

Summary
Although the mammalian microbiota is well contained within the intestine, it profoundly shapes development and metabolism of almost every host organ. We questioned the range and depth of microbial metabolite penetration into the host, and how this is modulated by intestinal immunity. Chemically identical microbial and host metabolites were distinguished by stable isotope tracing from 13C-labeled live non-replicating Escherichia coli, differentiating 12C host isotopes with high-resolution mass spectrometry. Hundreds of endogenous microbial compounds penetrated 23 host tissues and fluids after intestinal exposure: subsequent 12C host metabolome signatures included lipidemia, reduced glycolysis, and inflammation. Penetrant bacterial metabolites from the small intestine were rapidly cleared into the urine, whereas induced antibodies curtailed microbial metabolite exposure by accelerating intestinal bacterial transit into the colon where metabolite transport mechanisms are limiting. Pervasive penetration of microbial molecules can cause extensive host tissue responses: these are limited by immune and non-immune intestinal mucosal adaptations to the microbiota.
Keywords: IgA, microbiota, microbial metabolites, germ free, colonization, metabolomics, 13C-isotope tracing, intestinal transit, lipidemia, inflammation
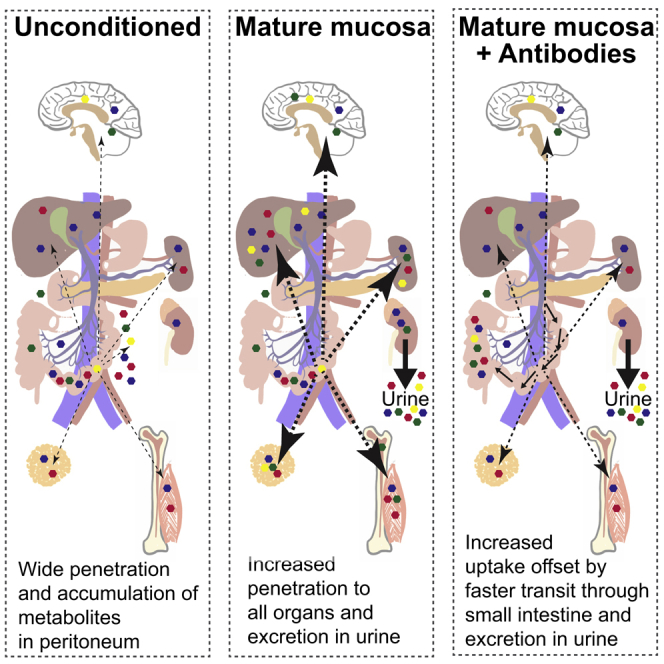
Graphical Abstract
Highlights
-
•
Metabolites from mutualistic bacteria broadly penetrate host tissues and organs
-
•
Bacterial metabolites induce widespread host metabolic and immunological responses
-
•
The small intestine is highly susceptible to host-microbial metabolomic exchange
-
•
Secretory immunoglobulins accelerate microbial clearance from the small intestine
Bacteria-derived metabolites pervade the mammalian host, shaping immunity and metabolism. Using stable isotope tracing, Uchimura and colleagues profile the scope and depth of host tissue penetration by bacterial metabolites. Extensive host immune and metabolic responses to microbial metabolite penetration are constrained by secretory antibodies that limit microbial small-intestinal dwell time.
Introduction
The relationship between mammals and their resident microbiota shapes both the microbial communities and every host organ system from conception until death. There are extensive differences in all body systems between animals of the same strain in a germ-free or colonized state (Smith et al., 2007); most effects of the microbiota are seen if a germ-free animal is colonized at any age, including but not limited to intestinal secretory immunoglobulin (Ig)A induction (Gensollen et al., 2016). Given these ubiquitous interactions over all body systems, it is unsurprising that almost every disease model has been reported to be sensitive to microbial colonization status (Belkaid and Harrison, 2017, Blumberg and Powrie, 2012, Sommer and Bäckhed, 2013).
Since the biomass of the microbiota is well separated from host tissues except during pathogenic infections, at first sight it is paradoxical that the presence of the microbiota shapes the cellular composition and function of non-intestinal mammalian organs so extensively, even when they are almost always sterile. The answer probably lies mainly in the penetration of microbial metabolites, which in some cases have been shown to exert powerful effects on host cell differentiation and metabolism. Microbial metabolite penetration of host tissues was first shown by detecting aromatic compounds in colonized but not germ-free animals more than 40 years ago (Cotran et al., 1960, Dayman and Jepson, 1969). Typically, metabolites are inferred to originate from microbial metabolism when they are present in host tissues only when animals are fully colonized or harbor specific compositions of microbial consortia (El Aidy et al., 2013, Nicholls et al., 2003, Williams et al., 2002). This approach is ambiguous for those compounds that can be synthesized either by host or microbial metabolism, because an increased metabolite concentration after microbial colonization may be due to the propagation from microbes or to the host metabolic response to their presence.
Stable isotope tracing to differentiate microbial or host origins of chemically identical metabolites has so far been used to follow gut microbial metabolism in an in vitro model where host effects are eliminated (de Graaf et al., 2010) or to show selective utilization of carbon sources resistant to host digestion by in vivo incorporation into microbial RNA of a limited number of species within the intestinal consortia (Tannock et al., 2014). These labeled nutrient supplementation approaches are thus limited to spotlighting the fate of individual nutrients within the microbial communities themselves.
Assessing overall penetration of host tissues with microbial metabolites requires selective labeling of all microbial (but not host) molecules, yet avoiding exponential dilution of the label as bacteria use host or nutritional carbon sources to divide in vivo. We developed a strategy where germ-free mice are gavaged with a dose of either 12C- or fully 13C-labeled live E. coli HA107 that cannot replicate in the germ-free intestine (Hapfelmeier et al., 2010), so the 13C-label is not immediately and exponentially diluted in vivo through bacterial replication using host- or food-derived 12C nutrients. High-resolution mass spectrometry (Fuhrer et al., 2017, Sévin et al., 2017) was then used to reveal both the 13C host penetration by microbial metabolites and the 12C host metabolome response to the presence of the microbes in fasting animals. Although E. coli is a minor constituent of the adult human microbiota, it is a prominent member of the early life microbiota at a time when diversity is limited (Bäckhed et al., 2015), and thus models some aspects of early-life host-microbial molecular interactions from a low-diversity microbiota.
A second problem in resolving the in vivo mechanisms that determine the molecular exchange between intestinal microbes and their mammalian host is that the intestinal mucosa itself is extensively reprogrammed by exposure to intestinal microbes through epithelial cell differentiation, secretion of antibodies, antibacterial polypeptides, microvascular development, and xenobiotic metabolic pathway induction (Gomez de Agüero et al., 2016, Hooper et al., 2001, Macpherson et al., 2000, Stappenbeck et al., 2002, Vaishnava et al., 2011). This means that microbes instruct the very mechanisms that build the dynamic interface separating the host, its intestinal microbes, and their metabolites. 13C-labeled bacterial challenge of a colonized animal would largely obscure distinction of microbial and host compounds based on the observed isotopes, because of the much larger load of natural 12C metabolites from the resident microbiota. We have resolved this problem using a system that allows maturation of the intestinal barrier in germ-free animals through transient intestinal live microbial preconditioning without permanent colonization (Hapfelmeier et al., 2010). This approach allowed us to compare microbial-host metabolite exchange directly in fasting germ-free mice, with or without prior microbial-driven mucosal adaptations including Ig induction. The combination of this isotope tracing approach and the transient colonization model has provided a unique insight into how microbial transit through the small intestine and propagation of microbial metabolites throughout the host are checked by secretory and metabolic adaptations of the intestinal mucosa.
Results
A Broad Scope of Intestinal Bacterial-Derived Metabolites Penetrates Host Systemic Organs
While a few microbial metabolites, such as short chain fatty acids or aryl hydrocarbon receptor ligands, are well known to shape host metabolism and immune functions (Arpaia et al., 2013, Gomez de Agüero et al., 2016, Perry et al., 2016, Smith et al., 2013b), we do not know the full extent of microbial metabolite penetration, nor the entire scope of consequent host metabolic responses. To assess the propagation of host-microbial mutualism in 16 mouse tissues and 7 fluids systematically, we gavaged fully 13C-labeled or 12C live E. coli HA107, which cannot replicate in vivo, into fasting C57BL/6 germ-free mice (Figure S1A). By employing high-resolution untargeted mass-spectrometry with high throughput methodology (Fuhrer et al., 2011, Fuhrer et al., 2017, Sévin et al., 2017), we resolved more than 1,000 annotated compounds (Mr 50–1,000 Da at 1 mD resolution) and determined the mass shift showing 13C or 12C proportions within each molecule (Figures S1B and S1C). Molecules from fully 13C-labeled bacteria (Figures S1D and S1E) showed bacterial metabolite exposure (Figures S1B and S1F). However, if the molecule concerned came from the 12C host response rather than the bacteria (whether or not the challenge bacteria were 13C-labeled), it gave an appropriate mass/charge ratio signal, but without the 13C mass shift (Figure S1C, illustrated for a hypothetical C4 compound). These experiments can distinguish host and pre-existing endogenous microbial compounds in fasting mice, rather than products of bacterial metabolism from dietary carbon sources.
Two hours after gavage, a wide range of 13C-labeled bacterial metabolites had already propagated into the host to reach almost all fluids and organs (Figures 1, 2A–2C, and S2A–S2C). However, the distribution of different compound classes varied. Bacterial amino acids (including essential amino acids) penetrated very widely over all host tissues (Figures 1 and S2A). Other bacterial metabolites, including five- and six-membered ring and heterocyclic compounds, were present in the serum and rapidly excreted in the urine (Figures 2B and S2C). Urinary lipid amounts were generally low, consistent with known lipoprotein exclusion from glomerular filtration (Figures 2B and S2B). Bacterial fatty acids and glycerolipids were present mainly in the small intestinal tissue and mesenteric lymph nodes (Figure 2C), consistent with lymphatic uptake. Most compound classes apart from lipids were able to penetrate into the peritoneal fluid of these naive germ-free mice (compare analysis of different compound classes from Figure 2A shown in Figures S2A, S2C, with S2B).
Figure 1.

Depth of Penetration of 13C-Labeled Bacterial Amino Acids from the Intestine to Systemic Sites of the Body
Germ-free C57BL/6 mice were gavaged with 13C- or 12C-labeled HA107 or PBS as control (n = 3–4 mice per group). Differential ions detected in different host tissues after 2 hr annotated as fully 13C-labeled amino acids are indicated where the identical mass did not exhibit any significant change in the 12C to PBS comparison (positive Log2-fold change cutoff of 1 and p value < 0.05). Results are representative of three experiments. See also Figure S1.
Figure 2.

Penetration of 13C-Labeled Bacterial Metabolites from the Intestine to Host Tissues and Fluids
Germ-free C57BL/6 mice were gavaged with 13C-labeled HA107 or PBS as control (n = 3–4 mice per group). Fluid samples were collected 2 or 18 hr later and analyzed with Q-TOF mass spectrometry. All ions which were annotated as metabolite isotopes ≥75% 13C labeled are plotted.
(A) Left panel shows samples from small intestinal fluid, peritoneal fluid, serum, and urine at 2 hr; lines connect identical metabolites. Inset shows small intestinal, cecal, and colon fluids also at 2 hr. Right panel shows 18 hr cecal, colon, peritoneal, serum, and urine fluid samples with (inset) small intestinal, cecum, and colon fluids.
(B) Cluster analysis heatmap of all 13C-labeled annotated metabolites (p < 0.05, ≥3 mice/group) in different host fluids at 2 hr and 18 hr after gavage. Metabolite classes are shown in the lower color bar and legend. Results are representative of three experiments.
(C) As for (B), except 13C-labeled annotated metabolites in host tissues are shown.
See also Figure S2.
By 18 hr, the front of detected 13C-labeled metabolites had moved along the intestine to reach the cecum and the colon (compare Figure 2A insets and S2A–S2F insets at 2 and 18 hr), consistent with the known murine intestinal transit times (Padmanabhan et al., 2013). Although bacterial metabolites had been cleared from the small intestinal fluid and peritoneum by 18 hr (Figure S2D-S2F), they were still present in small intestinal tissues (Figure 2C) and being excreted at low amounts in the urine and the bile (Figure 2B). In many cases the bacterial metabolites detected in these unconditioned germ-free mice constituted a high proportion of the total (bacterial+host) concentration for that metabolite across different fluids and tissues of the mouse (Figure S2G).
Since the HA107 system depends on cell wall auxotrophy to abrogate in vivo replication, we asked whether increased bacterial fragility of the HA107 system generates increased systemic exposure of bacterial metabolites by comparing penetration of compounds from HA107 or its non-auxotrophic parent strain. For maximum sensitivity, challenge bacteria were labeled by growth in 14C glucose. Although 14C metabolic labeling reveals only the bacterial origin of the radiolabel but not the original molecular identity of the bacterial compound, it had the advantage that we could measure overall 14C transfer, whereas bacterial replication in vivo would dilute the proportion of 13C-label and potential underestimation of penetration. We found overlapping ranges of radiolabel recovery in serum, urine, and host tissues and no significant differences at most time points, regardless of whether or not the test bacteria were engineered with cell wall auxotrophy (Figures 3A, 3B, and S3A–S3C).
Figure 3.

Kinetics of Metabolite Penetration and Durable Intestinal Conditioning Responses to E. coli HA107
(A and B) 14C-metabolically labeled E. coli HA107 was administered into the stomach (closed circles) or mixed directly with cecal contents (open circles) of germ-free C57BL/6 mice. For comparison, 14C-labeled E. coli JM83 (replication competent parent strain of HA107; red triangles) was administered into the stomach in a separate group. 14C-radioactivity in the urine (A) or serum (B) at 2, 8, and 18 hr. Results are expressed as a percentage of the gavage dose/g fluid over background.
(C) RNA-seq analysis of small intestinal tissue from C57BL/6 germ-free mice primed with HA107 transitory colonization 2 weeks earlier, compared with control germ-free mice. Upper panel: volcano plot shows significantly altered gene expression (padj < 0.05, n ≥ 3) in HA107-preconditioned germ-free mice or in sham-treated germ-free mice. Immunoglobulin genes are annotated. Expanded box shows a STRING-DB representation of significantly upregulated non-immunoglobulin genes in preconditioned mice.
(D) Expression of transcripts coding microbiocidal peptides ( ± SEM, n ≥ 3).
(E) Live intestinal microvascular endomicroscopy imaging. FITC-dextran (70 kD) fluorescence was visulized after i.v. injection into HA107-preconditioned C57BL/6 germ-free and control germ-free mice in the small intestinal villus microvasculature with live Cellvizio endomicroscopy. The ± SEM of endothelial/epithelial ratios of each group (n = 6) is indicated (t test p < 0.02). Results are representative of six experiments.
See also Figure S3.
We also confirmed that our method of bacterial gavage into the stomach in buffer did not result in gastric microbial death. A single bacterium of the replication competent parent strain of E. coli HA107 was sufficient to permanently colonize the intestine (Figure S3D). Second, direct measurement recovery of a dose of 1 × 107 CFU HA107 from the luminal contents and mucosal scrapings of the intestine showed that 85.6% ± 4.4% ( ± SD, n = 6) of the gavage dose was present in the small intestine 90 min after administration and 73.3% ± 5.1% from the small intestine and the cecum 150 min after administration. Third, radiolabel penetration from 14C HA107 or its non-auxotrophic parent strain was equivalent but substantially less than the same dose of 14C HA107 material that had been lysed by sonication (Figure S3E). Hence there was no evidence for immediate gastric lysis, nor that potential fragility of the employed HA107 strain would liberate more endogenous metabolites than wild-type E. coli.
We concluded that there is extensive penetration of an extremely broad range of bacterial-derived molecules into host tissues from the germ-free intestine, even though live bacteria penetrate only as far as the local (mesenteric) lymph nodes (Hapfelmeier et al., 2010). These results did not distinguish the relative penetration through the small or large intestinal surfaces, nor show how prior mucosal adaptation of the germ-free intestine through secretory Ig induction, antibacterial peptides, and a consolidated barrier function (Hapfelmeier et al., 2010, Petersson et al., 2011, Salzman et al., 2010, Vaishnava et al., 2011) could shape microbial-host molecular exchange at both small and large intestinal surfaces.
The Small Intestine Is the Predominant Interface for Metabolite Penetration of Intestinal Microbes
The small intestine has minimal mucus coverage and is highly adapted for molecular uptake, whereas the large intestine has a thick layer of protective mucus overlying the epithelium (Johansson et al., 2008). We compared uptake of 14C HA107 after delivery into the stomach or after direct in vivo mixing with cecal contents. 14C labeling was also used in this experiment to detect host exposure to microbial metabolites with maximum sensitivity, even if they underwent secondary metabolism in the host. There was substantial radiolabel uptake from the upper intestine and recovery in the urine, but little uptake from cecal bacteria (Figures 3A, 3B, and S3A–S3C). This verified that the small intestine was the main absorptive surface for extensive microbial molecular exchange with host organ systems, and supported results with the specific molecular 13C system that the urine is a highly effective immediate elimination route.
The lower small intestine is normally colonized with intestinal microbes, so microbial molecular exposure has potentially important biomedical consequences. Mice are naturally coprophagic, but even adult humans or infants with reduced intestinal motility can have high densities of microbes in the proximal small intestine (Donowitz et al., 2016, Jones et al., 2011). In the developing world, small intestinal microbes cause childhood environmental enteropathy with malnutrition and growth stunting, affecting up to 25% of children aged under 5 years (Korpe and Petri, 2012).
Maturation of the Intestinal Mucosal Barrier after Transient Live Microbial Exposure Is Durable and Involves Immune and Non-immune Transcriptional Reprogramming
Secretory IgA is known to be induced by small intestinal microbes (Bunker et al., 2015), and environmental enteropathy is associated with compensatory secretory IgA responses against microbes responsible for the loss of intestinal function (Beatty et al., 1983, Kau et al., 2015). We therefore addressed whether antibodies or other barrier functions that are induced as a result of exposure to intestinal microbes constrain microbial metabolite penetration and how this impacts the body metabolome of the host response.
To assess how mucosal protective factors affect bacterial metabolite exchange in the intestine, we needed to experimentally uncouple maturation of the mucosa and its barrier functions from permanent colonization. To confirm the extent of mucosal adaptations after live reversible E. coli HA107 colonization, we carried out transcriptomic analysis of the intestinal mucosa 2 weeks later, once mice had returned to germ-free status (Figure 3C). In line with our earlier results demonstrating a persistent IgA response (Hapfelmeier et al., 2010), we found a series of significantly upregulated Ig transcripts 2 weeks after transitory exposure to HA107. In addition, there was upregulation of genes with known functions in xenobiotic and metal metabolism, mitochondrial function, ion channels, angiogenesis, and epithelial microbiocidal peptide expression (Figures 3C and 3D). In a complementary approach, in vivo endomicroscopy was used to compare the endothelial to epithelial ratios in the small intestine of transiently colonized and sham control animals (Figure 3E), consistent with earlier observations of an increased microvascular density induced by permanent colonization with Bacteroides thetaiotaomicron (Stappenbeck et al., 2002).
We concluded that even transitory mucosal conditioning by intestinal bacteria results in extensive re-programming of immune, epithelial, and stromal elements of the intestinal mucosa of adult animals that is durable after the mice have returned to germ-free status. Thus, in principle we could evaluate the barriers to microbial molecular penetration through the intestine by comparing secondary bacterial challenge in germ-free mice that had previously been preconditioned by transient colonization with unconditioned germ-free controls.
Immunoglobulin Induction Accelerates Small Intestinal Transit of Bacteria and Their Metabolites
To determine how mucosal adaptation or antibody induction, or both, affect the depth of metabolite penetration, we compared germ-free C57BL/6 wild-type with Igh-J-deficient mice lacking all antibody isotypes due to deletion of the J-segments of the Ig heavy chain locus (Chen et al., 1993). This avoided the confounding effects of compensatory intestinal IgM and overinduction of systemic IgG in animals selectively deficient for the IgA isotype (Harriman et al., 1999).
Groups (≥18) of germ-free wild-type C57BL/6 and Igh-J−/− mice were preconditioned with HA107; separate groups of each strain were treated in parallel with sterile PBS as controls. Preliminary experiments established that 14C HA107-treated mice had no residual detectable radiolabel 14 days after treatment. Two weeks after the preconditioned HA107-treated mice had returned to germ-free status, they were divided into 3 groups of ≥6 animals of each strain to be challenged with a single dose of 13C- or 12C-HA107, or sterile PBS, administered directly into the stomach (Figure S1Aii). To investigate how antibodies modulated the effect of preconditioning, we compared the relative difference of positive fold-changes due to propagation of 13C-labeled compounds between pre- and unconditioned germ-free mice from the C57BL/6 and Igh-J−/− strains (Figures S1B, S1C, 4A, and 4B).
Figure 4.

Transient Intestinal Microbial Conditioning Enhances Clearance of Bacteria and Their Metabolites from the Intestine of C57BL/6 Mice Compared with Antibody-Deficient Igh-J−/− Mice
Germ-free C57BL/6 mice (red) or antibody-deficient Igh-J−/− mice (blue) were preconditioned with HA107 or control treated. Twelve days after all groups had returned to germ-free status, they were gavaged with 13C-labeled HA107 or PBS as control (n = 3–4 per group) and Log2 fold-changes were calculated (Figure S1Aii).
(A) Samples from small intestinal, cecal, and colon fluids were collected at 2 hr and analyzed with Q-TOF mass spectrometry. Differences between the Log2-fold values of preconditioned and unconditioned mice are shown for ions annotated as metabolite isotopes with ≥75% 13C labeled. Fold-changes based on low abundant ions (counts < 200) were excluded. Mann-Whitney U test; ∗∗∗p < 0.001.
(B) As for (A), but the plot shows individual compound classes.
(C) Small intestinal transit of replication-deficient bacteria in wild-type mice and antibody-deficient mice. HA107 preconditioned C57BL/6 mice (red symbols) were compared with HA107 preconditioned antibody-deficient Igh-J−/− mice (blue) 2 hr after challenging with 107 colony forming units (CFU) HA107 delivered into the stomach. The small intestine was divided into 10 sections and CFU in luminal contents of each section were determined with auxtrophic supplements ( ± SD n = 3, ∗p < 0.05).
(D) Whole intestinal transit of replication-deficient bacteria in wild-type mice and antibody-deficient mice. HA107 preconditioned C57BL/6 mice (red symbols) or antibody-deficient Igh-J−/− mice (blue) were gavaged with 108 CFU HA107. Feces were plated with auxotrophic supplements (∗p < 0.05). Results are representative of two experiments with ≥6 mice/group.
See also Figure S4.
Two hours after bacterial challenge, conditioned C57BL/6 mice showed increased bacterial metabolites in the cecal fluid and colonic fluid compared with the antibody-deficient Igh-J−/− strain (p < 0.001 for all measured compounds and for different compound classes considered individually in colonic fluid; Figures 4A and 4B). In contrast, bacterial compounds were enriched in the small intestinal fluid of unconditioned mice compared to conditioned mice (Figure 4A). Since these results were generalized across compound classes, we hypothesized that induced secretory Ig accelerated the transit of intact intestinal bacteria, either through limiting bacterial motility, reducing adherence, or exclusion effects.
We verified in our reversible colonization system that maximum concentrations of secretory IgA directed against intestinal bacteria were expressed in the small intestine (Figure S4A; Bunker et al., 2015) and that these IgA antibodies coated the HA107 challenge organisms and their flagella (Figures S4B–S4D).
The hypothesis that induced immunoglobulins directly accelerate the small intestinal transit of intestinal bacteria was confirmed by showing faster small intestinal transit (Figure 4C) and fecal shedding (Figure 4D) of a test dose of HA107 from preconditioned germ-free C57BL/6 mice compared with preconditioned germ-free Igh-J−/− controls. By 18 hr, most of the load of challenge bacteria had been cleared from the colon of the C57BL/6 wild-type mice. Similar results were obtained comparing pretreated and non-treated control C57BL/6 mice (not shown).
Given that isolated loop experiments had showed no difference in the survival of the bacteria in the intestine whether or not the animals could express antibodies (Figure S4E), we considered whether IgA coating of flagella (Figure S4B, enlarged area) could explain rapid clearance of bacteria from the small intestine. Small intestinal secretions contained anti-flagellin antibodies of the IgA isotype (Figure S4F) and intestinal fluid from preconditioned C57BL/6 but not Igh-J-deficient mice attenuated the motility of the bacterial strain (Figure S4G and Videos S1 and S2). Precoating with intestinal fluid from preconditioned C57BL/6 mice, but not Igh-J-deficient mice, also limited the bacterial motility in a motility agar assay (Figure S4H), consistent with reported monoclonal anti-flagellin inhibition of bacterial motility (Cullender et al., 2013). While size-exclusion effects or reduced adherence may also contribute to accelerated antibody-dependent clearance from the small intestine, increased bacterial motility in the absence of secreted small intestinal antibodies potentially allows bacteria to move against the flow of small intestinal secretions, and hence increases exposure of the host to their small intestinal metabolites.
Small intestinal wash were made by flushing freshly isolated small intestine with 5ml ice-cold PBS. 108/ml GFP expressing E. coli MG1655 were incubated with intestinal wash prepared from HA107 preconditioned Igh-J−/− mice (Video S1) or C57BL/6 (Video S2) for 30 min on ice prior to confocal microscopy acquisition. The bacteria were then loaded to micro-fluid chip (Absis). Zeiss LSM 700 confocol microscopy were used to acquire pictures and videos were re-constructed from pictures using Volocity software.
Non-immune Mechanisms of Prior Intestinal Conditioning with Live Microbes Determine the Bodily Distribution and Excretion of Microbial Metabolites
So far, we had found that without conditioning 13C bacterial metabolites appear in the peritoneal fluid of C57BL/6 and antibody-deficient mice early after the bacterial challenge (Figures 2A, 2B, S2A–S2C, 5A, and S5A). However, instead of accumulation in the peritoneum, there was preferential elimination in the urine after preconditioning: this effect was antibody independent as it was seen in both wild-type and antibody-deficient animals (Figure 5A). At later times there was persistent uptake of bacterial molecules from the intestine into the serum of conditioned antibody-deficient mice (Figure 5B), reflecting the increased dwell time of bacteria in the small intestine (Figures 4C and 4D). Since metabolites were very effectively filtered into and concentrated within the urine, and the lower serum concentrations remain constant until at least 18 hr (compare Figures 3A and 3B), we concluded that the general preconditioning response improved early clearance of metabolites into the urine. Increased cardiac output and peripheral perfusion have been observed by comparing germ-free and colonized mice (Smith et al., 2007), and bacterial pre-conditioning also increases intestinal microvascular density (Figure 3E) which likely favors early metabolite uptake through the intestinal microvasculature and rapid clearance into the urine as an antibody-independent effect (Figure 5A).
Figure 5.

Transient Intestinal Microbial Conditioning Enhances Antibody-Independent Early Clearance of Bacterial Metabolites into the Urine
Germ-free C57BL/6 mice (red symbols) or antibody-deficient Igh-J−/− mice (blue) were preconditioned with HA107 or control-treated, returning to germ-free status 12 days prior to re-challenge with a gastric dose of fully 13C-labeled or 12C- HA107 as in the Figure 4 legend.
(A and B) Log2 fold differences between HA107-preconditioned and unconditioned germ-free mice for individual annotated ions representing metabolites containing ≥75% 13C are shown for peritoneal fluid, serum, or urine 2 hr (A) or 18 hr (B) after challenge.
(C and D) As for (B) but showing persistent bacterial metabolites in intestinal (C) or systemic (D) tissues of antibody-deficient mice at 18 hr.
(E) Pairwise Tanimoto chemical similarity indices of more than 1.5-fold increased bacterial metabolites (≥75% 13C) in ileal tissues at 18 hr of conditioned antibody-deficient mice. Numbers in squared brackets show mass shifts of each metabolite and in round brackets denote chemical annotations for each metabolite in Table S1. Metabolites with Tanimoto chemical similarity ≥ 0.7 are interconnected and node size shows fold-increase. The paired Mann-Whitney U test was applied to calculate the p value of each network.
(F) As (E), except analysis of blood metabolite networks. Results are representative of three experiments.
See also Figure S5.
From the results showing increased dwell time of bacteria in the small intestine of antibody-deficient mice, we expected to detect increased bacterial metabolites in intestinal tissues. This was found in the small intestine, mesenteric lymph nodes, and some systemic tissues (Figures 5C and 5D). Specifically, we found that these bacterial metabolites in the ileum of conditioned antibody-deficient mice included potentially inflammatory biogenic amine indole derivates such as methoxytryptamine, methoxytryptophol, methoxydimethyltryptamine, and serotonin (Figure 5E). In some cases, there was evidence of 12C incorporation into the dominantly 13C-labeled metabolites (Table S1 specifies molecular mass-shift results from Figure 5E), indicating formation after additional host or bacterial metabolism in vivo. There was also evidence of bacterial saccharides such as arabitol and galactose-xylose normally associated with bacterial but not host metabolism (Figure 5E). The serum of conditioned antibody-deficient mice contained increased bacterial non-esterified fatty acids, most of which were fully 13C-labeled (Figure 5F and Table S1). Increased lipid exposure as a function of colonization was verified in permanently colonized chow-fed animals (Figure S5B). It is therefore likely that the bacterial lipid load makes a substantial contribution to host lipids, particularly in the context of defective intestinal barrier function.
Natural Antibodies Binding to Microbial Metabolites Are Induced by Transient Colonization of Germ-free Mice
Given that we had shown binding of induced IgA against the HA107 transient colonizer, we also addressed whether binding to any of its bacterial metabolites could be detected. Using pure compounds selected from the results of the 13C mass spectrometric identifications, we carried out ELISA binding studies comparing intestinal IgA from C57BL/6 mice and antibody-deficient controls, with and without prior transient colonization. This showed antibody binding against identified authentic metabolites (Figures 5E and 5F), including heptadecanoic acid, pentadecanoic acid, scopolamine, and N-acetylaspartylglutamic acid, with E. coli DNA as positive control and maltose as negative control (Figure S5C). These are likely amplified natural specificities (Bunker et al., 2017) as the binding curves of germ-free and transiently colonized mice superimposed when corrected for IgA concentration (Figure S5C insets). Nevertheless, natural IgA per se was insufficient to block the responsiveness of the mucosa independently of small intestinal bacterial clearance. Cytokine secretion induced in ileal explants by the presence of bacteria was affected neither by specific IgA added to the explant system, nor when the explants themselves were from animals expressing mucosal IgA (Figure S5D). We concluded that the principal effect of antibody induction on metabolite exposure is to reduce the bacterial dwell time of the source microbe, rather than preventing adherence or access of the released metabolites to the intestinal mucosa.
Persisting Exposure of Antibody-Deficient Mice to Bacterial Metabolites Results in Increased Host Cytokine and Metabolomic Responses
Given the pervasive penetration of bacterial-derived metabolites in many different host tissues in antibody-deficient animals, including inflammatory triggers such as biogenic amines and lipids (Figures 5B and 5C), a metabolic and immune host response was expected. To address this in molecular terms, we exploited the discrimination power of the stable isotope tracing system by analyzing the changes in 12C metabolite amounts across different host tissues that were independent of whether a 13C or 12C bacterial challenge dose had been given (Figure S1C).
Tissue 12C-metabolite measurements 18 hr after a transient dose of either 13C-labeled or 12C HA107 showed both increases and loss of different specific metabolites in intestinal and systemic tissues of conditioned antibody-deficient compared with wild-type mice (Figures 6A–6C).
Figure 6.

Exaggerated Host Metabolite Responses in Antibody-Deficient Mice after Microbial Challenge
Germ-free C57BL/6 mice (black symbols) or antibody-deficient Igh-J−/− mice (green) were preconditioned with HA107 or control treated, returning to germ-free status 12 days prior to re-challenge with a gastric dose of fully 13C-labeled or 12C HA107. Tissue samples were collected after 18 hr for Q-TOF mass spectrometry. Annotated ions representing metabolites containing concordant changes from 13C and 12C pulsed mice without mass shift were plotted.
(A and B) Log2 fold-change differences between preconditioned and control mice for each metabolite is shown for (A) duodenal, jejunal, and ileal tissues; (B) mesenteric lymph nodes, blood, muscle, and adipose tissues.
(C) Heatmap showing cluster analysis of concordant increases (red) or losses (blue) of 12C-host annotated metabolites (p < 0.05, ≥3 mice/group) where the identical metabolite change was seen without a mass shift in both 13C- and 12C-HA107 challenged antibody-deficient mice. Lower color bar and legend shows metabolite classes. Results are representative of two experiments.
(D) Preconditioned germ-free C57BL/6 mice or antibody-deficient Igh-J−/− mice were either re-challenged with a gastric dose of 1010 HA107 or left untreated. Total cells isolated from different lymphoid tissues were cultured for 2 hr prior to supernatant cytokine analysis. Results are calculated from two independent experiments with 3–4 mice/group.
See also Figures S6 and S7.
The hypothesis that antibody-dependent clearance of bacteria from the small intestine as a mechanism of limiting microbial metabolite exposure predicted that resulting immune responses would also be attenuated in vivo. Leukocyte cytokine release from different immune compartments in preconditioned wild-type and antibody-deficient mice showed that bacterial rechallenge generated inflammatory and non-inflammatory cytokine release and that this effect was generally less in conditioned C57BL/6 wild-type than in antibody-deficient mice (Figure 6D). Direct stimulation of leukocyte preparations with pure compounds that had been identified earlier as being of bacterial origin (Figures 5E and 5F) reproduced similar cytokine responses (Table S2). These results verify clear immunostimulatory effects from the penetration of the identified bacterial metabolites and that the functional consequences of exposure are limited by antibody secretion.
As many of the compounds shown by their mass spectrometric signatures to be of host response origin in the in vivo experiments are not annotated in metabolic pathways, we used chemical similarity clustering analysis. For example, in antibody-deficient ileal tissues, the Tanimoto network of ≥2-fold changes comparing HA107-challenged Igh-J−/− antibody-deficient mice with unchallenged Igh-J−/− controls showed increased host lysophospholipids, phosphatidic acids, and non-esterified fatty acids with free head-groups such as arachidonic acid (Figure 7A, Table S3). Increased arachidonic acid-derived host prostaglandins and leukotrienes including leukotriene E3 and prostaglandins A4, E2, F1a, and F2a were consistent with innate immune activation (Figures S6A and 7A).
Figure 7.

Tanimoto Chemical Similarity Networks of Increased Metabolite Responses in Conditioned Antibody-Deficient Mice after Microbial Challenge
Tanimoto chemical similarity indices of increased host response metabolites in both 13C- and 12C-pulsed mice without mass shift of the annotated ions were analyzed as described in Figure 5. Networks from the ileum (A), the pancreas (B), or the blood (C) are shown with interconnected chemical similarities ≥0.7. Numbers in squared brackets correspond to chemical annotations for each metabolite in Table S3. Node size represents the absolute value of Δ-fold change difference between conditioned and control germ-free Igh-J−/−. Paired Mann-Whitney U test was applied to calculate p value of each network. Results are representative of two independent experiments with 3–4 mice/group. See also Figures S6 and S7.
Increased prostaglandins and leukotrienes were also seen in the pancreas of conditioned antibody-deficient mice at 18 hr, following likely phospholipid breakdown (Figures 6C, S6A, and 7B). In the blood of antibody-deficient mice, there were also increased host non-esterified fatty acids, as well as glycinated fatty acids (Figure 7C), normally associated with lipid overload and deficient mitochondrial metabolism (Vianey-Liaud et al., 1987). Therefore the lipidemia as a result of bacterial exposure in fasting mice is (1) of both bacterial and host origin and (2) can be triggered by antibody-unprotected exposure of the host to bacterial metabolites in the small intestine.
In contrast, the muscle and the spleen of antibody-deficient animals showed a loss of esterified and non-esterified lipids and increased saccharide intermediates (Figures S6B, S7A, and S7B, Table S3). Fat tissue also showed accumulation of saccharide intermediates under these conditions (Figure S7C). These results are consistent with the established effects of lipotoxicity on lipid and carbohydrate metabolism triggered by circulating non-esterified fatty acids and eicosanoid signaling (Ertunc and Hotamisligil, 2016).
To verify the protective importance of the antibody effect on small intestinal dwell time, we bypassed the effects of microbial conditioning of the intestinal mucosa and antibody control of bacterial transit by direct intraperitoneal injection of identified pure bacterial metabolites. This showed that there were clear responses in different lymphoid compartments, but these no longer consistently differed according to whether or not the mice secrete antibodies, or whether the intestinal mucosa had been preconditioned (Figure S6C and Table S2).
Discussion
The intestinal microbiota is commonly characterized in terms of its composition of different taxa: host responses or a disease phenotype are then correlated with specific taxa or a restricted range of microbial metabolites. Here we have presented a systematic analysis of the potential extent and reach of microbial-host molecular cross-talk from pre-formed endogenous microbial molecules and show that the microbiota itself induces immune and non-immune boundaries for microbial-host metabolite penetration. A few microbial taxa, such as segmented filamentous bacteria, deliver direct signaling through intimate apposition or attachment to the epithelial suface. Yet most of the microbial biomass exists in a highly dynamic system with constant microbial division and death, in the context of continuous transverse and longitudinal flow along the gastrointestinal tract.
Substantial rates of bacterial cell death have been described under steady-state colonization conditions (Maurice et al., 2013). We did not find evidence that the cell wall auxotrophy of HA107 increased the fragility of the organism compared with its non-auxotrophic parent strain, and we could recover viable bacteria from fecal samples. Although challenge HA107 bacteria in these experiments were efficiently recovered from the feces, bacterial death in the mouse intestine certainly contributes to the metabolite load. This is not restricted to the HA107 model: in vivo replication rates and overall dynamics indicate high rates of bacterial death in the intestine (Li et al., 2015, Myhrvold et al., 2015). Partitioning of bacterial molecules from effete cells into the host has parallels with symbiosis in the rhizosphere. Nevertheless, given the inflammatory potential of bacterial molecules, the vulnerability of the host is reduced by antibodies that likely limit its exposure to metabolites from dead bacteria.
Exposure to intestinal microbes drives durable adaptation of the intestinal mucosa through immune and non-immune mechanisms. Beneficial effects of intestinal Ig secretion include toxin and virus neutralization, blocking excessive live bacterial translocation, clearance of unwanted macromolecular structures at the epithelial surface, and directed sampling of luminal antigen (Macpherson et al., 2018). Lack of specific IgA has been shown to increase transcriptional evidence of innate immune system activation in the intestine (Peterson et al., 2007). We now show that antibody induction and secretion decreases microbial motility, accelerates in vivo intestinal transit, and limits exposure of the small intestinal mucosa to the pro-inflammatory molecular constituents of intestinal microbes. Only extremely low titers of specific serum IgG occur after intestinal preconditioning (Gomez de Agüero et al., 2016); we therefore tested the elimination of all antibody isotypes to avoid the compensatory effects of IgM secretion and increased IgG induction in IgA deficiency (Harriman et al., 1999) or the enteropathy that occurs with polymeric immunoglobulin receptor deficiency (Johansen et al., 1999). It is likely that affinity matured secretory IgM can compensate IgA, and a role for IgG isotypes is not excluded.
These effects are poised on the small intestinal mucosa, where limiting bacterial metabolite uptake must be set against the necessity for nutrient absorption. In humans in the developed world, the lower small intestine has a microbial biomass of up to 107 organisms/mL intestinal contents. Globally, microbial colonization of the small intestine remains a severe problem. One of the most urgent biomedical problems of our age is to understand why the presence of high densities of small intestinal microbes in children from the undeveloped world causes intestinal dysfunction and infant malnutrition that cannot be adequately treated with caloric increases (Kau et al., 2015, Smith et al., 2013a). Compensatory IgA that limits microbial metabolite exposure by decreasing the small intestinal dwell time may contribute to the protective effect for taxa that do not attach to or invade the epithelium. Reduction in an intestinal clearance effect is also consistent with the small intestinal lymphonodular hyperplasia in the setting of IgA and affinity matured IgM deficiency, owing to a deletion of activation-induced cytidine deaminase (Fagarasan et al., 2002).
We have distinguished bacterial metabolite exposure and resultant host responses with extremely wide coverage, even where the same molecule can be derived from either bacterial or host metabolism. The results attest to pervasive microbial metabolite exposure, over many classes of metabolites, including increased bacterial lipid penetration. A high proportion of the penetrant microbial metabolites were very rapidly excreted in the urine, although there were also durable increases, some of which benefit the host, such as essential amino acids. To be able to interrogate bacterial metabolite penetration at the same time as the host molecular response using stable isotope tracing, the model was limited to a single taxon; metabolite exposure from other taxa may vary according to their frequencies, flagellate statuses, and metabolic pathway differences.
While the breadth of metabolic exchange allows a broad portfolio of metabolic responses, the predominant effect in antibody-deficient fasting mice was lipidemia of both microbial and host origin, and host lipid breakdown in the intestine and the muscle. Breakdown of phospholipid components in the intestine was also accompanied by prostaglandin and leukotriene synthesis. Extending our model could potentially separate host or dietary molecular effects from microbial molecular effects in the fed as well as the fasting state.
A substantial investment of metabolic energy is made in the secretory IgA response which comprises approximately 75% of all immunoglobulin production in mammals (Macpherson et al., 2008), most of which is secreted in the small intestine. The small intestine is a delicately positioned interface with the environment. On one hand, it is specialized for uptake of nutrients with minimal mucus covering and a combination of digestive and secretory mechanisms. Some of these nutrients are products of microbial metabolism, including vitamins, short chain fatty acids, and compounds capable of receptor signaling mechanisms that mature host organ systems (Gomez de Agüero et al., 2016). On the other hand, the small intestine must handle the inevitable contamination with intestinal microbes that expose the host to a range of compounds that potentially result in innate immune activation: this it does with a combination of physiological adaptations to clear microbial compounds quickly into the urine and secretory immunoglobulins accelerating transit away from the susceptible small intestinal mucosa, thereby modulating both microbial penetration into the host and the resultant host immune and metabolic responses.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-mouse IgA | Bio-Rad / AbD Serotec | Cat# STAR137; RRID: AB_2075638 |
| HRP-conjugated anti-mouse IgA | Sigma-Aldrich | Cat# A4789; RRID: AB_258201 |
| Mouse IgA, k antibody | BD Biosciences | Cat# 553476; RRID: AB_479590 |
| FITC Rat anti-mouse IgA | BD Biosciences | Cat# 559354; RRID: AB_397235 |
| Bacterial and Virus Strains | ||
| E. coli MG1655 | ATCC | ATCC 47076 |
| E. coli JM83 | ATCC | ATCC 35607 |
| E. coli HA107 | Hapfelmeier et al., 2010 | This strain is available from A.J.M. under a material transfer agreement with the University of Bern |
| sDMDMm2 microbiota | Brugiroux et al., 2016, Uchimura et al., 2016 | DSM 26114, 26090, 26085, 28989, 26109, 26113, 26115, 26117, 26074, 32035, 26127, 32036 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 13C-D-glucose | Cambridge Isotope Laboratories | Cat#CLM-1396 |
| 2-[1-14C]-deoxy-D-glucose | Perkin Elmer | Cat#NEC495A001MC |
| Fluorescein isothiocyanate-dextran average molecular weight 70 kDa | Sigma-Aldrich | Cat#46945 |
| Deposited Data | ||
| Raw and analyzed RNA-seq data | This paper | GEO: GSE102543 |
| Experimental Models: Organisms/Strains | ||
| Mus musculus: Strain C57BL/6(J) | University of Zürich | The colony has been bred in the Macpherson laboratory for more than 12 years. |
| Mus musculus: Strain Igh-J−/− | Chen et al., 1993 | The original Igh-J−/− was backcrossed with C57BL/6(J). |
| Software and Algorithms | ||
| R and RStudio | R Consortium | https://www.rstudio.com/ |
| ggplot2 in R | Hadley Wickham | http://ggplot2.org/ |
| ComplexHeatmap in R | Gu et al., 2016 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| GraphPad PRISM Version 6.0h | GraphPad Software | https://www.graphpad.com/ |
| FastQC version 0.11.2 | Simon Andrews | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trimmomatic version 0.33 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| TopHat version 2.0.13 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Samtools version 1.3 | Li et al., 2009 | http://www.htslib.org/ |
| HTSeq version 0.6.1 | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.9.1/ |
| DESeq2 in R | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| STRING version 10.5 | Szklarczyk et al., 2015 | https://string-db.org/ |
| Metamapp | Barupal et al., 2012 | https://github.com/barupal/metamapp |
| Cytoscape 3.4.0 | Smoot et al., 2011 | http://www.cytoscape.org/ |
| Other | ||
| Metabolomics data | This paper | http://www.mucosalimmunology.ch/de/The-lab-University-of-Bern/Supplementary-resources-of-Publications |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Contact, Andrew J. Macpherson (andrew.macpherson@insel.ch).
Experimental Model and Subject Details
Mice
Germ-free C57BL/6(J) and Igh-J−/− (Chen et al., 1993) mice were born and maintained in flexible-film isolators in the Clean Mouse Facility, University of Bern, Switzerland. Age and gender-matched mice were used at 10-12 weeks for the metabolomics samplings in this study. All mice were independently confirmed to be pathogen-free. They were also verified germ-free within the breeding isolators and during experiments (apart from the transient colonization and the colonized experiment shown in Figures S5B and S5C) by culture-dependent and -independent methods. All mouse experiments were performed in accordance with Swiss Federal and Cantonal regulations.
Microbe strains
Escherichia coli HA107 was constructed as an auxotrophic E. coli strain lacking synthetic capabilities of two amino acids, meso-DAP and D-alanine (Hapfelmeier et al., 2010). It grows equivalent to wild-type E. coli via supplementing with 100 μg/mL meso-DAP and 400 μg/mL D-alanine in vitro, and only transiently colonizes germ-free mice due to its auxotroph (Hapfelmeier et al., 2010).
Method Details
Preconditioning the intestinal mucosa through transient bacterial colonization in germ-free mice
E. coli HA107 bacterial overnight cultures were centrifuged at 3480g for 10 min at 4°C. After washing twice with sterile PBS, the required dose of bacteria was resuspended in PBS (1010 CFU per 500 μl). The bacterial solution was gavaged directly into the mouse stomach on alternate days four times. One week after the last HA107 gavage, the feces from the mice were routinely verified by culture-dependent and -independent methods to be germ-free. Control germ-free mice were similarly treated in parallel throughout, although gavaged with an equivalent volume of sterile PBS. Preconditioned mice were studied further 14 days after the last conditioning dose.
Preparation of 13C-labeled bacteria
To prepare 13C-labeled HA107, sterile 13C-labeled bacterial extracts from E. coli MG1655 were used as source of 13C-labeled amino acids and vitamins. For this, E. coli MG1655 was initially cultured in M9 medium using 13C-D-glucose (Cambridge Isotope Laboratories) as sole carbon source. After three sequential subcultures, fully 13C-labeled MG1655 bacterial extracts were obtained and sterilized by filtration (0.22 μm). E. coli HA107 was then cultured in M9 medium containing 13C-D-glucose and 13C-labeled bacterial extracts from E. coli MG1655. After three sequential cultures, the HA107 bacterial culture was centrifuged at 3480 g for 10 min at 4°C. After washing twice with sterile PBS, the required dose of HA107 bacteria was suspended in PBS (1010 CFU per 500 μl). We verified that this protocol achieved essentially complete (> 90%) 13C-labeling of the carbon chains of all bacterial molecules across all compound classes (Figures S1D and S1E).
Mass spectrometry
Samples were snap-frozen in liquid nitrogen. To prepare fluid samples for mass spectrometry, weighed fluid samples (50 μg) were precipitated with final 80% methanol for 1 hour at −20°C. After centrifugation at 20,000 g for 10 min at room temperature, the supernatant was transferred to Q-TOF plates. To prepare tissue samples for mass spectrometry, metabolites from weighed tissue samples (50 μg) were extracted with 20 times weight of Millipore water (80°C) followed by homogenization at 25 Hz for 2 min in a tissue homogenizer (Retsch MM 400) with a clean steel ball (Berani). After centrifugation at 20,000 g for 10 min at room temperature, supernatants were transferred to Q-TOF plates.
Quantification of relative metabolites concentrations was carried out with an Agilent 6550 Q-TOF mass spectrometer (Agilent Technologies Inc., Santa Clara, CA) by non-targeted flow injection analysis as described previously (Fuhrer et al., 2011). Profile spectra with high mass accuracy were recorded from 50 to 1000 m/z in negative ionization mode. The common mass axis after sample alignment was recalibrated using known frequently occurring metabolite ions. Ions were finally annotated based on accurate mass comparison using 1 mDa mass tolerance against 9261 unique metabolites present in a pooled list of compounds derived from the Human Metabolome Database (Wishart et al., 2013), E. coli genome scale-model (Orth et al., 2011) and the E. coli Metabolome Database (Sajed et al., 2016). All potentially possible 13C isotopes were annotated taking into acount the respective sum mass formula.
Determining the extent of 13C-labeling in E. coli HA107 used for stable isotope tracing
The free intracellular metabolites were extracted from the cell pellet with Millipore water at 80°C and analyzed by non-targeted flow injection analysis as described above. Proteinogenic amino acids were obtained from the extracted pellet after hydrolysis with 6 M HCl and subsequently derivatized with N-methyl-N-(tert-butyldimethylsilyl)-trifluoroacetamide as described previously (Nanchen et al., 2007). The 13C-labeling patterns were then determined on a 6890N Network GC system with a 5975 inert XL mass selective detector (Agilent Technologies Inc., Santa Clara, CA). Fractional label for both intracellular metabolites and proteinogenic amino acids was then estimated from the relative abundances of m0 and mmax ion counts as mmax / (m0 + mmax).
Metabolomics data analysis
Log2 fold-changes of 13C-labeled compounds were calculated from the division of [positive ion counts at m+x isotope annotation from 13C-HA107 gavaged samples] by [ion counts at m+x isotope annotation from PBS gavaged samples]. ΔLog2 fold-change was calculated from the subtraction of [Log2 fold-change of germ-free control mice] from [Log2 fold-change of conditioned mice]. Log2 fold-changes of 12C compounds in 13C-HA107 gavaged samples were calculated from the division of [ion counts at m0 annotation from 13C-HA107 gavaged samples] by [ion counts at m0 annotation from PBS gavaged samples]. In the same way, Log2 fold-change of 12C compounds in 12C-HA107 gavaged samples were calculated from the division of [ion counts at m0 annotation from 12C-HA107 gavaged samples] by [ion counts at m0 annotation from PBS gavaged samples]. For the host response of endogenous 12C compounds, compounds with the same direction of Log2 fold-change in both Log2 fold-change of 12C compounds in 13C-HA107 gavaged samples and Log2 fold-change of 12C compounds in 12C-HA107 gavaged samples were taken. Where shown, the Metamapp program was used to evaluate the Tanimoto chemical similarity index on compounds selectively present in one condition applying Cytoscape software for network visualization. Mann Whitney U test was performed using wilcox.test function in R (∗ = p < 0.05, ∗∗ = p < 0.01, ∗∗∗ = p < 0.001).
Preparation of 14C-labeled bacteria
M9 medium supplemented with 100 μg/mL meso-DAP, 400 μg/mL D-alanine, 10 μg/mL lysine, 10 μg/mL threonine, 10 μg/mL methionine, 10 μg/mL proline, 4 μg/mL thiamine, 2 mM L-arabinose, and 5 mM succinate was inoculated with 1/100th amount of E. coli HA107 (or JM83) overnight culture grown in LB medium. The culture was incubated with shaking at 180 rpm at 37°C. When the culture reached OD600 0.28 ± 0.05, 2-[1-14C]-deoxy-D-glucose (Perkin Elmer) was added. The bacterial culture was further incubated for 12-14 hours at 37°C. After harvesting the bacterial culture, the bacterial pellet was washed twice with PBS to remove unincorporated 14C-radioactive materials and resuspended prior to gavage.
Cecal injection
For cecal injection mice were anaesthetized with isoflurane by inhalation (3.0 to 4.0% v/v isoflurane from a vaporizer with O2 flow at 1-2 L/min). An incision (∼1 cm) was made with curved scissors at left side of lower abdomen, and the cecum was located. 14C-labeled HA107 was injected into the middle of cecum and mixed thoroughly with the contents. The abdominal incision was closed with AUTOCLIP Wound Clip System (BD). Temgesic (0.05-0.1 μg/g body weight) was injected subcutaneously for analgesia.
Liquid scintillation counting
Weighed samples were incubated with 1 mL of NCS Tissue Solubilizer (GE Healthcare) at 56°C until completely dissolved. The pH was neutralized with 200 μl of 100% acetic acid. Ultima Gold liquid scintillation cocktail (18 ml, PerkinElmer) was added to each sample. The 14C-radioactivity in each sample was measured in a TRI-CARB 2300TR Liquid Scintillation Analyzer (Packard). Scintillation background for each fluid or tissue type was subtracted using matched samples from control germ-free mice that had not received 14C-labeled bacteria.
RNA isolation
Tissue samples were snap-frozen in TRIzol reagent (Invitrogen) using liquid nitrogen. Thawed tissues in TRIzol reagent were immediately homogenized at 25 Hz for 2 min in a tissue homogenizer (Retsch MM 400) with sterile steel balls (Berani). Total RNA was isolated following the standard protocol from Invitrogen. The total RNA was further purified with RNeasy MinElute kit (QIAGEN). RNA quality was assessed on a Bioanalyzer 2100 (Agilent). Ribosomal RNA was removed with Ribominus Transcriptome Isolation Kit (Invitrogen). RNA libraries were prepared with TruSeq RNA sample preparation v2 kit (TruSeq Stranded mRNA Sample Preparation, Illumina). RNA sequencing was performed with Illumina HiSeq 2500 using the 150bp paired-end mode. Read quality control was with FastQC. Each read was mapped onto the mouse genome using TopHat2. Counts per gene were assessed using HTSeq-count software. Differential gene expression analysis was performed using DESeq2. Adjusted Log2 fold-change and adjusted p-values (corrected for multiple testing) were used to determine significant differences in gene expression. Functional protein association networks of genes of adjusted p-value of < 0.05 were analyzed using the STRING database (version 10) taking network confidence scores ≥ 0.4.
Live intestinal endomicroscopy
Mice investigated with endomicroscopy were anaesthetized with isoflurane inhalation as described for cecal injection on a heating pad at 37°C. The abdomen of the anaesthetized mouse was opened and a Cellvizio confocal miniprobe (Mauna Kea) was inserted into the intestinal ileal lumen. Fluorescein isothiocyanate (FITC)-dextran (0.5 mg, 70 kDa, Sigma) in PBS was injected intravenously to provide real-time vascular contrast. The fluorescence of FITC in the intestinal vasculature was recorded with Cellvizio software (Mauna Kea). Metrics of the endothelial and epithelial layers were evaluated with ImageJ software (NIH).
Enzyme-linked immunosorbent assay (ELISA)
The gastrointestinal tracts from HA107 conditioned germ-free C57BL/6 mice were separated according to section (duodenum, jejunum, cecum and colon). Isolated luminal contents from each section were weighed. IgA concentration was measured with ELISA using goat anti-mouse IgA (Bio-Rad Antibodies) coating antibody. The detection antibody was HRP-conjugated anti-mouse IgA (Sigma). Standards were myeloma-derived purified IgA (Hycult). The IgA concentration in the contents of each germ-free intestinal segment was plotted.
Bacterial immunofluorescent staining
E. coli HA107 was cultured without shaking at 37°C for overnight. The bacteria were then washed in PBS and resuspended at 107 per ml. Portions (50 μl) of the bacterial suspension were dropped onto glass slides and the bacterial drop was encircled using a wax pen. Bacteria were allowed to sediment onto the glass slides and to dry. Transiently flamed bacteria on glass slides were stained with intestinal wash prepared from small intestine of HA107 conditioned germ-free C57BL/6 mice. The IgA antibodies bound to bacteria were visualized with FITC-conjugated anti-mouse IgA (BD Bioscience). DAPI (4’, 6-diamidino-2-phenylindole) was used to counterstain the bacterial DNA. Immunofluorescent pictures were taken using Flash Slide Scanner (PerkinElmer) with 40 magnifications.
Bacterial motility assay
E. coli expressing GFP constitutively were resuspended at 108 CFU/mL with PBS-based intestinal wash from either HA107 conditioned C57BL/6 or Igh-J −/− mice. Bacteria were then loaded to microfluid chips. Serial pictures were taken every second over 5 min by Zeiss LSM 700 confocol microscope. The images were transformed into time-lapse videos with Volocity software (PerkinElmer).
Bacterial intestinal transit time
Germ-free C57BL/6 and Igh-J −/− mice were conditioned with HA107 as described previously. For small intestinal transit, 107 CFU of HA107 were introduced directly into the stomachs of mice. Two hours after the intragastric gavage, small intestines were collected and separated into 10 sections of identical length from duodenum to ileum. All luminal contents in each section were plated on LB agar plates with the auxotrophic supplements and results were evaluated for each segment. To measure whole intestinal transit time, 1010 CFU of HA107 were introduced to the stomachs of mice. Feces were sequentially collected and cultured as described previously.
Metabolite-specific ELISA
Metabolite-specific ELISA was performed to address the binding of intestinal IgA to purified identified metabolites including pentadecanoic acid (Sigma); 5-methoxytryptamine (Sigma); N-acetylaspartylglutamic acid (Sigma); scopolamine hydrochloride (Sigma); D-maltose (VWR) and heptadecanoic acid (Sigma). E.coli LPS (Sigma) and DNA from E. coli were used as controls. ELISA plates (Milian) were coated with 50μl/well of 10μg/mL antigen overnight (ON) using coating buffer (0.1M NaHCO3) except pentadecanoic acid and heptadecanoic acid coated using 100% ethanol. After blocking (PBS with 2% BSA, RT, 2h), serial diluted intestinal washes containing IgA were added to the plates and incubated for 2 hours. To reveal the IgA binding affinity to metabolites, HRP conjugated anti-IgA antibody (1:100, Sigma) was added to each well and incubated for 45 min. Plates were then washed (4X, PBS containing 0.5% Tween-20) and developing reagent (100 μL per well, 1mg ABTS plus 5μl H2O2/10ml 0.1 M NaH2PO4) was added. Samples then were read at OD415 using an ELISA plate reader.
Cytokine release assays
Mice used in cytokine release assays were preconditioned with HA107 as described in Method Preconditioning. Peritoneal cells were collected by gentle flushing of the peritoneal cavity with RPMI 1640 medium; splenocytes, mesenteric lymph node cells (MLN cells) and bone marrow cells harvested from mouse femur were isolated using a 100μm cell strainer. After red blood cell lysis, 2 × 106 cells/mL were prepared in complete medium (RPMI 1640 with 10% FBS, 50 μM β-mercaptoethanol, 100 U/mL penicillin/streptomycin and 2mM HEPES). A 100μl suspension was seeded in each well of 96-well cell culture plates (Thermo Fisher). The natural cytokine release from cells was determined after incubating at 37°C supplemented with 5% CO2 for 2 hours. For in vitro stimulation with metabolites/bacteria-derived components, 250μg/mL of each compound as listed was added to the culture and incubation was carried out for 6 hours. The culture supernatants were collected for further analysis.
Intraperitoneal challenge with pentadecanoic acid or 5-methoxytryptamine
Mice were preconditioned with HA107 or left untreated. Two weeks after the last oral bacterial priming, mice were intraperitoneally injected with 1mg/kg body weight pentadecanoic acid or 5-methoxytryptamine. 6 hours after injection, cells were isolated from indicated lymphoid organs and cultured for 2 hours. The supernatants were collected for further analysis.
Cytokine quantification
Cytokine measurements were performed using the Meso Scale Discovery (MSD) system. Samples were initially diluted 1:1 with commercial MSD diluent buffer and added into Meso Scale 19-plex plates. Serial titrations of cytokine standards were also added to the plates. After overnight incubation, plates were washed (3X, PBS with 0.5% Tween-20) and a panel of commercial 19 secondary antibodies was added to plates and incubated for 2 hours following the manufacturer’s protocol and instructions. Plates were then washed (3X, PBS containing 0.5% Tween-20), and 150μl substrate buffer was added into each well. Plates were then read on Meso Scale plate reader.
Quantification and Statistical Analysis
Statistical parameters including the definitions and exact values of n (e.g., number of experiments, number of mice, etc) and distributions are reported in the Figure Legends. Mann-Whitney U test was used for all metabolomic comparisons and unpaired student-t test was performed in intestinal transit tests and microbial culture related tests. Statistical analysis was performed in GraphPad Prism and R software. In all figures: ∗ = p < 0.05, ∗∗ = p < 0.01, ∗∗∗ = p < 0.001.
Data and Software Availability
The RNA sequencing data files have been deposited in the NCBI Gene Expression Omnibus under accession number GEO:GSE102543. String analysis (https://string-db.org/) has been used to elucidate the RNA sequencing data. All original metabolomic data are available at http://www.mucosalimmunology.ch/de/The-lab-University-of-Bern/Supplementary-resources-of-Publications.
Acknowledgments
We thank the staff of the clean mouse facility of the University of Bern and Professors Wolf Hardt and Christoph Mueller for their comments. We thank Jamie Macpherson, Dr. Irene Keller, Dr. Fabian Blank, and Carlos A. Wotzkow for assistance. The work was supported by the Genaxen Foundation, the Swiss National Science Foundation (SNSF 310030B_160262 and SNF Sinergia CRSII3_136286 to A.J.M. and SNSF Sinergia CRSII3_154414 to A.J.M. and U.S.), and the Systems X program (GutX A.J.M. and U.S.).
Author Contributions
A.J.M. and U.S. conceived and designed the project. A.J.M., U.S., and K.D.M. supervised experiments. Y.U., H.L., M.A.L., A.J.M., M.G.A., B.Y., F.R., S.C.G.-V., and M.S. performed metabolomics sampling and bacterial experiments. T.F. and M.Z. performed mass spectrometry and data processing. Y.U., T.F., and A.J.M. performed bioinformatics analysis. H.L. and M.A.L. performed intestinal bacterial transit experiments. H.L., J.Z., and B.Y. performed cytokine experiments. S.H. provided E. coli HA107. A.J.M., U.S., Y.U., H.L., and T.F. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 4, 2018
Footnotes
Supplemental Information includes seven figures, three tables, and two videos and can be found with this article online at https://doi.org/10.1016/j.immuni.2018.08.004.
Contributor Information
Uwe Sauer, Email: sauer@imsb.biol.ethz.ch.
Andrew J. Macpherson, Email: andrew.macpherson@insel.ch.
Supplemental Information
References
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N., Campbell C., Fan X., Dikiy S., van der Veeken J., deRoos P., Liu H., Cross J.R., Pfeffer K., Coffer P.J., Rudensky A.Y. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F., Roswall J., Peng Y., Feng Q., Jia H., Kovatcheva-Datchary P., Li Y., Xia Y., Xie H., Zhong H. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:690–703. doi: 10.1016/j.chom.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Barupal D.K., Haldiya P.K., Wohlgemuth G., Kind T., Kothari S.L., Pinkerton K.E., Fiehn O. MetaMapp: mapping and visualizing metabolomic data by integrating information from biochemical pathways and chemical and mass spectral similarity. BMC Bioinformatics. 2012;13:99. doi: 10.1186/1471-2105-13-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty D.W., Napier B., Sinclair-Smith C.C., McCabe K., Hughes E.J. Secretory IgA synthesis in Kwashiorkor. J. Clin. Lab. Immunol. 1983;12:31–36. [PubMed] [Google Scholar]
- Belkaid Y., Harrison O.J. Homeostatic immunity and the microbiota. Immunity. 2017;46:562–576. doi: 10.1016/j.immuni.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg R., Powrie F. Microbiota, disease, and back to health: a metastable journey. Sci. Transl. Med. 2012;4:137rv7. doi: 10.1126/scitranslmed.3004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugiroux S., Beutler M., Pfann C., Garzetti D., Ruscheweyh H.J., Ring D., Diehl M., Herp S., Lötscher Y., Hussain S. Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat. Microbiol. 2016;2:16215. doi: 10.1038/nmicrobiol.2016.215. [DOI] [PubMed] [Google Scholar]
- Bunker J.J., Flynn T.M., Koval J.C., Shaw D.G., Meisel M., McDonald B.D., Ishizuka I.E., Dent A.L., Wilson P.C., Jabri B. Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity. 2015;43:541–553. doi: 10.1016/j.immuni.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunker J.J., Erickson S.A., Flynn T.M., Henry C., Koval J.C., Meisel M., Jabri B., Antonopoulos D.A., Wilson P.C., Bendelac A. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science. 2017;358:358. doi: 10.1126/science.aan6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Trounstine M., Alt F.W., Young F., Kurahara C., Loring J.F., Huszar D. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int. Immunol. 1993;5:647–656. doi: 10.1093/intimm/5.6.647. [DOI] [PubMed] [Google Scholar]
- Cotran R., Kendrick M.I., Kass E.H. Role of intestinal bacteria in aromatization of quinic acid in man and guinea pig. Proc. Soc. Exp. Biol. Med. 1960;104:424–426. doi: 10.3181/00379727-104-25860. [DOI] [PubMed] [Google Scholar]
- Cullender T.C., Chassaing B., Janzon A., Kumar K., Muller C.E., Werner J.J., Angenent L.T., Bell M.E., Hay A.G., Peterson D.A. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe. 2013;14:571–581. doi: 10.1016/j.chom.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayman J., Jepson J.B. The metabolism of caffeic acid in humans: the dehydroxylating action of intestinal bacteria. Biochem. J. 1969;113:11P. doi: 10.1042/bj1130011p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf A.A., Maathuis A., de Waard P., Deutz N.E., Dijkema C., de Vos W.M., Venema K. Profiling human gut bacterial metabolism and its kinetics using [U-13C]glucose and NMR. NMR Biomed. 2010;23:2–12. doi: 10.1002/nbm.1418. [DOI] [PubMed] [Google Scholar]
- Donowitz J.R., Haque R., Kirkpatrick B.D., Alam M., Lu M., Kabir M., Kakon S.H., Islam B.Z., Afreen S., Musa A. Small intestine bacterial overgrowth and environmental enteropathy in Bangladeshi children. MBio. 2016;7:e02102–e02115. doi: 10.1128/mBio.02102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Aidy S., Merrifield C.A., Derrien M., van Baarlen P., Hooiveld G., Levenez F., Doré J., Dekker J., Holmes E., Claus S.P. The gut microbiota elicits a profound metabolic reorientation in the mouse jejunal mucosa during conventionalisation. Gut. 2013;62:1306–1314. doi: 10.1136/gutjnl-2011-301955. [DOI] [PubMed] [Google Scholar]
- Ertunc M.E., Hotamisligil G.S. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016;57:2099–2114. doi: 10.1194/jlr.R066514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagarasan S., Muramatsu M., Suzuki K., Nagaoka H., Hiai H., Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. 2002;298:1424–1427. doi: 10.1126/science.1077336. [DOI] [PubMed] [Google Scholar]
- Fuhrer T., Heer D., Begemann B., Zamboni N. High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Anal. Chem. 2011;83:7074–7080. doi: 10.1021/ac201267k. [DOI] [PubMed] [Google Scholar]
- Fuhrer T., Zampieri M., Sévin D.C., Sauer U., Zamboni N. Genomewide landscape of gene-metabolome associations in Escherichia coli. Mol. Syst. Biol. 2017;13:907. doi: 10.15252/msb.20167150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensollen T., Iyer S.S., Kasper D.L., Blumberg R.S. How colonization by microbiota in early life shapes the immune system. Science. 2016;352:539–544. doi: 10.1126/science.aad9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez de Agüero M., Ganal-Vonarburg S.C., Fuhrer T., Rupp S., Uchimura Y., Li H., Steinert A., Heikenwalder M., Hapfelmeier S., Sauer U. The maternal microbiota drives early postnatal innate immune development. Science. 2016;351:1296–1302. doi: 10.1126/science.aad2571. [DOI] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Hapfelmeier S., Lawson M.A., Slack E., Kirundi J.K., Stoel M., Heikenwalder M., Cahenzli J., Velykoredko Y., Balmer M.L., Endt K. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. 2010;328:1705–1709. doi: 10.1126/science.1188454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriman G.R., Bogue M., Rogers P., Finegold M., Pacheco S., Bradley A., Zhang Y., Mbawuike I.N. Targeted deletion of the IgA constant region in mice leads to IgA deficiency with alterations in expression of other Ig isotypes. J. Immunol. 1999;162:2521–2529. [PubMed] [Google Scholar]
- Hooper L.V., Wong M.H., Thelin A., Hansson L., Falk P.G., Gordon J.I. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- Johansen F.E., Pekna M., Norderhaug I.N., Haneberg B., Hietala M.A., Krajci P., Betsholtz C., Brandtzaeg P. Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J. Exp. Med. 1999;190:915–922. doi: 10.1084/jem.190.7.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M.E., Phillipson M., Petersson J., Velcich A., Holm L., Hansson G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones H.F., Davidson G.P., Brooks D.A., Butler R.N. Is small-bowel bacterial overgrowth an underdiagnosed disorder in children with gastrointestinal symptoms? J. Pediatr. Gastroenterol. Nutr. 2011;52:632–634. doi: 10.1097/MPG.0b013e31820d5c16. [DOI] [PubMed] [Google Scholar]
- Kau A.L., Planer J.D., Liu J., Rao S., Yatsunenko T., Trehan I., Manary M.J., Liu T.C., Stappenbeck T.S., Maleta K.M. Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci. Transl. Med. 2015;7:276ra24. doi: 10.1126/scitranslmed.aaa4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpe P.S., Petri W.A., Jr. Environmental enteropathy: critical implications of a poorly understood condition. Trends Mol. Med. 2012;18:328–336. doi: 10.1016/j.molmed.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Limenitakis J.P., Fuhrer T., Geuking M.B., Lawson M.A., Wyss M., Brugiroux S., Keller I., Macpherson J.A., Rupp S. The outer mucus layer hosts a distinct intestinal microbial niche. Nat. Commun. 2015;6:8292. doi: 10.1038/ncomms9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson A.J., Gatto D., Sainsbury E., Harriman G.R., Hengartner H., Zinkernagel R.M. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288:2222–2226. doi: 10.1126/science.288.5474.2222. [DOI] [PubMed] [Google Scholar]
- Macpherson A.J., McCoy K.D., Johansen F.E., Brandtzaeg P. The immune geography of IgA induction and function. Mucosal Immunol. 2008;1:11–22. doi: 10.1038/mi.2007.6. [DOI] [PubMed] [Google Scholar]
- Macpherson A.J., Yilmaz B., Limenitakis J.P., Ganal-Vonarburg S.C. IgA function in relation to the intestinal microbiota. Annu. Rev. Immunol. 2018;36:359–381. doi: 10.1146/annurev-immunol-042617-053238. [DOI] [PubMed] [Google Scholar]
- Maurice C.F., Haiser H.J., Turnbaugh P.J. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50. doi: 10.1016/j.cell.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhrvold C., Kotula J.W., Hicks W.M., Conway N.J., Silver P.A. A distributed cell division counter reveals growth dynamics in the gut microbiota. Nat. Commun. 2015;6:10039. doi: 10.1038/ncomms10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanchen A., Fuhrer T., Sauer U. Determination of metabolic flux ratios from 13C-experiments and gas chromatography-mass spectrometry data: protocol and principles. Methods Mol. Biol. 2007;358:177–197. doi: 10.1007/978-1-59745-244-1_11. [DOI] [PubMed] [Google Scholar]
- Nicholls A.W., Mortishire-Smith R.J., Nicholson J.K. NMR spectroscopic-based metabonomic studies of urinary metabolite variation in acclimatizing germ-free rats. Chem. Res. Toxicol. 2003;16:1395–1404. doi: 10.1021/tx0340293. [DOI] [PubMed] [Google Scholar]
- Orth J.D., Conrad T.M., Na J., Lerman J.A., Nam H., Feist A.M., Palsson B.O. A comprehensive genome-scale reconstruction of Escherichia coli metabolism--2011. Mol. Syst. Biol. 2011;7:535. doi: 10.1038/msb.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan P., Grosse J., Asad A.B., Radda G.K., Golay X. Gastrointestinal transit measurements in mice with 99mTc-DTPA-labeled activated charcoal using NanoSPECT-CT. EJNMMI Res. 2013;3:60. doi: 10.1186/2191-219X-3-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry R.J., Peng L., Barry N.A., Cline G.W., Zhang D., Cardone R.L., Petersen K.F., Kibbey R.G., Goodman A.L., Shulman G.I. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature. 2016;534:213–217. doi: 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson D.A., McNulty N.P., Guruge J.L., Gordon J.I. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe. 2007;2:328–339. doi: 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Petersson J., Schreiber O., Hansson G.C., Gendler S.J., Velcich A., Lundberg J.O., Roos S., Holm L., Phillipson M. Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011;300:G327–G333. doi: 10.1152/ajpgi.00422.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajed T., Marcu A., Ramirez M., Pon A., Guo A.C., Knox C., Wilson M., Grant J.R., Djoumbou Y., Wishart D.S. ECMDB 2.0: A richer resource for understanding the biochemistry of E. coli. Nucleic Acids Res. 2016;44(D1):D495–D501. doi: 10.1093/nar/gkv1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman N.H., Hung K., Haribhai D., Chu H., Karlsson-Sjöberg J., Amir E., Teggatz P., Barman M., Hayward M., Eastwood D. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sévin D.C., Fuhrer T., Zamboni N., Sauer U. Nontargeted in vitro metabolomics for high-throughput identification of novel enzymes in Escherichia coli. Nat. Methods. 2017;14:187–194. doi: 10.1038/nmeth.4103. [DOI] [PubMed] [Google Scholar]
- Smith K., McCoy K.D., Macpherson A.J. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 2007;19:59–69. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Smith M.I., Yatsunenko T., Manary M.J., Trehan I., Mkakosya R., Cheng J., Kau A.L., Rich S.S., Concannon P., Mychaleckyj J.C. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science. 2013;339:548–554. doi: 10.1126/science.1229000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P.M., Howitt M.R., Panikov N., Michaud M., Gallini C.A., Bohlooly-Y M., Glickman J.N., Garrett W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer F., Bäckhed F. The gut microbiota--masters of host development and physiology. Nat. Rev. Microbiol. 2013;11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- Stappenbeck T.S., Hooper L.V., Gordon J.I. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc. Natl. Acad. Sci. USA. 2002;99:15451–15455. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock G.W., Lawley B., Munro K., Sims I.M., Lee J., Butts C.A., Roy N. RNA-stable-isotope probing shows utilization of carbon from inulin by specific bacterial populations in the rat large bowel. Appl. Environ. Microbiol. 2014;80:2240–2247. doi: 10.1128/AEM.03799-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura Y., Wyss M., Brugiroux S., Limenitakis J.P., Stecher B., McCoy K.D., Macpherson A.J. Complete genome sequences of 12 species of a stable defined moderately diverse microbiota 2. Genome Announc. 2016;4 doi: 10.1128/genomeA.00951-16. e00951–e00916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S., Yamamoto M., Severson K.M., Ruhn K.A., Yu X., Koren O., Ley R., Wakeland E.K., Hooper L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vianey-Liaud C., Divry P., Gregersen N., Mathieu M. The inborn errors of mitochondrial fatty acid oxidation. J. Inherit. Metab. Dis. 1987;10(Suppl 1):159–200. doi: 10.1007/BF01812855. [DOI] [PubMed] [Google Scholar]
- Williams R.E., Eyton-Jones H.W., Farnworth M.J., Gallagher R., Provan W.M. Effect of intestinal microflora on the urinary metabolic profile of rats: a (1)H-nuclear magnetic resonance spectroscopy study. Xenobiotica. 2002;32:783–794. doi: 10.1080/00498250210143047. [DOI] [PubMed] [Google Scholar]
- Wishart D.S., Jewison T., Guo A.C., Wilson M., Knox C., Liu Y., Djoumbou Y., Mandal R., Aziat F., Dong E. HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Small intestinal wash were made by flushing freshly isolated small intestine with 5ml ice-cold PBS. 108/ml GFP expressing E. coli MG1655 were incubated with intestinal wash prepared from HA107 preconditioned Igh-J−/− mice (Video S1) or C57BL/6 (Video S2) for 30 min on ice prior to confocal microscopy acquisition. The bacteria were then loaded to micro-fluid chip (Absis). Zeiss LSM 700 confocol microscopy were used to acquire pictures and videos were re-constructed from pictures using Volocity software.