

Abstract
New applications of bioluminescence imaging require an expanded set of luciferase enzymes and luciferin substrates. Here we report two novel luciferins for use in vitro and in cells. These molecules comprise regioisomeric pyridone cores that can be accessed from a common synthetic route. The analogs exhibited unique emission spectra with firefly luciferase, although photon intensities remained weak. Enhanced light outputs were achieved using mutant luciferase enzymes. One of the luciferin-luciferase pairs produced light on par with native probes in live cells. The pyridone analogs and complementary luciferases add to a growing set of designer probes for bioluminescence imaging.
Keywords: bioluminescence, imaging, pyridone, luciferin, luciferase
Introduction
Bioluminescence enables sensitive imaging of biological events in vitro and in vivo.[1–2] This method employs luciferase enzymes that catalyze the oxidation of small molecule luciferins, releasing visible light in the process[3–4] (Scheme 1A). Several luciferase-luciferin pairs have been identified in nature,[5] but only a handful have been optimized for routine use in cells and living organisms.[6–10] Among the most popular is firefly luciferase (Fluc) and D-luciferin (1). This bioluminescent pair emits the largest percentage of tissue-penetrant light among well characterized enzymes and substrates.[11] Consequently, Fluc and D-luciferin have been widely used to track cell movements,[12–13] gene expression patterns[14–15] and other activities[16–18] in a variety of pre-clinical models.
Scheme 1.
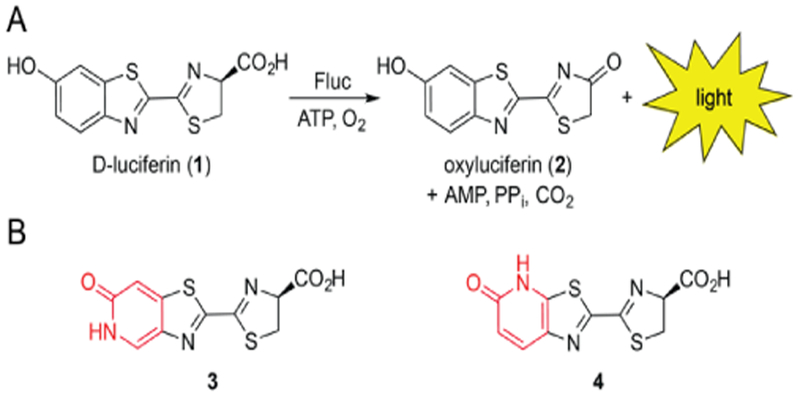
A) Firefly luciferase catalyzes the oxidation of D-luciferin, producing a photon of light. B) Luciferin analogs 3 and 4 bearing pyridone moieties (red) were examined in this study.
While versatile, bioluminescence imaging with Fluc/D-luciferin has some notable shortcomings. D-Luciferin is not optimally cell permeant,[19] requiring large doses to saturate Fluc and achieve robust photon production in cultured cell and tissue models.[20] Imaging with Fluc has also been historically limited to visualizing one feature at a time owing to a lack of distinguishable probes.[2, 21] To craft improved bioluminescent tools, many groups have relied on Fluc mutagenesis to isolate “brighter” and spectrally resolved luciferases.[22–24] A complementary and potentially more general approach involves modulating the luciferin itself. The small molecule is the effective light emitter; thus, modifications to the scaffold can, in principle, impact both pharmacokinetic and spectral properties. There have been many notable successes in engineering unique luciferins in recent years.[25–37] However, many of the scaffolds exhibit reduced photon outputs with luciferase,[25,30,32–37] limiting their use in some cellular and tissue environments.
To further expand the scope of bioluminescence imaging, we aimed to develop a new class of spectrally resolved luciferins for cellular application. We initially focused on scaffolds comprising a pyridone moiety (3 and 4, Scheme 1B). Pyridones are found in a number of drug molecules,[38–40] suggesting that they would be sufficiently stable under physiological conditions. We further reasoned that 3 and 4 would be good substrates for Fluc or related mutants, given their structural similarity to the native substrate. Pyridones can also tautomerize in the excited state and thus potentially provide unique colors of light (Figure S1). Well-behaved luciferins with distinguishable spectra would be useful additions to the bioluminescence toolkit.
Results and Discussion
Computational analysis of light-emitting molecules
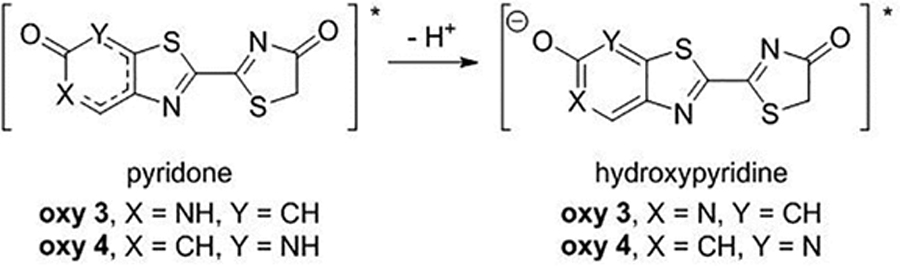
We reasoned that the Fluc-catalyzed oxidation of 3-4 would provide either a pyridone (neutral) or a deprotonated hydroxypyridine (from the corresponding tautomer, Table 1) as the functional light emitter. Luciferins and related chromophores are known to undergo proton transfers[41–43] in the excited state. In the case of the pyridones, deprotonation would provide fully aromatic architectures. Such species would likely emit visible light conducive to cellular imaging applications. To gain insight on the predicted wavelengths, we performed density functional theory (DFT) calculations. The relative HOMO and LUMO energies of the oxyluciferin emitters (Table S1) were used to approximate bioluminescent wavelengths.[44] The predicted emission for the pyridone structures were >100-nm blue-shifted compared to the corresponding hydroxypyridines. Luciferin oxy3 was also predicted to release ~50 nm more red-shifted light than oxy4 (Tables 1 and S1). A differential of this magnitude can easily be resolved with optical filters,[45–46] further suggesting that 3 and 4 could be viable candidates for multispectral imaging.
Table 1.
Predicted ΔEHOMO-LUMO and observed bioluminescent λem for the pyridone and hydroxypyridine luciferin tautomers.
 | |||
|---|---|---|---|
| compound | pyridone (nm) | hydroxypyridine (nm) | λem(nm) |
| oxy3 | 403 | 582 | 570 |
| oxy4 | 349 | 510 | 530 |
Wavelengths were calculated from predicated HOMO/LUMO energies from DFT calculations (B3LYP functional and 6–311+G** basis set)
Synthesis of isomeric pyridone luciferins
We prepared the luciferins using a route previously developed in our group[25, 47] (Scheme 2). This method relies on an auxiliary dithiazolium reagent (Appel’s salt, 5)[48] to access the annulated thiazole of the luciferin core. Appel’s salt adducts can be readily formed and fragmented to provide cyanothioformamides[48] in a single pot; these latter intermediates are good candidates for cyclization via C-H activation.[49] To prepare the adduct en route to the pyridone luciferins, aniline 6 was first condensed with Appel’s salt 5. The resulting product (7) was subsequently fragmented[48] to access cyanothioformamide 8. Oxidative cyclization of 8 with a Pd catalyst[49] provided intermediates 9a-b. These regioisomers could be readily separated via flash column chromatography. The cyclized isomers were then subjected to pyridine hydrochloride at elevated temperatures to remove the methyl protecting groups. Some undesired N-methylation was observed during this process. Fortunately, the unwanted byproducts could be readily separated from the desired cyanobenzothiazoles upon treatment with acetic anhydride and subsequent chromatography. Condensation of 10 and 11 with D-cysteine (under basic conditions) ultimately afforded luciferins 3 and 4.
Scheme 2.
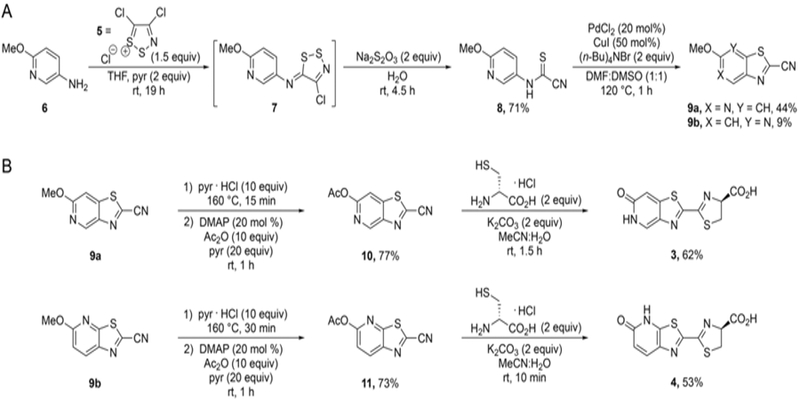
Synthesis of pyridone luciferins. A) Formation of the thiazolo-pyridyl core. B) Condensations with D-cysteine provided the desired analogs.
Bioluminescence assays with luciferin analogs
With the desired pyridone luciferins in hand, we evaluated their light-emitting properties. Analogs 3 and 4 were first incubated with Fluc in the presence of ATP. Light emission was measured using a cooled CCD camera (Figure 1A-B). Dose-dependent photon production was observed with both analogs, although the outputs were lower than that achieved with the native substrate, D-luciferin (Figure S2A). At saturating doses of 3 or 4, light emission reached ~1–3% of output achieved with the native pair. While weak, such outputs are on par with other luciferins comprising altered heterocyclic cores.[25, 30, 34–35]
Figure 1.
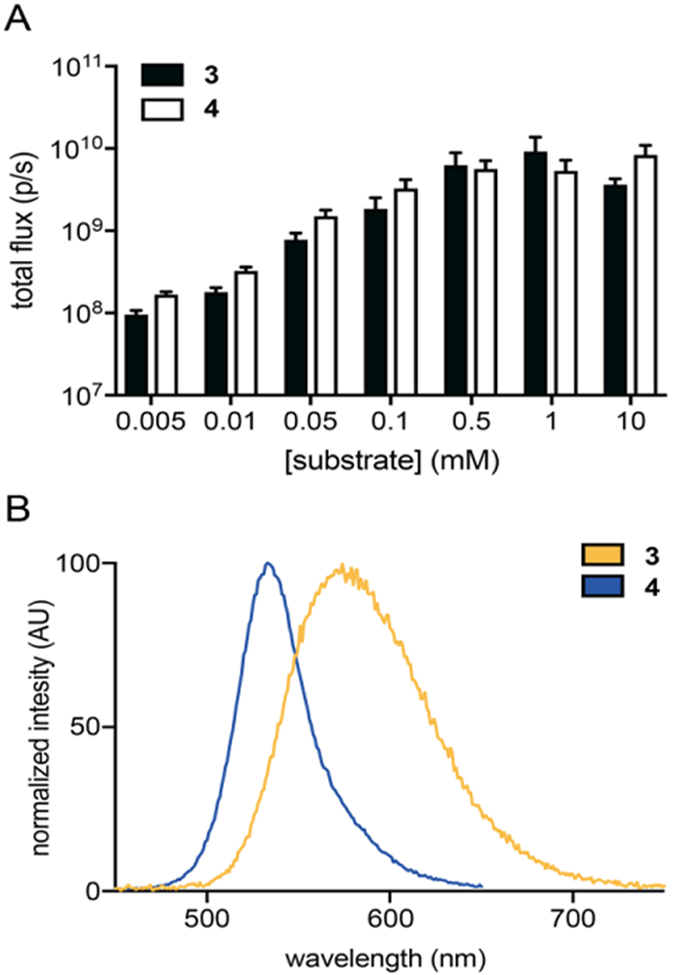
Fluc catalyzes light emission with pyridone analogs. A) Pyridone analogs exhibit dose-dependent light emission. Analogs 3 (black) and 4 (white) (5 μM – 10 mM) were incubated with Fluc (1 μg) in imaging buffer. Images were acquired immediately post-Fluc addition. Error bars represent the standard error of the mean for n ≥ 3 experiments. B) Bioluminescence emission spectra for 3 (λmax = 570 nm) and 4 (λmax = 530 nm). Each analog was incubated with Fluc in imaging buffer and emission spectra were acquired over a range of wavelengths.
As predicted, the luciferin analogs exhibited distinct emission profiles. The bioluminescence spectra for 3 and 4 are shown in Figure 1B. The peak emission for pyridone 3 (λem = 570 nm) was ~40 nm red-shifted compared to analog 4 (λem = 530 nm). These wavelengths flank the emission spectrum for D-luciferin[50] (λem = 566 nm, Figure S2B), providing further evidence that modifications to the luciferin heterocycle can impact the color of bioluminescent light. The emission maxima also matched the values predicted for the hydroxypyridine (anionic) emitters shown in Table 1. The spectra did not vary from pH 7–9 (Figure S3), further suggesting that the light emitter is a single species over this physiological range. The emission wavelength of D-luciferin, by contrast, varies under these conditions.[50]
While photophysical data suggested that the oxidized forms of 3 and 4 emit light as hydroxypyridines, the molecules appear to exist as pyridones in the ground state (Figures S4-S5). NMR analyses were used to compare 3 and 4 to methylated and tautomerically locked analogs (12 and 13, Schemes S1-S2). The aryl proton resonances of 3 and 4 were shifted upfield relative to the locked analogs, consistent with the pyridone assignment[51] (Figures S4-S5). Deprotonation of the analogs was observed at pH 9.5 (Figure S6), though, suggesting that N-H proton is susceptible to transfer in the excited state similar to the phenolic O-H in D-luciferin.[41–42]
Cellular bioluminescence imaging
The pyridone luciferins were sufficiently cell permeable for bioluminescence imaging applications. Analogs 3 and 4 (0.01–5 mM) were incubated with Fluc-expressing HEK293 cells. Photon emission was measured over time. Stronger light outputs were observed with increasing concentrations of both analogs (Figure 2). At saturating doses (~ 5 mM), maximum light emission was achieved 20 minutes post-luciferin administration (Figure S7). The intensities were ~3–20% of the total photon flux observed with D-luciferin in the same cells (Figure S7A). Light emission was also sustained for 1 h, suggesting that the pyridone analogs are suitable for a variety of cellular assays (Figure S7B). Analogs 3 and 4 could also be distinguished in cells based on their unique spectra (Figure S8A). As noted earlier, the emission maxima for 3 and 4 are ~50 nm resolved, which is on par with some luciferases used for dual color imaging.[45, 52]
Figure 2.
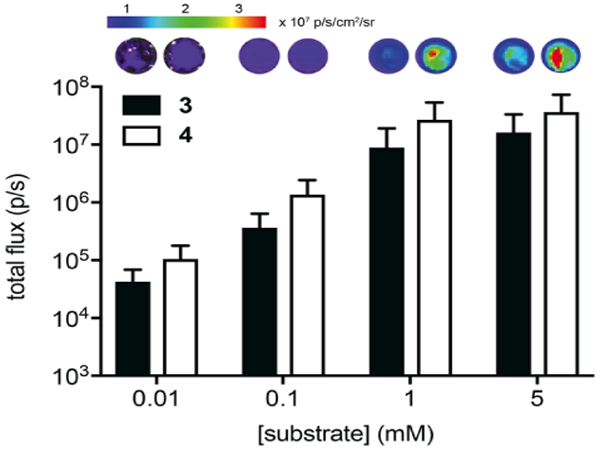
Pyridone analogs emit light in cells. Analogs 3 and 4 (0.01–5 mM) were incubated with Fluc-expressing HEK293 cells in PBS (pH 7.6). Images were acquired 10 min post-luciferin addition. Error bars represent the standard error of the mean for n ≥ 3 experiments. Sample images are shown.
Improved imaging with mutant luciferases
While the pyridone analogs were sufficiently robust for use in cellular imaging experiments, biochemical analyses suggested room for improvement. The apparent Km values measured for both luciferins were in the low millimolar range (Figure S9 and Table S2). These values are >100-fold greater than the Km for D-luciferin,[53] suggesting that the pyridones are poor binders of Fluc. To identify enzymes that could better process the analogs, we incubated luciferin 4 with a panel of 166 mutant luciferases in bacterial lysate.[54] These enzymes were previously identified from screens with sterically modified luciferins[54] and were thus known to be functionally active. Upon treatment with 4, ten of the mutant enzymes exhibited 10–80-fold brighter outputs compared to Fluc (Figure S10) in lysate.
The two “brightest” mutants (24 and 166) were selected for isolation and further characterization (Table S3). Upon purification from bacterial expression systems, the luciferases maintained enhanced photon outputs with analog 4 (Figure 3A and S11). Interestingly, increased light emission was not observed with isomeric luciferin 3 (Figure 3A). Enzymes 24 and 166 comprise M249L, S314T and G316T mutations. S314T and G316T have been previously shown to improve the catalytic efficiency of Fluc.[54] Furthermore, the G316A mutation is known to affect bioluminescent color,[53] which may suggest a change in the polarity of the active site.[55] Mutant 166 further comprised an F295L mutation. This mutation is found in thermostable luciferases.[23] The Km values measured for luciferin 4 and each mutant were similar to that of native Fluc, suggesting no improvement in substrate affinity (Tables S2, 4 and 5). Thus, the mutated residues are likely contributing to a change in active site architecture that promotes more favorable substrate turnover.
Figure 3.
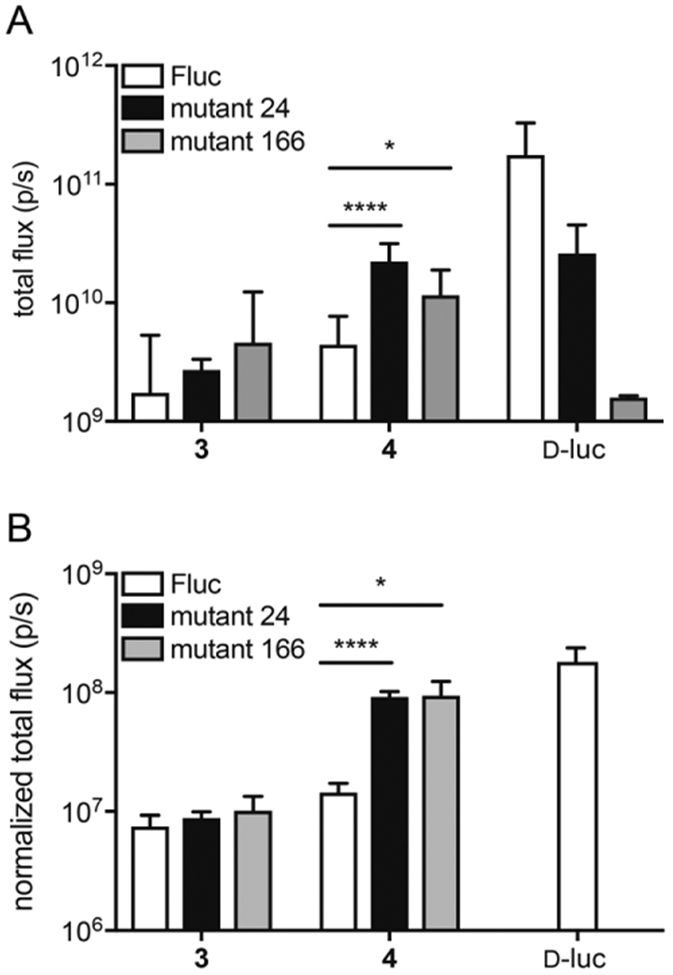
Bioluminescent photon production from mutant luciferase enzymes and luciferin analogs. A) Analogs 3-4 (1000 μM) or D-luciferin (D-luc) were incubated with mutant luciferases (24 or 166) or native Fluc and ATP. Light emission was measured and quantified. Error bars represent the standard error of the mean for n ≥ 3 experiments. B) HEK 293 cells expressing mutant luciferases (24 or 166) or native Fluc were incubated with analogs 3-4 (1000 μM) or D-luciferin (D-luc). Error bars represent the standard error of the mean for n ≥ 3 experiments. For A) and B), p-values (from one-tailed unpaired t tests) are annotated by asterisks (*p < 0.05, **** p < 0.0001).
Encouraged by the in vitro findings, we analyzed mutants 24 and 166 in cellular studies. Plasmids encoding the luciferases were transfected into HEK293 cells. Luciferase expression was verified and normalized to a fluorescent reporter (GFP) in each case (Figure S12). The cells were incubated with either analog 3 or 4 and bioluminescent photon production was measured. The light output obtained from cells expressing mutant 24 or 166 (in the presence of analog 4) was on par with bioluminescence levels obtained from conventional imaging tools (Fluc/D-luciferin, Figure 3B). Similar to the in vitro studies, no improvement in the light output with analog 3 was obtained. The bioluminescence emission spectra from both pyridone analogs could still be resolved in this setting (Figure S8B-C), though, suggesting that the engineered pairs will be useful for multicomponent imaging.
Conclusions
In summary, two regioisomeric pyridone luciferins were designed as new bioluminescent probes. Computational studies suggested that these molecules would emit unique colors of light that could be spectrally resolved in conventional imaging experiments. The regioisomeric scaffolds were synthesized using a common route and shown to be capable of light emission with the bioluminescent enzyme, firefly luciferase (Fluc). The pyridones are capable of tautomerization during the light emitting process, but under the conditions examined, the emission spectra remained constant. The luciferin analogs also are sufficiently cell permeant to enable bioluminescence in Fluc-expressing cells. Further improvements in light output were achieved using mutant luciferase enzymes. The light levels achieved were on par with native bioluminescent tools. These novel luciferase-luciferin pairs complement a growing number of unique bioluminescent tools, including luciferases that use orthogonal substrates.[29,54,56] A collection of bioluminescent probes will enable a variety of multicellular and other multicomponent imaging applications.
Experimental Section
Computational methods
Calculations were performed with Spartan Student Edition Version 6. Organic structures were modeled and energy minimized. ΔEHOMO-LUMO values were determined using the B3LYP level of theory and basis set 6–311+G**.
Recombinant protein expression and purification
Plasmids encoding Fluc or Fluc mutants (pET vectors) were transformed into chemically competent E. coli BL21 cells. Cultures (1 L) were grown in LB media (containing 40 μg/mL kanamycin, LB-Kan) at 37 °C (with shaking). Cultures were grown to mid-log phase (OD600 ~ 0.8 – 1.0), induced with 0.5 mM IPTG, and incubated at 22 °C for 16 – 18 h. Cells were harvested by centrifugation (4000 xg, 4 °C), then resuspended in 20 mL of 50 mM NaPO4, 300 mM NaCl, 1 mM DTT, and 1 mM PMSF (pH 7.4). Lysozyme (1 mg) was added, and the mixtures were sonicated and centrifuged (10000 xg, 1 h at 4 °C). Luciferases were purified from the clarified supernatants using nickel-affinity chromatography (BioLogic Duo Flow Chromatography System, Bio-Rad). Protein isolates were dialyzed into 25 mM Tris-acetate buffer (pH 7.8) containing 1 mM EDTA and 0.2 mM ammonium sulfate (4 °C). DTT (1 mM) and 15% glycerol were added to the dialyzed samples prior to storage at −20 °C. Final protein concentrations were determined via BCA assay or UV spectroscopy (NanoDrop 2000c). Samples were also analyzed by SDS-PAGE and Coomassie staining.
General bioluminescence imaging
Samples were imaged using an IVIS Lumina (Xenogen) system equipped with a cooled CCD camera. Images were acquired using small to medium binning. Exposure times varied from 1 s to 10 min. All acquisition parameters were controlled using Living Image software. Living Image was also used to measure photon flux values from defined regions of interest. These data were exported to Microsoft Excel for further analyses.
Mutant luciferase library generation
Luciferase libraries were generated using mutagenic primers and gene assembly as previously described.54 All PCR reactions were performed using Q5 Hot-start DNA polymerase (New England BioLabs).
Mutant luciferase screening
E. coli BL21 cells (in 50% glycerol) expressing Fluc or mutant luciferases were added to 5 mL of LB-Kan. The cultures were incubated at 37 °C for 2–5 h (with shaking) until OD600 values reached ~0.8. A portion of each culture (0.5 mL) was reserved for sequencing analysis. IPTG (0.5 mM) was added to the remainder of the cultures and protein expression was induced for 2 h at 30 °C or 12 h at 25 °C. After induction, the cultures were centrifuged for 10 min at 3400 xg (4 °C). The supernatant was removed, and the pellet was resuspended in 0.4 – 0.6 mL of lysis buffer (50 mM Tris•HCl, 500 mM NaCl, 0.5% (v/v) Tween, 5 mM MgCl2, pH 7.4). Lysates were added to black 96-well plates (90 μL/well, in triplicate). Lysates were imaged using a stock solution of 10X luciferin and 10X ATP in PBS (1 mM luciferin, 10 mM ATP). The stock solution (10 μL) was added to each well, 6 wells at a time (final [luciferin] = 100 μM and [ATP] = 1 mM). Images were acquired as described above. Mutants with >10-fold light output compared to Fluc (in the same assay) were further analyzed. The reserved cultures for these mutants were added to 3.5 mL of LB-Kan and grown overnight in a shaking incubator (37 °C). Plasmid DNA was then harvested and sequenced.
Light emission assays with recombinant luciferase
All bioluminescence assays were carried out in triplicate, using black, flat-bottom, 96-well plates (Greiner). Each well contained purified Fluc or a mutant luciferase (0–1 μg), luciferin substrate (0–10 mM), ATP (Sigma, 1 mM), coenzyme-A (trilithium salt, LiCoA Calbiochem, 0.5 mM), and reaction buffer (50 mM Tris-HCl, 0.5 mg/ml BSA, 0.1 mM EDTA, 1 mM TCEP, 2 mM MgSO4), totaling 100 μL. Additionally, all non-enzyme assay components were premixed in the wells prior to luciferase addition. Wells containing 0–10 mM substrate were prepared at pH 7.6 or 9. Images for all assays were acquired as described above.
Cellular bioluminescence assays
HEK293 cells were grown in DMEM media supplemented with fetal bovine serum (FBS, 10%), penicillin (10 U/mL), and streptomycin (10 μg/mL). The cells were cultured in a 5% CO2 humidified incubator at 37 °C. Fluc-expressing HEK cells were provided by the Contag lab (Stanford). Transient transfections (with pcDNA-IRES-GFP vectors) were performed using cationic lipid formulations (Lipofectamine 2000, Invitrogen). For light emission assays, approximately 100,000 cells were plated in black 96 well plates in PBS (pH 7.6) containing 1% BSA. The cells were then incubated with luciferin analogs (0.01–5 mM) in PBS (pH 7.6) containing 1% BSA. Bioluminescence images were acquired as above.
Flow cytometry
GFP expression was verified in transiently transfected cells via flow cytometry. Cells were trypsinized and washed with FACS buffer (PBS with 1% BSA) prior to analysis. For each sample, 10,000 cells were collected and data were analyzed using FloJo software (Tree Star, Inc.).
Bioluminescence emission spectra
Emission spectra for 3, 4 and D-luc (1) were recorded on a Horiba Jobin-Yvon FluoroMax®−4 spectrometer. Each luciferin (1 mM) was incubated in a cuvette (10-mm path length) with ATP (1 mM), LiCoA (0.5 mM) and reaction buffer totaling 900 μL. Purified Fluc (10 μg) was added immediately prior to data acquisition. The excitation and emission slits on the instrument were adjusted to 0 and 5 nm, respectively. Emission data were collected at 1 nm intervals from 400–700 nm at ambient temperature. The acquisition times of 1 s per wavelength were used. Light emission values were measured in relative luminescence units (RLU), and the intensities were normalized to the peak emission intensity for each analog.
Bioluminescence kinetic measurements
Measurements were acquired on a Tecan F200 PRO luminometer with a neutral density filter. Reactions were performed in black 96-well, flat-bottom plates (Greiner). Solutions of luciferin (20 μM-4 mM analog) in bioluminescence buffer (20 mM Tris-HCl pH 7.6, 2 mM MgSO4, 2 mM ATP, 0.1 mM EDTA, 1 mM TCEP, 0.5 mg/mL BSA) were prepared, and 50 L was added to each well. The luminescence from each well was measured for 1.5 s prior to the addition of Fluc in bioluminescence buffer (50 L). For wells containing D-luc, a 0.002 mg/mL solution of enzyme was used. For all other compounds, a 0.02 mg/mL solution of enzyme was used. Luminescence values were recorded every 0.1 s over a 13.5 s period. Samples were analyzed in triplicate and three runs of each compound-enzyme pair were performed. Emission maxima were determined by averaging the five maximum photon outputs per run. Photon outputs were then normalized and plotted as a function of concentration. Km values were determined using nonlinear regression analyses and robust fit outlier removal in GraphPad Prism (version 6.0f for Macintosh, GraphPad Software).
Absorbance and fluorescence emission spectra
Absorbance spectra were acquired using a Cary 50 absorption spectrometer. Fluorescence spectra were acquired using a Cary Eclipse spectrometer (with 1 nm excitation and emission slit widths). Solutions were prepared via a 1:100 dilution of luciferin stock solution (10 mM in DMSO). To analyze the pH dependence of absorbance and fluorescence, compound stocks were diluted into BIS-TRIS propane buffer (20 mM, pH 6.5–9.5).
NMR analyses
Luciferin analogs (1 mM) were dissolved in deuterated phosphate buffer saline (20 mM phosphate, pH 7.4, containing 4.5 mM KCl and 228 mM NaCl). 1H-NMR spectra were acquired on Bruker Advanced spectrometers at ambient temperature.
Synthetic procedures
General synthetic methods
All reagents purchased from commercial suppliers were of analytical grade and used without further purification. Anhydrous solvents were dried by passage over neutral alumina. Reaction vessels were either flame or oven dried prior to use. Appel’s salt48 5 (4,5-dichloro-1,2,3-dithiazolium chloride), and 5-amino-6-methoxypyridine57 6 were prepared according to literature procedure. Reaction progress was monitored by thin-layer chromatography on EMD 60 F254 plates and visualized with UV light. Compounds were purified via flash column chromatography using Sorbent Technologies 60 Å, 230–400 mesh silica gel, unless otherwise stated. HPLC purifications were performed on a Varian ProStar equipped with 325 Dual Wavelength UV-Vis Detector. Semi-preparative runs were performed using an Agilent Prep-C18 Scalar column (9.4 × 150 mm, 5 μm) with a 4.2 mL/min flow rate, eluting with a gradient of 5–95% MeOH in ammonium acetate buffer (25 mM, pH 8). NMR spectra were acquired with Bruker Advanced spectrometers at 298 K. 1H-NMR spectra were acquired at either 500 or 400 MHz, and 13C-NMR spectra were acquired at 126 or 101 MHz. Chemical shifts are reported in ppm relative to residual non-deuterated NMR solvent, and coupling constants (J) are provided in Hz. Low and high-resolution electrospray ionization (ESI) mass spectra were collected at the University of California-Irvine Mass Spectrometry Facility. Infrared spectra were collected using a Nicolet iS5 FT-IR Spectrometer (Thermo Scientific). The abbreviations used can be found in the document JOC Standard Abbreviations and Acronyms, http://pubs.acs.org/page/joceah/submission/authors.html
N-(6-Methoxy-pyridin-3-yl)cyanothioformamide (8):
Appel’s salt (5, 31.3 g, 150 mmol) was added to a suspension of amino pyridine 6 (12.4 g, 100 mmol) in THF (300 mL). The mixture was stirred under N2. The mixture turned grey/green within 10 min. After 1.5 h, pyridine (16.6 mL, 205 mmol) was added. The mixture immediately turned dark red, and a yellow precipitate formed. After 19 h, a solution of sodium thiosulfate (31.6 g, 200 mmol) in 150 mL of H2O was added. The reaction was stirred for an additional 4.5 h. The mixture was then filtered, and the organic phase was removed in vacuo. The remaining aqueous solution was extracted with EtOAc (3 × 100 mL), and the combined organic fractions were washed with water (3 × 50 mL), and dried with MgSO4. The organics were then filtered and concentrated in vacuo. The residue was purified by flash column chromatography (eluting with a gradient of 10:1 hexanes:EtOAc to 4:1 hexanes:EtOAc), affording 8 as a light orange solid (13.7 g, 71%). 1H NMR (400 MHz, acetone-d6, mixture of tautomers) δ 8.66 (d, J = 4.0, 0.8H), 8.41(d, J = 4.0, 0.2H), 8.23 (dd, J = 8.0, 4.0, 0.8H), 7.92 (dd, J = 8.0, 4.0, 0.2H), 6.94 (d, J = 8.0, 0.2H), 6.90 (d, J = 8.0, 0.8H), 3.95 (s, 0.6H), 3.93 (s, 2.4H). 13C NMR (101 MHz, DMSO-d6) (mixture of tautomers, not all tautomeric carbon signals were distinct) δ 162.0, 161.7, 141.7, 134.4, 128.9, 113.8, 110.6, 53.6. HRMS (ESI+) calcd for C8H8N3OS [M + H]+ 194.0338, found 194.0394.
2-Cyano-6-methoxy-thiazolo[4,5-c]pyridine (9a) and 2-Cyano-5-methoxy-thiazolo[5,4-b]pyridine (9b):
Palladium chloride (0.354 g, 2.00 mmol), CuI (0.956 mg, 0.502 mmol), TBAB (6.45 g, 20.0 mmol), and 8 (1.94 g, 10.0 mmol) were suspended in anhydrous 1:1 DMF:DMSO (160 mL). The resultant red-brown mixture was placed under N2 and stirred at 120 °C for 1 h. The reaction was then diluted with H2O (100 mL) and extracted with EtOAc (7 × 100 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography (eluting with 4:1 hexanes:EtOAc). Fractions containing 9b were combined and concentrated, affording 9b (0.17 g, 8.6%) as a white solid. Fractions containing regioisomer 9a were combined and concentrated in vacuo. The resulting solid was triturated with cold hexanes, and the precipitate was collected to afford regioisomer 9a (0.85 g, 44%). 9a: 1H NMR (400 MHz, DMSO-d6) δ 9.21 (s, 1H), 7.73 (s, 1H), 3.98 (s, 3H). 13C NMR (126 MHz, acetone-d6) δ 163.91, 147.54, 145.78, 145.42, 136.76, 113.54, 102.58, 54.99. HRMS (ESI+) calcd for C8H6N3OS [M + H]+ 192.0232, found 192.0224. 9b: 1H NMR (400 MHz, DMSO-d6) δ 8.55 (d, J = 9.0, 1H), 7.22 (d, J = 9.0, 1H), 4.01 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.3, 155.4, 140.7, 135.0, 132.4, 113.3, 113.1, 54.6. HRMS (ESI+) calcd for C8H6N3OS [M + H]+ 192.0232, found 192.0233.
2-Cyano-6-acetoxy-thiazolo[4,5-c]pyridine (10):
Pyridine hydrochloride (1.8 g, 16 mmol) and 9a (300 mg, 1.6 mmol) were combined in a septum-sealed flask and stirred at 160 °C for 15 min. The resulting red-brown liquid was left to cool to room temperature. Acetic anhydride (1.5 mL, 16 mmol), pyridine (2.6 mL, 32 mmol) and DMAP (0.037 g, 0.32 mmol) were added to the flask and stirred for 1 h. The beige-colored heterogeneous mixture was then extracted with EtOAc (20 mL). The organics were washed with sat. NaHSO4 (3 × 5 mL), dried with MgSO4, filtered, and then concentrated in vacuo. The crude mixture was purified by flash column chromatography (3:1 hexanes:EtOAc to 1:1 hexanes:EtOAc) affording 10 as a white solid (270 mg, 77%). 1H NMR (500 MHz, acetone-d6) δ 9.31 (s, 1H), 8.10 (s, 1H), 2.36 (s, 3H). 13C NMR (126 MHz, acetone-d6) δ 169.4, 157.0, 148.6, 147.2, 146.4, 140.3, 113.3, 110.6, 21.0. HRMS (ESI-) calcd for C7H2N3SO [M – CH3CO]- 175.9919, found 175.9927. IR (dry film) νmax (cm-1): 3106 (CH, aromatic), 2946 (CH, aromatic), 2236 (CN, nitrile), 1744 (CO, ester).
2-Cyano-5-acetoxy-thiazolo[5,4-b]pyridine (11):
Pyridine hydrochloride (1.3 g, 1.2 mmol) and 9b (190 mg, 1.0 mmol) were combined in a sealed tube and stirred at 160 °C for 30 min. The resulting red-brown liquid was left to cool. Acetic anhydride (1.0 mL, 10 mmol), pyridine (1.6 mL, 20 mmol) and DMAP (0.026 g, 0.20 mmol) were added to the flask and stirred for 1 h. The beige-colored heterogeneous mixture was extracted with EtOAc (20 mL). The organics were washed with sat. NaHSO4 (3 × 5 mL), dried with MgSO4, filtered, then concentrated in vacuo. The crude mixture was purified by flash column chromatography (3:1 hexanes:EtOAc to 1:1 hexanes:EtOAc) affording 11 as a white solid (0.16 g, 73%) 1H NMR (400 MHz, CDCl3) δ 8.55 (d, J = 8.0 Hz, 1H), 7.40 (d, J = 8.0, 1H), 2.41 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 168.6, 158.1, 155.9, 144.4, 137.6, 135.7, 117.1, 112.6, 21.4. HRMS (ESI-) calcd for C7H2N3SO [M – CH3CO]- 175.9919, found 175.9916. IR (dry film) νmax (cm-1): 3073 (CH, aromatic), 2233 (CN, nitrile), 1758 (CO, ester).
(S)-2-(Thiazolo[4,5-c]pyridin-6(5H)-one-2-yl)-4,5-dihydrothiazole-4-carboxylic acid (3):
D-Cysteine hydrochloride monohydrate (0.211 g, 1.20 mmol) and K2CO3 (0.166 g, 1.20 mmol) were dissolved in H2O (1.6 mL). The mixture was added to a suspension of 10 (0.142 g, 0.648 mmol) in MeCN (8.8 mL) and stirred. Yellow precipitate began to form within minutes. After 1.5 h, TLC (1:1 hexanes:EtOAc) revealed complete consumption of 10, and the reaction mixture was then filtered. The precipitate was purified by HPLC as described in the general synthetic methods. The desired fractions were combined and concentrated to provide 3 as a yellow solid (0.11 g, 62%). 1H NMR (400 MHz, D2O) δ 8.35 (d, J = 0.8, 1H), 7.03 (d, J = 0.8, 1H), 5.31 (dd, J = 10.0, 8.0, 1H), 3.88 (dd, J = 11.2, 10.0, 1H), 3.69 (dd, J = 11.2, 8.1, 1H). 13C NMR (126 MHz, D2O) δ 179.8, 167.6, 164.3, 164.2, 155.9, 141.8, 134.5, 110.9, 83.0, 39.4. HRMS (ESI-) calcd for C9H6N3OS2 [M - COOH]- 235.9952, found 235.9949.
(S)-2-(Thiazolo[5,4-b]pyridin-5(4H)-one-2-yl)-4,5-dihydrothiazole-4-carboxylic acid (4):
D-Cysteine hydrochloride monohydrate (0.462 g, 2.63 mmol) and K2CO3 (0.363 g, 2.63 mmol) were dissolved in H2O (2.6 mL). The mixture was added to a suspension of 11 (0.291 g, 1.33 mmol) in MeCN (5.3 mL) and stirred. Yellow precipitate began to form within minutes. After 10 min, TLC (3:2 hexanes:EtOAc) revealed complete consumption of 11 and the reaction mixture was filtered. The precipitate was purified by HPLC as described in the general synthetic methods. The desired fractions were combined and concentrated to provide 4 as a yellow solid (0.235 g, 53%). 1H NMR (500 MHz, D2O) δ 7.99 (d, J = 9.2, 1H), 6.63 (d, J = 9.2, 1H), 5.24 (dd, J = 9.8, 7.9, 1H), 3.84 (dd, J = 11.2, 9.8, 1H), 3.63 (dd, J = 11.2, 7.9, 1H). 13C NMR (126 MHz, D2O) δ 180.6, 174.0, 168.9, 159.6, 154.4, 139.9, 135.6, 118.6, 82.5, 38.9. HRMS (ESI-) calcd for C10H6N3O3S2 [M - H]- 279.9850, found 279.9846.
(S)-2-(6-Methoxy-thiazolo[4,5-c]pyridin-2-yl)-4,5-dihydrothiazole-4-carboxylic acid (12):
D-Cysteine hydrochloride monohydrate (34.7 mg, 0.219 mmol) and 9a (39.9 mg, 0.209 mmol) were suspended in 30:1 MeCN:DMF (1.8 mL) in a 20 mL vial. A solution of K2CO3 (29.2 mg, 0.211 mmol in 0.42 mL H2O) was added, and the mixture was stirred for 3 h. The reaction mixture was then filtered, providing 12 as a white solid (50 mg, 75%). 1H NMR (400 MHz, D2O) δ 8.80 (d, J = 0.8, 1H), 7.46 (d, J = 0.8, 1H), 5.32 (dd, J = 10.0, 8.4, 1H), 4.01 (s, 3H), 3.91 (dd, J = 11.2, 10.0, 1H), 3.71 (dd, J = 11.6, 8.4, 1H). 13C NMR (126 MHz, D2O) δ 177.3, 165.2, 161.7, 161.3, 147.5, 144.8, 142.3, 100.6, 80.3, 55.2, 36.7. HRMS (ESI-) calcd for C11H9N3O3S2 [M - COOH]- 250.0109, found 250.0119.
(S)-2-(6-Methoxy-thiazolo[5,4-b]pyridin-2-yl)-4,5-dihydrothiazole-4-carboxylic acid (13):
D-Cysteine hydrochloride monohydrate (0.0181 g, 0.103 mmol) and K2CO3 (0.0145 g, 0.105 mmol) were dissolved in H2O (0.4 mL). The mixture was added to a suspension of 9b (0.0198 g, 0.104 mmol) in MeCN (0.5 mL) and DMF (0.03 mL). The resulting mixture was stirred. After 10 min, TLC revealed complete consumption of 9b, and the reaction mixture was filtered. The filtrate was collected and concentrated in vacuo. The crude mixture was washed with MeOH (3 × 5 mL), affording 13 as a white solid (29 mg, 83%). 1H NMR (400 MHz, D2O) δ 8.11 (d, J = 12.0, 1H), 7.00 (d, J = 8.0, 1H), 5.29 (t, J = 12.0, 1H), 4.00 (s, 3H), 3.88 (t, J = 12.0, 1H), 3.68 (t, J = 8.0, 1H). 13C NMR (126 MHz, D2O) δ 177.4, 165.7, 163.5, 157.6, 154.5, 141.1, 133.9, 112.0, 80.2, 54.7, 36.4. HRMS (ESI-) calcd for C11H9N3O3S2 [M - COOH]- 250.0109, found 250.0115.
Supplementary Material
Acknowledgements
This work was supported by a grant from the National Institutes of Health (R01-GM107630 to J.A.P.) and the UC Irvine School of Physical Sciences. B.S.Z. was supported by a GAANN Fellowship. Some experiments were performed at the Laser Spectroscopy Labs (LSL) at the University of California, Irvine (UCI). We also thank members of the Chamberlin, Pedersen, Martin, Nowick, and Weiss laboratories for providing reagents and experimental assistance. We also thank members of the Prescher lab for experimental advice and helpful discussions.
References
- [1].Massoud TF, Gambhir SS, Genes Dev. 2003, 17, 545–580. [DOI] [PubMed] [Google Scholar]
- [2].Paley MA, Prescher JA, MedChemComm 2014, 5, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Branchini BR, Behney CE,Southworth TL, Fontaine DM, Gulick AM, Vinyard DJ, Brudvig GW, J. Am. Chem. Soc 2015, 137, 7592–7595. [DOI] [PubMed] [Google Scholar]
- [4].Fraga H, Fernandes D, Novotny J, Fontes R, da Silva JCG, ChemBioChem 2006, 7, 929–935. [DOI] [PubMed] [Google Scholar]
- [5].Kaskova ZM, Tsarkova AS, Yampolsky IV, Chem. Soc. Rev 2016, 45, 6048–6077. [DOI] [PubMed] [Google Scholar]
- [6].Contag CH, Spilman SD, Contag PR, Oshiro M, Eames B, Dennery P, Stevenson DK, Benaron DA, Photochem. Photobiol 1997, 66, 523–531. [DOI] [PubMed] [Google Scholar]
- [7].Bhaumik S, Gambhir SS, Proc. Natl. Acad. Sci. U. S. A 2002, 99, 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tannous BA, Kim DE, Fernandez JL, Weissleder R, Breakefield XO, Mol. Ther 2005, 11, 435–443. [DOI] [PubMed] [Google Scholar]
- [9].Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV, ACS Chem. Biol 2012, 7, 1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chu J, Oh Y, Sens A, Ataie N, Dana H, Macklin JJ, Laviv T, Welf ES, Dean KM, Zhang FJ, Kim BB, Tang CT, Hu M, Baird MA, Davidson MW, Kay MA, Fiolka R, Yasuda R, Kim DS, Ng HL, Lin MZ, Nat. Biotechnol 2016, 34, 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhao H, Doyle TC, Coquoz O, Kalish F, Rice BW, Contag CH, J. Biomed. Opt 2005, 10, 41210. [DOI] [PubMed] [Google Scholar]
- [12].Liu H, Patel MR, Prescher JA, Patsialou A, Qian D, Lin J, Wen S, Chang YF, Bachmann MH, Shimono Y, Dalerba P, Adorno M, Lobo N, Bueno J, Dirbas FM, Goswami S, Somlo G, Condeelis J, Contag CH, Gambhir SS, Clarke MF, Proc. Natl. Acad. Sci. U. S. A 2010, 107, 18115–18120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sweeney TJ, Mailander V, Tucker AA, Olomu AB, Zhang W, Cao Y, Negrin RS, Contag CH, Proc. Natl. Acad. Sci. U. S. A 1999, 96, 12044–12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sanada Y, Yamamoto T, Satake R, Yamashita A, Kanai S, Kato N, van de Loo FAJ, Nishimura F, Scherer PE, Yanaka N, Sci. Rep 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Thorne SH, Contag CH, Proc. IEEE 2005, 93, 750–762. [Google Scholar]
- [16].Fan F, Binkowski BF, Butler BL, Stecha PF, Lewis MK, Wood KV, ACS Chem. Biol 2008, 3, 346–351. [DOI] [PubMed] [Google Scholar]
- [17].Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D, Proc. Natl. Acad. Sci. U. S. A 2004, 101, 12288–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sellmyer MA, Bronsart L, Imoto H, Contag CH, Wandless TJ, Prescher JA, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 8567–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Evans MS, Chaurette JP, Adams ST Jr, Reddy GR, Paley MA, Aronin N, Prescher JA, Miller SC, Nat. Methods 2014, 11, 393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Keyaerts M, Verschueren J, Bos TJ, Tchouate-Gainkam LO, Peleman C, Breckpot K, Vanhove C, Caveliers V, Bossuyt A, Lahoutte T, Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 999–1007. [DOI] [PubMed] [Google Scholar]
- [21].Rathbun CM, Prescher JA, Biochemistry 2017, 56, 5178–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Branchini BR, Ablamsky DM, Rosenman JM, Uzasci L, Southworth TL, Zimmer M, Biochemistry 2007, 46, 13847–13855. [DOI] [PubMed] [Google Scholar]
- [23].Branchini BR, Ablamsky DM, Murtiashaw MH, Uzasci L, Fraga H, Southworth TL, Anal. Biochem 2007, 361, 253–262. [DOI] [PubMed] [Google Scholar]
- [24].Li X, Nakajima Y, Niwa K, Viviani VR, Ohmiya Y, Protein Sci. 2010, 19, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McCutcheon DC, Paley MA, Steinhardt RC, Prescher JA, J. Am. Chem. Soc 2012, 134, 7604–7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Steinhardt RC, O’Neill JM, Rathbun CM, McCutcheon DC, Paley MA, Prescher JA, Chem. – Eur. J 2016, 22, 3671–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Steinhardt RC, Rathbun CM, Krull BT, Yu JM, Yang Y, Nguyen BD, Kwon J, McCutcheon DC, Jones KA, Furche F, Prescher JA, ChemBioChem 2017, 18, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Reddy GR, Thompson WC, Miller SC, J. Am. Chem. Soc 2010, 132, 13586–13587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mofford DM, Reddy GR, Miller SC, J. Am. Chem. Soc 2014, 136, 13277–13282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Woodroofe CC, Meisenheimer PL, Klaubert DH, Kovic Y, Rosenberg JC, Behney CE, Southworth TL, Branchini BR, Biochemistry 2012, 51, 9807–9813. [DOI] [PubMed] [Google Scholar]
- [31].Conley NR, Dragulescu-Andrasi A, Rao JH, Moerner WE, Angew. Chem. Int. Ed. Engl 2012, 51, 3350–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Takakura H, Sasakura K, Ueno T, Urano Y, Terai T, Hanaoka K, Tsuboi T, Nagano T, Chem. - Asian J 2010, 5, 2053–2061. [DOI] [PubMed] [Google Scholar]
- [33].Kojima R, Takakura H, Ozawa T, Tada Y, Nagano T, Urano Y, Angew. Chem. Int. Ed. Engl 2013, 52, 1175–1179. [DOI] [PubMed] [Google Scholar]
- [34].Iwano S, Obata R, Miura C, Kiyama M, Hama K, Nakamura M, Amano Y, Kojima S, Hirano T, Maki S, Niwa H, Tetrahedron 2013, 69, 3847–3856. [Google Scholar]
- [35].Miura C, Kiyama M, Iwano S, Ito K, Obata R, Hirano T, Maki S, Niwa H, Tetrahedron 2013, 69, 9726–9734. [Google Scholar]
- [36].Jathoul AP, Grounds H, Anderson JC, Pule MA, Angew. Chem. Int. Ed. Engl 2014, 53, 13059–13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Anderson JC, Grounds H, Jathoul AP, Murray JAH, Pacman SJ, Tisi L, Rsc Adv. 2017, 7, 3975–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bisai V, Sarpong R, Org. Lett 2010, 12, 2551–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].de Candia M, Fossa P, Cellamare A, Mosti L, Carotti A, Altomare C, Eur. J. Pharm. Sci 2005, 26, 78–86. [DOI] [PubMed] [Google Scholar]
- [40].Dragovich PS, Prins TJ, Zhou R, Brown EL, Maldonado FC, Fuhrman SA, Zalman LS, Tuntland T, Lee CA, Patick AK, Matthews DA, Hendrickson TF, Kosa MB, Liu B, Batugo MR, Gleeson JPR, Sakata SK, Chen LJ, Guzman MC, Meador JW, Ferre RA, Worland ST, J. Med. Chem 2002, 45, 1607–1623. [DOI] [PubMed] [Google Scholar]
- [41].Erez Y, Huppert D, J. Phys. Chem. A 2010, 114, 8075–8082. [DOI] [PubMed] [Google Scholar]
- [42].Erez Y, Presiado I, Gepshtein R, Pinto da Silva L, Esteves da Silva JC, Huppert D, J. Phys. Chem. A 2012, 116, 7452–7461. [DOI] [PubMed] [Google Scholar]
- [43].Rini M, Magnes BZ, Pines E, Nibbering ET, Science 2003, 301, 349–352. [DOI] [PubMed] [Google Scholar]
- [44].Mcelroy WD, Seliger HH, White EH, Photochem. Photobiol 1969, 10, 153–170. [DOI] [PubMed] [Google Scholar]
- [45].Mezzanotte L, Que I, Kaijzel E, Branchini B, Roda A, Lowik C, PLoS One 2011, 6, e19277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Krishnamoorthy A, Robertson JB, Appl. Environ. Microbiol 2015, 81, 6484–6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].McCutcheon DC, Porterfield WB, Prescher JA, Org. Biomol. Chem 2015, 13, 2117–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Michaelidou SS, Koutentis PA, Synthesis 2009, 2009, 4167–4174. [Google Scholar]
- [49].Inamoto K, Hasegawa C, Hiroya K, Doi T, Org. Lett 2008, 10, 5147–5150. [DOI] [PubMed] [Google Scholar]
- [50].Branchini BR, Murtiashaw MH, Magyar RA, Portier NC, Ruggiero MC, Stroh JG, J. Am. Chem. Soc 2002, 124, 2112–2113. [DOI] [PubMed] [Google Scholar]
- [51].Forlani L, Cristoni G, Boga C, Todesco PE, Del Vecchio E, Selva S, Monari M, ARKIVOC 2002, 198–215. [Google Scholar]
- [52].Gammon ST, Leevy WM, Gross S, Gokel GW, Piwnica-Worms D, Anal. Chem 2006, 78, 1520–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Branchini BR, Southworth TL, Murtiashaw MH, Boije H, Fleet SE, Biochemistry 2003, 42, 10429–10436. [DOI] [PubMed] [Google Scholar]
- [54].Jones KA, Porterfield WB, Rathbun CM, McCutcheon DC, Paley MA, Prescher JA, J. Am. Chem. Soc 2017, 139, 2351–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Viviani VR, Neves DR, Amaral DT, Prado RA, Matsuhashi T, Hirano T, Biochemistry 2014, 53, 5208–5220. [DOI] [PubMed] [Google Scholar]
- [56].Rathbun CM, Porterfield WB, Jones KA, Sagoe MJ, Reyes MR, Hua CT, Prescher JA, ACS Cent. Sci, [Online early access] DOI: 10.1021/acscentsci.7b00394. Published Online: November 15, 2017. http://pubs.acs.org/doi/abs/10.1021/acscentsci.7b00394 (accessed November 21, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ahmad Y, Hey DH, J. Chem. Soc 1954, 0, 4516–4523. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.