0) | 13 (5.9) | 51 (51.0)** | 10.1 [2.0–26.6]†† | 12.0 [6.0–41.0] | 6.4 [1.9–25.4] |
Abbreviations: IQR, Interquartile range; IV, intravenous; DMARDs, disease-modifying anti rheumatic drugs; Ever used, total patients who received each medication at least once; Before 1997, patients diagnosed before 1997; After 1997, patients diagnosed after 1997
Time to start medication from start of initial treatment for Oral prednisone, IV methylprednisolone and 1 major agent are shown in days instead of months.
P <0.05,
P <0.01, vs Before 1997 patients,
P <0.01 vs Oral Prednisone,
P <0.01 vs IV Methylprednisolone,
P <0.01 vs Methotrexate,
P <0.01 vs IV Immunoglobulin,
P <0.01 vs Other DMARDs,
P <0.01 vs Cytotoxic/Biologics Agents,
P <0.01 vs 1 major agent
The median dose of IV methylprednisolone (IVMP) initial pulse therapy was 30.0 [IQ range 22.1–30.0] mg/kg/dose, and the median dose for other intervals was 22.0 [14.7–30.0] mg/kg/dose. IVMP was given as an initial pulse, primarily for 3 days, in 31.3% who received IVMP, and as weekly infusions in 24.8%, every 2 week infusions in 11.4%, and monthly infusions in 19.4%.
Among the patients who received methotrexate (MTX), 99.2% received it weekly. The median initial dose of MTX was 0.50 [ IQ range 0.40–0.76] mg/m2/week. The administration route of MTX was: PO in 47.5%, SQ in 35.3%, IV in 12.6 and IM in 4.6% of patients receiving MTX; 40.4% of patients changed the route of MTX administration later.
The median dose of IV immunoglobulin (IVIG) was 2 [IQR 1–2] g/kg/dose. The Interval of IVIG was monthly in 71.7% who received IVIG, every 2 weeks in 10.6% and every 3 weeks in 10.3%.
Other DMARDs included cyclosporine in 59.1% of patients who received other DMARDs, mycophenolate mofetil 30.3%, azathioprine 28.8%, tacrolimus 12.1%, and leflunomide 3.0%. The percentages are >100 because 15 patients received more than 1 other DMARD.
Cytoxic/Biologic therapies included intravenous cyclophosphamide in 50.0% of patients who received cytotoxic/biologic agents, and of these, 75.8% received it monthly with median dose of 750 mg/m2, rituximab 32.5%, etanercept 27.5%, infliximab 22.5%, oral cyclophosphamide 15.0% with median dose of 1mg/kg/day; 14 patients received more than 1 cytotoxic/biologic therapy. For the 13 patients who received rituximab, the median number of IV rituximab infusions was 4.0 [IQ range 2.5–4.0]; doses ranged from 500 mg/m2 to 1000 mg per infusion, and 6 patients received 4 weekly IV infusions and 6 received 2 weekly IV infusions(13).
Antimalarial drugs included hydroxychloroquine in 100% of patients who received antimalarial drugs, chloroquine 1.3%, and quinacrine 1.3%; 4 patients received more than 1 antimalarial drug.
The most frequent 2 medication combinations were prednisone and methotrexate in 84.6%, and prednisone and IV immunoglobulin in 12.3% among 228 patients who received 2 medications in combination.
The most frequent 3 medication combinations were: prednisone, methotrexate and IV methylprednisolone in 35.4% of patients, prednisone, methotrexate and IV immunoglobulin in 35.4%, prednisone, IV methylprednisolone and IV immunoglobulin in 10.0%, and prednisone, methotrexate and cyclosporine 9.2% among 130 patients who received 3 medications in combination.
IVMP was used more frequently in patients who had severe disease at onset compared to those with moderate illness severity at onset (75.8% vs. 48.2%, P=0.0001), including for patients diagnosed before and after 1997. IVMP was also used earlier after the start of initial treatment in patients who were severe compared to those who were mild at diagnosis (median 0 days [IQR 0–79 days] vs. 161 days [IQR 11–728 days after initial treatment; P=0.022). There were no significant differences in the frequency of other medications by disease severity at onset. Among MSA groups, patients with anti-MJ (NXP2) autoantibodies used IVIG earlier than the patients with anti-p155/140 (TIF1) autoantibodies (median 1.0 months [IQR 0.2–13.8 months] vs. 10.3 months [IQR 3.0–27.0 months] after initial treatment; P=0.037), but the frequency of usage of IVIG was similar in both autoantibody groups (41.4 and 40.2%, respectively). Antimalarial drugs were more frequently received by patients with anti-p155/140 (TIF1) autoantibodies than anti-MJ (NXP2) autoantibodies (65.8% vs. 39.1%, P=0.002) or patients who were MSA/MAA-negative (65.8% vs. 27.3%, P=0.0002), including before and after the diagnosis year of 1997. There was no difference in the timing of administration by illness severity or MSAs for other medications, including IVMP, MTX, other DMARDs, IVIG and antimalarial drugs.
Combinations of medications were prescribed more frequently in patients diagnosed after 1997 (Table 2). The median number of major medications together in combination was 2.0, and patients diagnosed before 1997 were given fewer combinations of major medications than those diagnosed after 1997 (median 2.0 vs. 4.0, P<0.001). As the number of medications in combination increased, the time from initial treatment to the start of the combination therapy progressively increased (Table 2). Patients with severe illness at onset received a greater number of major medications in combination than patients who had moderate disease severity at illness onset (median 3.0 [IQR 1.0–4.0] vs. 2.0 [IQR 1.0–3.0], P=0.009). Patients with severe illness at onset more frequently received a combination of ≥ 4 major medications compared to patients with moderate illness severity (29.7% vs. 14.5%, p=0.040). Patients with anti-p155/140 (TIF1) autoantibodies were found to have received a greater number of major medications in combination than patients who were MSA-negative (median 2.0 [IQR 2.0–3.0] vs. 2.0 [IQR 1.0–2.8], P=0.040). There were no other significant differences in the maximum combination of major medications among MSA groups.
The median time to half the initial oral prednisone dose was shorter in patients diagnosed after 1997 compared to patients diagnosed before 1997 (10 vs. 19 months, P<0.0001) (Figure 2A). The median time to discontinuation of oral prednisone was 41 months [IQR 24–89], and this did not differ by diagnosis year (Figure 2B). The median daily maximum dose of oral prednisone in the initial 6 months of treatment did not differ by diagnosis year (median daily dose of 2.0 mg/kg/day for patients diagnosed before 1997 vs. 1.6 mg/kg/day for patients diagnosed after 1997 [Figures 2C]). However, the maximum daily oral prednisone dose was lower in months 6–24 in patients diagnosed after 1997 compared to those diagnosed before 1997 (Figure 2C).
Figure 2.
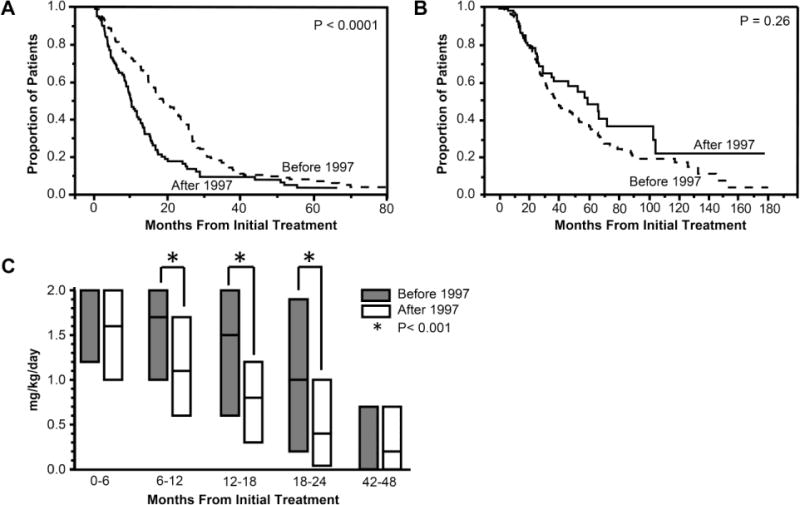
A, Kaplan-Meier analysis plot showing time to half of initial dose of oral prednisone in juvenile dermatomyositis patients diagnosed before 1997 versus after 1997. The Y axis shows the proportion of patients who were still receiving more than 50% of the initial dose of prednisone at any point in time. X axis shows months from initial treatment.
B, Kaplan-Meier analysis plot showing time to final discontinuation of oral prednisone in juvenile dermatomyositis patients diagnosed before 1997 versus after 1997. The Y axis shows the proportion of patients who were still receiving prednisone at any point in time. X axis shows months from initial treatment.
C, Median daily maximum dose of oral prednisone in each 6 month treatment period. The Y axis shows the median daily maximum dose of oral prednisone. X axis shows the months from initial treatment. The bars show the median prednisone dose with 25% and 75%. For 0–6 months, the median dose of prednisone before 1997 is the same as the 75%, and for 42–48 months, the median dose of prednisone before 1997 is the same as the 25%. The median daily maximum dose for patients diagnosed before 1997 and after 1997 was 1.7 [IQR 1.0–2.0] vs. 1.1 [IQR 0.6–1.7] mg/kg per day for 6–12 months, 1.5 [0.6–2.0] vs. 0.8 [0.3–1.2] mg/kg per day for 12–18 months, and 1.0 [0.2–1.9] vs. 0.4 [0.04–1.0] mg/kg per day for 18–24 months.
The median time to half of the initial dose of oral prednisone did not differ between patients with moderate and severe onset severity (median 16 and 14 months, respectively), and did not differ between patients with anti-p155/140 (TIF1) autoantibodies and MSA negative patients (median time 14 and 18 months, respectively). The maximum dose of prednisone over time did not differ between onset severity or MSA groups (data not shown). The time to discontinuation of oral prednisone was not different between patients with moderate vs. severe illness onset (41 vs. 38 months). The time to discontinuation of oral prednisone was shorter in patients with anti-MJ (NXP2) autoantibodies (median 36 months) compared to anti-p155/140 (TIF1) autoantibodies (median 58 months, p=0.028), but neither group differed from MSA negative patients (median 31 months).
Time to discontinuation of other major therapies was shorter in patients diagnosed before 1997 compared to those diagnosed after 1997 (Figure 3). The median time to discontinuation of IVMP by Kaplan-Meier time-to-event analysis was 5 months from the initiation of IVMP treatment in patients diagnosed before 1997 vs. 25 months in patients diagnosed after 1997, P<0.001 (Figure 3A). The median time to discontinuation of MTX was 46 months from MTX treatment initiation in patients diagnosed before 1997 vs. 81 months in patients diagnosed after 1997, P=0.006 (Figure 3B), and the median time to discontinuation of IVIG was 13 months after IVIG treatment initiation in patients diagnosed before 1997 vs. 53 months in patients diagnosed after 1997, P=0.001 (Figure 3C). There was no significant difference in time to discontinuation of antimalarial drugs before or after 1997 (median of 37 months before 1997 vs. 39 months after 1997, P=0.91).
Figure 3.
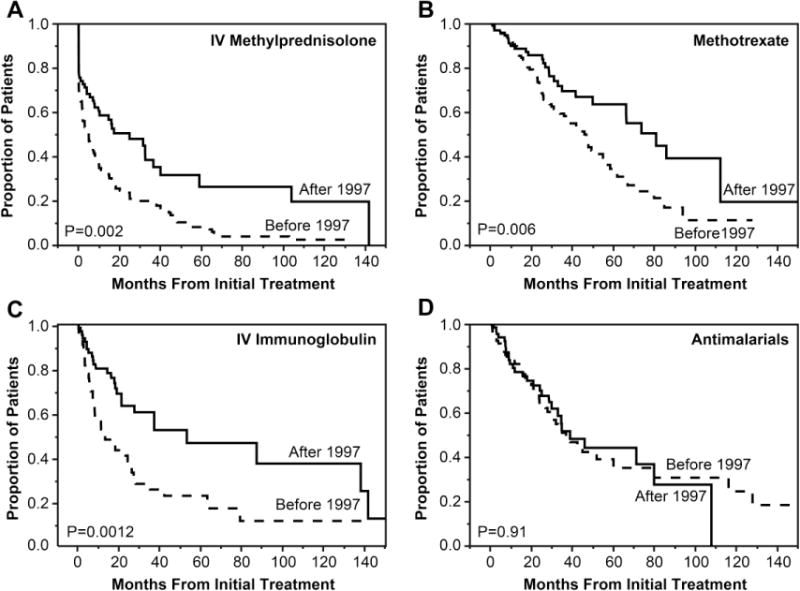
Kaplan-Meier analysis plots showing time to discontinuation of each medication. The Y axis shows the proportion of patients who were still receiving each medication at any point in time. The X axis shows the months from initial treatment.
A, Intravenous methylprednisolone (IVMP). The median time to discontinuation of IVMP was 5 months from the initiation of IVMP treatment in patients diagnosed before 1997 vs. 25 months in patients diagnosed after 1997.
B, Methotrexate. The median time to discontinuation of methotrexate was 46 months from methotrexate treatment initiation in patients diagnosed before 1997 vs. 81 months in patients diagnosed after 1997.
C, Intravenous immunoglobulin (IVIG). The median time to discontinuation of IVIG was 13 months after IVIG treatment initiation in patients diagnosed before 1997 vs. 53 months in patients diagnosed after 1997.
D, Antimalarial drugs. The median time to discontinuation of antimalarial drugs was 37 months after initiation of antimalarial drugs in patients diagnosed before 1997 vs. 39 months in patients diagnosed after 1997.
The patients who had moderate severity at disease onset were given IVMP and IVIG longer than severe/very severe patients (IVMP median 10 months vs. 4 months, P=0.021; IVIG median 35 months vs. 17 months, P=0.030) (Supplementary Figure). There were no other differences in time to discontinuation of IVMP, MTX, IVIG or antimalarial drugs for moderate vs. severe/very severe illness at onset or for patients with anti-p155/140 (TIF1) vs. MJ (NXP2) autoantibodies. In multivariable analysis, only diagnosis date after 1997 was associated with a longer time to discontinuation of IVMP (RR 0.48, 95% CI 0.31–0.72, p=0.0003) and severe illness onset was associated with shorter time to discontinuation of IVMP (RR 1.55, 95%CI 1.0–2.38, p=0.48). For MTX and IVIG, only diagnosis date after 1997 was associated with a longer time to medication discontinuation (RR 0.29–0.51, p<0.005). MSA group, age at first treatment, race, gender, and initial muscle and cutaneous symptom scores were not associated with time to discontinuation of these medications, and there were no associations with time to discontinuation of antimalarial drugs.
We examined factors associated with use of IVMP, MTX, IVIG, antimalarial drugs and combination therapy by logistic regression analysis, with significant factors from univariable analysis included in multiple logistic regression (Table 3). A diagnosis date after 1997 was significantly associated with usage of each of these medications. IVMP was also associated with severe illness onset and with anti-p155/140 (TIF1) and anti-MJ (NXP2) autoantibodies. MTX was frequently received by patients with anti-MJ (NXP2) autoantibodies and older age at first treatment. IVIG usage had no other associations beyond year of diagnosis. Antimalarial therapy was associated with mild illness onset and with anti-p155/140 (TIF1) autoantibodies. The combination of ≥ 2 major medications was associated with older age at first treatment and with a higher skin symptoms score at illness onset. Race, gender and the muscle symptom score were not associated with specific medications received.
Table 3.
Factors associated with medications received by patients with juvenile dermatomyositis by logistic regression analysis.
| IV Methylprednisolone (N=182) |
Methotrexate (N=225) | IV Immunoglobulin (N=119) |
Antimalarial drugs (N=153) | 2 or more major medication combination (N=242) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Univariabl e analysis OR (95% CI) |
Multivariabl e analysis OR (95% CI) |
Univariabl e analysis OR (95% CI) |
Multivaria ble analysis OR (95% CI) |
Univariable analysis OR (95% CI) |
Multivaria ble analysis OR (95% CI) |
Univariable analysis OR (95% CI) |
Multivariabl e analysis OR (95% CI) |
Univariabl e analysis OR (95% CI) |
Multivaria ble analysis OR (95% CI) |
|
| Year of Diagnosis | ||||||||||
| Before 1997 | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) |
| After 1997 | 2.25 (1.37–3.76)† | 2.38 (1.32–4.38)† | 13.15 (5.65–38.41)§ | 14.73 (5.62–50.93)§ | 5.33 (3.23–8.96)§ | 5.46 (3.05–10.00)§ | 3.85 (2.34–6.47)§ | 3.22 (1.79–5.94)‡ | 9.44 (4.04–27.62)§ | 10.75 (4.00–37.87)§ |
| Age at first treatment | 1.04 (0.98–1.10) | 1.05 (0.99–1.13) | 1.09 (1.02–1.16)† | 1.09 (1.01–1.18)* | 1.04 (0.99–1.10) | 1.03 (0.96–1.10) | 1.04 (0.98–1.10) | 1.05 (0.98–1.13) | 1.11 (1.04–1.20)† | 1.13 (1.04–1.23)† |
| Gender | ||||||||||
| Male | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | |||||
| Female | 1.21 (0.75–1.95) | 1.01 (0.60–1.70) | 0.83 (0.51–1.36) | 1.15 (0.71–1.86) | 1.03 (0.58–1.78) | |||||
| Race | ||||||||||
| Caucasian | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | |||||
| Other Races | 1.12 (0.69–1.82) | 1.22 (0.72–2.11) | 0.93 (0.57–1.53) | 1.04 (0.64–1.67) | 1.24 (0.71–2.23) | |||||
| Onset Severity | ||||||||||
| Moderate | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) |
| Mild | 1.34 (0.66–2.78) | 1.20 (0.53–2.75) | 1.08 (0.51–2.41) | 0.75 (0.29–1.97) | 0.89 (0.40–1.88) | 0.82 (0.32–2.02) | 2.95 (1.40–6.56)† | 2.93 (1.19–7.73)* | 0.88 (0.41–1.98) | 0.62 (0.24–1.65) |
| Severe/Very Severe | 3.37 (1.96–5.99)§ | 3.00 (1.59–5.90)‡ | 1.48 (0.85–2.66) | 0.90 (0.45–1.83) | 1.89 (1.13–3.14)* | 1.72 (0.91–3.25) | 1.21 (0.74–2.00) | 1.15 (0.62–2.15) | 2.33 (1.23–4.70)† | 1.99 (0.92–4.57) |
| MSA | ||||||||||
| All MSA negative | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) | 1.00 (reference) |
| Anti-p155/140 (TIF1) | 2.54 (1.26–5.26)† | 2.40 (1.10–5.35)* | 2.77 (1.34–5.76)† | 1.82 (0.80–4.16) | 1.79 (0.85–3.94) | 1.25 (0.53–3.04) | 5.13 (2.44–11.40)§ | 4.38 (1.93–10.51)‡ | 2.44 (1.14–5.20)* | 1.55 (0.65–3.64) |
| Anti-MJ (NXP2) | 2.36 (1.13–5.03)* | 2.51 (1.14–5.69)* | 2.40 (1.13–5.14)* | 2.59 (1.13–6.03)* | 1.88 (0.87–4.26) | 1.64 (0.69–4.03) | 1.71 (0.79–3.88) | 1.96 (0.85–4.77) | 1.86 (0.85–4.05) | 1.97 (0.83–4.70) |
| Anti-MDA5 | 2.54 (0.95–7.07) | 2.44 (0.85–7.32) | 1.46 (0.55–4.00) | 0.92 (0.29–2.89) | 1.67 (0.59–4.71) | 1.29 (0.40–4.15) | 2.67 (0.97–7.52) | 2.57 (0.86–7.95) | 1.71 (0.61–5.16) | 1.04 (0.32–3.53) |
| Muscle symptom score | 1.00 (0.99–1.02) | 0.99 (0.98–1.01) | 1.01 (1.00–1.02) | 0.99 (0.98–1.01) | 1.02 (1.01–1.03)‡ | 1.01 (0.99–1.03) | 1.00 (0.99–1.01) | 1.00 (0.98–1.01) | 1.01 (0.99–1.03) | 0.99 (0.97–1.01) |
| Skin symptom score | 1.02 (1.00–1.04)* | 1.01 (0.99–1.03) | 1.03 (1.01–1.05)† | 1.03 (0.99–1.05) | 1.03 (1.01–1.05)† | 1.01 (0.99–1.04) | 1.03 (1.01–1.05)‡ | 1.01 (0.99–1.04) | 1.04 (1.01–1.06)† | 1.04 (1.01–1.07)† |
Abbreviations: OR, Odds ratio; 95% CI, 95% Confidence interval; MSA, myositis autoantibodies; TIF1, transcription intermediary factor 1; NXP2, anti-nuclear matrix protein 2; Anti–MDA5, Anti–melanoma differentiation–associated gene 5. Age at first treatment and Symptoms scores were calculated by unit odds ratios (per unit change), and the range of variable were Age at first treatment; 1.3–18.8, Muscle symptoms score; 0–100, and Skin symptoms score; 0–61.
P <0.05,
P <0.01,
P <0.001,
P <0.0001.
Discussion
This study provides a description of medications received by a large number of JDM patients as part of a multi-center North American registry with a long study period. The medications that JDM patients have received over time has not been previously well documented (26). In this study, we evaluated the trends in medication usage and how treatment choices have changed over time, as well as factors associated with usage of major medications in the treatment of JDM. While there was a wide variation in medications received, certain common elements were observed.
Our results showed that almost all patients received oral prednisone therapy, similar to other large North American and European/South American observational cohort studies of JDM (25, 26). Our data also showed the median maximum daily prednisone dose in the initial 6 months was 1.6 mg/kg/day in patients diagnosed after 1997, similar to the European/South American multicenter cohort diagnosed after 2001, in which their median daily dose was 1.3 mg/kg/day (15). After 1997, the role of oral prednisone changed from monotherapy to part of combination therapy, in which oral prednisone was administered with other medications, such as MTX, IVIG, other DMARDs, cytotoxic agents and biologic agents. MTX was received by 95% of JDM patients diagnosed after 1997, and was the most common medication administered with prednisone. Our data are similar to the European/South American multicenter cohort in showing this increase in other non-corticosteroid medications over the past two decades (26). However, similar to the CARRA North American multicenter registry, which enrolled patients earlier this past decade, in our cohort, a substantially larger percentage of patients received IVMP, MTX, other DMARDs, and antimalarial drugs than in Europe or South America (25, 26). One difference between our cohort and the CARRA registry is that the CARRA registry patients received IVIG and cytotoxic drugs less frequently, but more frequently received other biologic therapies (25).
We demonstrated that using multiple medications in combination with oral prednisone as part of initial therapy was associated with a more rapid reduction of the oral prednisone dose, although the duration of oral prednisone therapy has remained unchanged, with a median time to discontinuation of 41 months. This duration of oral prednisone therapy is much longer than was recommended by consensus of a group of North American pediatric rheumatologists (27). Our data suggest that most patients will be unable to discontinue prednisone therapy within 1 year after diagnosis, but they will be able to achieve lower prednisone doses. Although the maximum daily dose of prednisone in the initial 6 months of treatment and the time to discontinuation of oral prednisone were not significantly different by year of diagnosis, the time to decrease to half of the initial dose of oral prednisone was more rapid in patients diagnosed after 1997 compared to those diagnosed before 1997.
In smaller cohorts, the combination of prednisone with MTX and other drugs has resulted in a lower cumulative exposure to prednisone. A cohort study from Norway with long-term follow-up reported that patients diagnosed before 1990 were less commonly treated with MTX, IVMP, or antimalarial drugs, and cumulative prednisone doses were higher for patients diagnosed before 1990 compared with those diagnosed after 1990 (28). In a retrospective single center study, addition of MTX to prednisone as part of the initial therapy shortened the duration of glucocorticoid therapy considerably, reducing the total cumulative glucocorticoid dose (6). The children who received combination therapy with MTX experienced fewer corticosteroid side effects, including greater height velocity, lower weight gain, and decreased risk of developing cataracts compared with the group of patients who received prednisone alone (6). In the present study, we do not have information on cumulative corticosteroid doses or steroid-related adverse events.
In addition, we observed that the frequency of receiving IVMP, MTX, IVIG, other DMARDs, and cytotoxic/biologics therapies increased after 1997 in JDM patients, but the treatment durations of several of these medications were also longer after 1997. The shift in treatment after 1997 may have been related in part to publication of case reports and small case series describing frequent clinical response to these medications in prednisone-refractory patients (reviewed in(10)). In small observational studies of children with JDM who had previously failed treatment with glucocorticoids, the use of IVIG often resulted in a reduction in glucocorticoid dose (8, 29, 30). A diagnosis date after 1997 was the major factor associated with a longer time to discontinuation of some of these medications, and may be related to the more rapid dose reduction of oral prednisone.
Several MSAs, including anti-p155/140 (TIF1), anti-MJ (NXP2), and anti-MDA5, have been identified in the majority of children with JDM, with each MSA associated with relatively homogeneous clinical, demographic and laboratory manifestations, and distinct outcomes (2, 24, 31, 32). A large cohort study from the UK reported histopathologic severity to be associated with the need for long-term continued treatment over time, and only anti-Mi-2 autoantibodies, among the MSAs, were also associated with longer treatment duration (33). In the present study, time to discontinuation of oral prednisone in patients with anti-p155/140 (TIF1) autoantibodies was longer than for patients with anti-MJ (NXP2) autoantibodies. Similarly, we found no association of MSAs with treatment duration of other medications, although we did not have an adequate number of patients to examine anti-Mi-2 or -MDA5 autoantibodies and the duration of therapies. However, we did find several differences in the types of medications received in patients with different MSAs: IVMP was more frequently used in patients with anti-p155/140 (TIF1) and anti-MJ (NXP2) autoantibodies compared to MSA/MAA-negative patients, MTX was more commonly prescribed in patients with anti-MJ (NXP2) autoantibodies, and antimalarial drugs were received more frequently in patients with anti-p155/140 (TIF1) autoantibodies. Patients with these autoantibodies may have more severe disease necessitating pulse steroid and MTX treatment, and antimalarial drugs have been recommended for the treatment of skin manifestations of JDM, which have been reported to be prominent in the anti-p155/140 (TIF1) autoantibody group (24, 34, 35). It is important to note that the MSA testing was performed as part of the research studies at the time of patient enrollment, and the results were generally not available to physicians to use in their decision-making about therapies. These findings related to MSA results are a correlation that has been found retrospectively. However, if MSAs are obtained at diagnosis, treatment may in part be determined by MSA status, and the findings of this and the UK studies may aide in guiding therapeutic choices in different autoantibody subgroups (33).
In addition to MSA group and year of diagnosis, we found illness severity, demographics and clinical features also to be associated with the usage of various medications in JDM patients. For example, severe illness at onset was associated with IVMP usage, patients with mild illness at onset received antimalarial drugs more frequently, and patients who were older at the start of treatment received MTX and combination therapy more frequently. A higher skin symptom score at diagnosis was associated with usage of combination therapy. Some of these factors differed from the survey of North American pediatric rheumatologists about preferences in therapeutic choices in JDM. In contrast to this survey, we did not find IVIG used more frequently in patients with severe disease (14). Some of the factors found to be associated with usage of various medications may correlate with disease outcomes: for example, older patients have been reported to develop calcinosis and a prolonged illness course more frequently, and more severe skin or muscle symptoms have been associated with increased organ damage (36, 37). Thus, therapies are tailored to individual patient features, as well as illness severity or MSAs, with several characteristics determining in part which medications patients receive. Future therapies for JDM may soon be more evidence-based, determined by the results of randomized trials and by expert consensus (13, 27, 38–40), but should include such factors. Studies are also needed to examine the effect of various therapies on patient outcomes.
There are some limitations to this study. This study was based on retrospective natural history data, and there was much variation in the treatments received, as well as unmeasured confounders that may have affected treatment decisions beyond what was assessed in this study. For example, we could not evaluate the availability of medications, including variations in health insurance coverage, and access to different treatments. Second, most medications were received as part of combination therapy; however, the duration of medications were evaluated as monotherapies. Small sample sizes also precluded evaluation of certain medications (e.g., less commonly used DMARDs and biologic therapies) and some patient subgroups, such as those with anti-MDA5, anti-Mi-2 and anti-synthetase autoantibodies. We also did not evaluate adverse events associated with each medication. Finally, the effects of these medications on clinical outcomes were not evaluated as part of this study, and myositis assessment tools were not available for the patients enrolled in earlier years. Despite these limitations, our results showed changes in medication usage over time and determined factors associated with patients receiving each of the major medications in a large population of JDM patients over a relatively long period.
Conclusions
We conclude that oral prednisone has been the mainstay of therapy in JDM patients and combination therapy with other medications such as MTX, IVIG, other DMARDs, cytotoxic and biologic therapies was more frequently given after 1997. The dose of oral corticosteroid decreased more quickly in patients treated with these additional medications, with an ability to maintain patients with lower doses of oral prednisone later in the illness course, but this did not result in a shortening of overall prednisone duration. Diagnosis year, MSA status, onset severity, age at first treatment, and symptom scores at diagnosis impact the specific medications JDM patients received as part of their treatment. Future studies must examine such factors in determining responses to therapy.
Supplementary Material
Supplementary Figure. Kaplan-Meier analysis plots showing time to discontinuation of each medication by disease severity at illness onset. The Y axis shows the proportion of patients who were still receiving each medication at any point in time. The X axis shows the months from initial treatment.
A, Intravenous methylprednisolone (IVMP). The median time to discontinuation of IVMP was 10 months after IVMP treatment initiation in patients with moderate illness onset vs. 4 months in patients with severe illness onset.
B, Methotrexate. The median time to discontinuation of methotrexate was 49 months after methotrexate treatment initiation in patients with moderate illness onset vs. 56 months in patients with severe illness onset.
C, Intravenous immunoglobulin (IVIG). The median time to discontinuation of IVIG was 35 months after IVIG treatment initiation in patients with moderate illness onset vs. 17 months in patients with severe illness onset.
D, Antimalarial drugs. The median time to discontinuation of antimalarial drugs was 45 months after initiation of antimalarial treatment in patients with moderate illness onset vs. 35 months in patients with severe illness onset.
Acknowledgments
We thank Dr. Laura Lewandowski and Dr. Peter Grayson for critical reading of the manuscript.
Grants and Financial supporters of the study: This research was supported in part by the Intramural Research Program of the National Institute of Environmental Health Sciences (project ES101081, Takayuki Kishi, Nastaran Bayat, Lisa Rider, and Frederick Miller) and National Institute of Arthritis and Musculoskeletal and Skin Diseases (Michael Ward), National Institutes of Health, and a statistical support contract with Social & Scientific Systems, Inc. (HHSN273201600011C). Takayuki Kishi was supported by a research fellowship of The Myositis Association and by Tokyo Women’s Medical University. Gulnara Mamyrova received funding through the Cure JM Foundation. Ira Targoff is a consultant to the Oklahoma Medical Research Foundation Clinical Immunology Laboratory regarding myositis autoantibody testing.
Members of the Childhood Myositis Heterogeneity Study Group who participated in the current study:
Leslie S. Abramson, Daniel A. Albert, Kathy Amoroso, Bita Arabshahi, Imelda M. Balboni, Susan Ballinger, Lilliana Barillas, C. April Bingham, John F. Bohnsack, Gilles Boire, Michael S. Borzy, Suzanne L. Bowyer, Ruy Carrasco, Chun Peng T. Chao, Randy Q. Cron, Rodolfo Curiel, Marietta M. DeGuzman, Kaleo Ede, Barbara Anne Eberhard, Terri H. Finkel, Robert C. Fuhlbrigge, Abraham Gedalia, Stephen W. George, Harry L. Gewanter, Ellen A. Goldmuntz, Donald P. Goldsmith, Beth Gottlieb, Thomas A. Griffin, Hilary M. Haftel, William Hannan, Melissa Hawkins, Teresa Hennon, Michael Henrickson, Gloria C. Higgins, J. Roger Hollister, Russell J. Hopp, Lisa Imundo, Jerry C. Jacobs, Anna Jansen, James Jarvis, Rita S. Jerath, Olcay Y. Jones, Lawrence K. Jung, Ankur Kamdar, Ildy M. Katona, James D. Katz, Susan Kim, Yukiko Kimura, Daniel J. Kingsbury, Steven J. Klein, C. Michael Knee, W. Patrick Knibbe, Bianca A. Lang, Carol B. Lindsley, Katherine L. Madson, Paul L. McCarthy, Stephen R. Mitchell, Jack Moallem, Chihiro Morishima, Frederick T. Murphy, Kabita Nanda, Judyann C. Olson, Elif A. Oral, Lauren M. Pachman, Christopher T. Parker, Murray H. Passo, Maria D. Perez, Donald A. Person, Egla C. Rabinovich, Linda I. Ray, Robert M. Rennebohm, Rafael F. Rivas-Chacon, Tova Ronis, Margalit Rosenkranz, Deborah Rothman, Bracha Shaham, Robert M. Sheets, David D. Sherry, Edward M. Sills, Sara H. Sinal, Abigail Smukler, Jennifer Soep, Sangeeta H. Sule, Robert P. Sundel, Elizabeth S. Taylor-Albert, Scott A. Vogelgesang, Carol A. Wallace, Jennifer C. Wargula, Patience H. White, Christianne M. Yung, and Lawrence S. Zemel.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures of conflict of interest: None
References
- 1.Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371:2201–12. doi: 10.1016/S0140-6736(08)60955-1. [DOI] [PubMed] [Google Scholar]
- 2.Rider LG, Nistala K. The juvenile idiopathic inflammatory myopathies: pathogenesis, clinical and autoantibody phenotypes, and outcomes. J Intern Med. 2016;280:24–38. doi: 10.1111/joim.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rider LG, Katz JD, Jones OY. Developments in the classification and treatment of the juvenile idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2013;39:877–904. doi: 10.1016/j.rdc.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tansley SL, Simou S, Shaddick G, Betteridge ZE, Almeida B, Gunawardena H, et al. Autoantibodies in juvenile-onset myositis: Their diagnostic value and associated clinical phenotype in a large UK cohort. J Autoimmun. 2017;84:55–64. doi: 10.1016/j.jaut.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowyer SL, Blane CE, Sullivan DB, Cassidy JT. Childhood dermatomyositis: factors predicting functional outcome and development of dystrophic calcification. J Pediatr. 1983;103:882–8. doi: 10.1016/s0022-3476(83)80706-9. [DOI] [PubMed] [Google Scholar]
- 6.Ramanan AV, Campbell-Webster N, Ota S, Parker S, Tran D, Tyrrell PN, et al. The effectiveness of treating juvenile dermatomyositis with methotrexate and aggressively tapered corticosteroids. Arthritis Rheum. 2005;52:3570–8. doi: 10.1002/art.21378. [DOI] [PubMed] [Google Scholar]
- 7.Laxer RM, Stein LD, Petty RE. Intravenous pulse methylprednisolone treatment of juvenile dermatomyositis. Arthritis Rheum. 1987;30:328–34. doi: 10.1002/art.1780300312. [DOI] [PubMed] [Google Scholar]
- 8.Lang BA, Laxer RM, Murphy G, Silverman ED, Roifman CM. Treatment of dermatomyositis with intravenous gammaglobulin. Am J Med. 1991;91:169–72. doi: 10.1016/0002-9343(91)90010-u. [DOI] [PubMed] [Google Scholar]
- 9.Al Mayouf SM, Laxer RM, Schneider R, Silverman ED, Feldman BM. Intravenous immunoglobulin therapy for juvenile dermatomyositis: efficacy and safety. J Rheumatol. 2000;27:2498–503. [PubMed] [Google Scholar]
- 10.Rider LG, Miller FW. Classification and treatment of the juvenile idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 1997;23:619–55. doi: 10.1016/s0889-857x(05)70350-1. [DOI] [PubMed] [Google Scholar]
- 11.Robinson AB, Reed AM. Clinical features, pathogenesis and treatment of juvenile and adult dermatomyositis. Nat Rev Rheumatol. 2011;7:664–75. doi: 10.1038/nrrheum.2011.139. [DOI] [PubMed] [Google Scholar]
- 12.Martin N, Li CK, Wedderburn LR. Juvenile dermatomyositis: new insights and new treatment strategies. Ther Adv Musculoskelet Dis. 2012;4:41–50. doi: 10.1177/1759720X11424460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65:314–24. doi: 10.1002/art.37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stringer E, Bohnsack J, Bowyer SL, Griffin TA, Huber AM, Lang B, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: The Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM Treatment Survey. J Rheumatol. 2010;37:1953–61. doi: 10.3899/jrheum.090953. [DOI] [PubMed] [Google Scholar]
- 15.Hasija R, Pistorio A, Ravelli A, Demirkaya E, Khubchandani R, Guseinova D, et al. Therapeutic approaches in the treatment of juvenile dermatomyositis in patients with recent-onset disease and in those experiencing disease flare: an international multicenter PRINTO study. Arthritis Rheum. 2011;63:3142–52. doi: 10.1002/art.30475. [DOI] [PubMed] [Google Scholar]
- 16.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two pars) N Engl J Med. 1975;292:344–7. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 17.Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts) N Engl J Med. 1975;292:403–7. doi: 10.1056/NEJM197502202920807. [DOI] [PubMed] [Google Scholar]
- 18.Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore) 2013;92:25–41. doi: 10.1097/MD.0b013e31827f264d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991;70:360–74. doi: 10.1097/00005792-199111000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Joffe MM, Love LA, Leff RL, Fraser DD, Targoff IN, Hicks JE, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med. 1993;94:379–87. doi: 10.1016/0002-9343(93)90148-i. [DOI] [PubMed] [Google Scholar]
- 21.Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O’Hanlon TP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006;54:3682–9. doi: 10.1002/art.22164. [DOI] [PubMed] [Google Scholar]
- 22.Habers GE, Huber AM, Mamyrova G, Targoff IN, O’Hanlon TP, Adams S, et al. Brief Report: Association of myositis autoantibodies, clinical features, and environmental exposures at illness onset with disease course in juvenile myositis. Arthritis Rheumatol. 2016;68:761–8. doi: 10.1002/art.39466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber AM, Mamyrova G, Lachenbruch PA, Lee JA, Katz JD, Targoff IN, et al. Early illness features associated with mortality in the juvenile idiopathic inflammatory myopathies. Arthritis Care Res (Hoboken) 2014;66:732–40. doi: 10.1002/acr.22212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rider LG, Shah M, Mamyrova G, Huber AM, Rice MM, Targoff IN, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore) 2013;92:223–43. doi: 10.1097/MD.0b013e31829d08f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillippi K, Hoeltzel M, Byun Robinson A, Kim S. Race, income, and disease outcomes in juvenile dermatomyositis. J Pediatr. 2017;184:38–44. doi: 10.1016/j.jpeds.2017.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guseinova D, Consolaro A, Trail L, Ferrari C, Pistorio A, Ruperto N, et al. Comparison of clinical features and drug therapies among European and Latin American patients with juvenile dermatomyositis. Clin Exp Rheumatol. 2011;29:117–24. [PubMed] [Google Scholar]
- 27.Huber AM, Robinson AB, Reed AM, Abramson L, Bout-Tabaku S, Carrasco R, et al. Consensus treatments for moderate juvenile dermatomyositis: beyond the first two months. Results of the second Childhood Arthritis and Rheumatology Research Alliance consensus conference. Arthritis Care Res (Hoboken) 2012;64:546–53. doi: 10.1002/acr.20695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanner H, Gran JT, Sjaastad I, Flato B. Cumulative organ damage and prognostic factors in juvenile dermatomyositis: a cross-sectional study median 16.8 years after symptom onset. Rheumatology (Oxford) 2009;48:1541–7. doi: 10.1093/rheumatology/kep302. [DOI] [PubMed] [Google Scholar]
- 29.Sansome A, Dubowitz V. Intravenous immunogloblin in juvenile dermatomyositis - four year review of nine cases. Arch Dis Child. 1995;72:25–8. doi: 10.1136/adc.72.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lam CG, Manlhiot C, Pullenayegum EM, Feldman BM. Efficacy of intravenous Ig therapy in juvenile dermatomyositis. Ann Rheum Dis. 2011;70:2089–94. doi: 10.1136/ard.2011.153718. [DOI] [PubMed] [Google Scholar]
- 31.Tansley SL, Betteridge ZE, Shaddick G, Gunawardena H, Arnold K, Wedderburn LR, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (Oxford) 2014;53:2204–8. doi: 10.1093/rheumatology/keu259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tansley S, Betteeridge Z, Gunawardena H, Jacques T, Owens C, Pilkington C, et al. Anti-MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: a prospective cohort study. Arthritis Res Ther. 2014;16:R138. doi: 10.1186/ar4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deakin CT, Yasin SA, Simou S, Arnold KA, Tansley SL, Betteridge ZE, et al. Muscle biopsy findings in combination with myositis-specific autoantibodies aid prediction of outcomes in juvenile dermatomyositis. Arthritis Rheumatol. 2016;68:2806–2816. doi: 10.1002/art.39753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rouster-Stevens KA, Gursahaney A, Ngai KL, Daru JA, Pachman LM. Pharmacokinetic study of oral prednisolone compared with intravenous methylprednisolone in patients with juvenile dermatomyositis. Arthritis Rheum. 2008;59:222–6. doi: 10.1002/art.23341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Femia AN, Vleugels RA, Callen JP. Cutaneous dermatomyositis: an updated review of treatment options and internal associations. Am J Clin Dermatol. 2013;14:291–313. doi: 10.1007/s40257-013-0028-6. [DOI] [PubMed] [Google Scholar]
- 36.Ravelli A, Trail L, Ferrari C, Ruperto N, Pistorio A, Pilkington C, et al. Long-term outcome and prognostic factors of juvenile dermatomyositis: a multinational, multicenter study of 490 patients. Arthritis Care Res(Hoboken) 2010;62:63–72. doi: 10.1002/acr.20015. [DOI] [PubMed] [Google Scholar]
- 37.Patwardhan A, Rennebohm R, Dvorchik I, Spencer CH. Is juvenile dermatomyositis a different disease in children up to three years of age at onset than in children above three years at onset? A retrospective review of 23 years of a single center’s experience. Pediatr Rheumatol Online J. 2012;10:34. doi: 10.1186/1546-0096-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruperto N, Pistorio A, Oliveira S, Zulian F, Cuttica R, Ravelli A, et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet. 2016;387:671–8. doi: 10.1016/S0140-6736(15)01021-1. [DOI] [PubMed] [Google Scholar]
- 39.Huber AM, Giannini EH, Bowyer SL, Kim S, Lang B, Lindsley CB, et al. Protocols for the initial treatment of moderately severe juvenile dermatomyositis: results of a Children’s Arthritis and Rheumatology Research Alliance Consensus Conference. Arthritis Care Res (Hoboken) 2010;62:219–25. doi: 10.1002/acr.20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enders FB, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76:329–40. doi: 10.1136/annrheumdis-2016-209247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure. Kaplan-Meier analysis plots showing time to discontinuation of each medication by disease severity at illness onset. The Y axis shows the proportion of patients who were still receiving each medication at any point in time. The X axis shows the months from initial treatment.
A, Intravenous methylprednisolone (IVMP). The median time to discontinuation of IVMP was 10 months after IVMP treatment initiation in patients with moderate illness onset vs. 4 months in patients with severe illness onset.
B, Methotrexate. The median time to discontinuation of methotrexate was 49 months after methotrexate treatment initiation in patients with moderate illness onset vs. 56 months in patients with severe illness onset.
C, Intravenous immunoglobulin (IVIG). The median time to discontinuation of IVIG was 35 months after IVIG treatment initiation in patients with moderate illness onset vs. 17 months in patients with severe illness onset.
D, Antimalarial drugs. The median time to discontinuation of antimalarial drugs was 45 months after initiation of antimalarial treatment in patients with moderate illness onset vs. 35 months in patients with severe illness onset.