

Abstract
The cellular prion protein (PrPc) is a surface adhesion molecule expressed at junctions of various cell types including brain microvascular endothelial cells (BMVEC) that are important components of the blood brain barrier (BBB). PrPc is involved in several physiological processes including regulation of epithelial cell barrier function and monocyte migration across BMVEC. BBB dysfunction and disruption are significant events in CNS inflammatory processes including HIV neuropathogenesis. TNF-α and VEGF are two inflammatory factors that have been implicated in the processes that affect BBB integrity. To examine the effect of inflammation on PrPc expression in BMVEC, we used these mediators and found that TNF-α and VEGF decrease surface PrPc on primary human BMVEC. We also showed that these factors decrease total PrPC protein as well as mRNA, indicating that they regulate expression of this protein by de novo synthesis. To determine the effect of PrPc loss from the surface of BMVEC on barrier integrity, we used small hairpin RNAs to knockdown PrPc. We found that the absence of PrPc from BMVEC causes increased permeability as determined by a FITC dextran permeability assay. This suggests that cell surface PrPc is essential for endothelial monolayer integrity. To determine the mechanism by which PrPc downregulation leads to increased permeability of an endothelial monolayer, we examined changes in expression and localization of tight junction proteins, occludin and claudin-5, and found that decreased PrPc leads to decreased total and membrane associated occludin and claudin-5. We propose that an additional mechanism by which inflammatory factors affect endothelial monolayer permeability is by decreasing cell associated PrPc. This increase in permeability may have subsequent consequences that lead to CNS damage.
Introduction
The cellular prion protein (PrPc) is the non-pathogenic cellular isoform of human prion protein that is constitutively expressed in CNS cells.1, 2 PrPC is an adhesion molecule and has several proposed functions including facilitating monocyte transmigration across endothelium and intracellular signal transduction.3–9 It is a glycosylphosphatidylinositol (GPI)-anchored glycoprotein found in membrane raft microdomains with several signaling molecules including Src-family kinases, suggesting that it might be part of a signaling complex.4, 10–13 This protein is present in unglycosylated, monoglycosylated, and diglycosylated forms and can have approximately sixty different sugars attached to it, enabling it to interact with several ligands for diverse functions.14, 15
In the CNS PrPc is abundantly expressed in neurons, microglia, and cells of the blood brain barrier (BBB), including astrocytes and brain microvascular endothelial cells (BMVEC).1, 9, 16, 17 It is present on brain endothelial cells of mouse, rat, and human origin, localized in raft/caveolae-like membrane microdomains and concentrated at intercellular junctions of cells.9 It is hypothesized that the junctional localization of PrPc is dependent upon homophilic interactions between PrPc on two adjacent cells.9 The interaction of PrPc between two endothelial cells as well as between endothelial cells and monocytes was also shown to be essential for the migration of monocytes across an endothelial monolayer.9, 18
BMVECs are a major component of the BBB. The integrity of the BBB is essential to CNS homeostasis by controlling the transmission of biochemical signals and the transmigration of leukocytes from blood into the CNS in a regulated manner.19–21 The BBB also excludes certain soluble factors from the CNS while allowing specific nutrients to transport in and out of the brain.22, 23 During neuroinflammation, the BBB responds by remodeling of junctional proteins in response to intracellular signaling that will result in endothelial retraction.24 Disruption of the BBB leads to increased transmigration of leukocytes, impaired CNS homeostasis, and pathogen entry that can lead to neurologic compromise.19, 25
In enterocytes, PrPC co-localizes with E-cadherin and interacts with several desmosomal proteins, suggesting that it contributes to the adhesion and barrier function of intestinal epithelial cells.26 Studies in these cells demonstrate that the absence of this protein leads to the mislocalization of tight junction (TJ) proteins that is accompanied by increased paracellular permeability.26
In this study we examined the effect of TNF-α and VEGF on BMVEC expression of PrPc and how changes in PrPc expression altered endothelial monolayer integrity and permeability. TNF-α is associated with BBB disruption and barrier permeability.27–29 It is increased in the CNS in response to various pathogens as well as in neurodegenerative diseases including Alzheimer’s (AD), Parkinson’s (PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS) and HIV infection of the CNS.30–35 TNF-α increases production of the cytokines IL-6, CCL2, and IL-8 from CNS cells and these dysregulate adhesion protein expression and increase leukocyte transmigration across the BBB and accumulation within the CNS.36–38 In endothelial monolayer BBB models, TNF-α has been shown to cause increased permeability by activating NF-κβ signaling, resulting in claudin-5, occludin and ZO-1 downregulation.39
Another factor implicated in BBB breakdown is vascular endothelial growth factor (VEGF).40, 41 VEGF is the most potent angiogenic factor and is involved in vasculogenesis during embryonic development and vascular injury.42 In addition to its importance in angiogenesis, it has been shown to be neuroprotective in rodent models of neurodegeneration, indicating that its role in the CNS is complex.43–45 In the inflamed CNS, VEGF is produced by reactive astrocytes and this growth factor has been shown to contribute to loss of brain microvascular integrity by decreasing the tight junction proteins claudin-5 and occludin.46, 47 VEGF promotes VE –cadherin endocytosis in a β-arrestin dependent manner as well as VE-cadherin phosphorylation, resulting in its disassembly.48, 49 This adherens protein is essential for BBB integrity and its dysregulation by VEGF can lead to increased BBB permeability.
We used these inflammatory factors in our study to examine the regulation of PrPc in the context of HIV CNS infection and its role in HIV neuropathogenesis. HIV enters the CNS within the first 2 weeks of primary infection mainly through infected monocytes crossing the BBB, and results in increased cytokine production, in particular TNF-α.50, 51 Increased VEGF has also been shown in reactive astrocytes during HIV neuropathogenesis.46, 52
In this study, we showed that TNF-α and VEGF decrease cell surface PrPc in BMVEC. These factors also decreased PrPc mRNA suggesting that they regulate de novo synthesis of this protein. We hypothesized that decreased PrPc expression will lead to endothelial monolayer integrity disruption and showed that PrPc knockdown in BMVEC leads to increased permeability in monolayers of BMVEC lacking PrPc. We also showed that loss of PrPc leads to decreased expression of tight junction proteins occludin and claudin-5, suggesting a mechanism by which PrPc knockdown leads to increased permeability of BMVEC monolayers. Therefore, we propose that during CNS disorders, inflammatory mediators that decrease PrPc expression in BMVEC contribute to CNS damage by increasing BBB permeability.
Materials and Methods
Brain microvascular endothelial cell culture and treatment
Human brain microvascular endothelial cells (BMVEC) (Applied Cell Biology Research Institute, Kirkland, WA) were grown in M199 media, supplemented with 20% heat inactivated newborn calf serum, 1% penicillin- streptomycin (all from Thermo Fisher Scientific, Waltham, MA), 0.8% heparin (Sigma, St. Louis, MO), 5% heat inactivated human serum AB (Lonza, Walkersville, MD), 0.1% ascorbic acid (Sigma, St. Louis, MO), 0.25% endothelial cell growth supplement (Sigma, St. Louis, MO), and 0.06% bovine brain extract (Clonetics, Walkersville, MD). Recombinant TNF-α (PeproTech, Rocky Hill, NJ) and recombinant VEGF (R&D, Minneapolis, MN) were added directly to the cell culture media to a final concentration of 10 ng/ml and 100 ng/ml, respectively.
Western blotting
BMVEC were grown to confluence on 0.2% gelatin (Fisher Scientific, Pittsburgh, PA) coated 60mm tissue culture dishes. Cells were lysed with cell lysis buffer (Cell Signaling, Boston, MA) supplemented with protease inhibitor cocktail (Roche, Indianapolis, MN). Total cellular protein concentration was determined by Bio-Rad protein assay (Bio-Rad, Hercules, CA). Forty micrograms of cell lysate were loaded onto 4–20% polyacrylamide gels (Bio-Rad), separated by electrophoresis, and transferred to nitrocellulose membranes (GE Healthcare Life Sciences, Pittsburgh, PA). Membranes were blocked for 2h at room temperature with 5% nonfat dry milk and 3% BSA in Tris-Buffered Saline and 0.1% Tween 20 (TBST). Blots were probed with antibody against PrPc (SAF 32 clone, 1:200 dilution, Cayman Chemicals, MI) overnight at 4°C, washed with TBST, and probed with anti-mouse IgG-HRP secondary antibody, 1:2000 dilution (Cell Signaling) for 1h at room temperature. Signal was detected using Western Lightning Plus-ECL (Perkin Elmer, Waltham, MA). Blots were stripped using Restore Plus Western Blot Stripping Buffer (Thermo Scientific), and reprobed with antibody against GAPDH, 1:500 dilution, (Cell Signaling), for 1h at room temperature, washed with TBST and probed with anti-mouse IgG-HRP secondary antibody (1:2000 dilution) for 1h at room temperature. Signal was detected using Western Lightning Plus-ECL. Data were quantified by densitometry using UN-SCAN-IT software (Silk Scientific, Orem, UT)
Flow cytometry
Surface PrPc was analyzed by flow cytometry using a fluorochrome-coupled mAb specific for human PrPc (eBioscience, San Diego, CA) and a corresponding isotype-matched (mouse IgG1K) negative control antibody (eBioscience). Antibodies were titered to determine optimal concentration for staining (0.25ug/100ul). BMVEC were dissociated with TrypLE (Thermo Scientific) and 1 ×105 cells were washed with FACS buffer, calcium and magnesium free PBS supplemented with 1% BSA (Thermo Fisher Scientific). Cells were incubated in the dark on ice for 30 min with antibodies. Following staining, cells were washed with FACS buffer and fixed with 2% paraformaldehyde. 1×105 events were acquired with a BD FACSCanto II flow cytometer and analyzed with FlowJo software (TreeStar, Ashland, OR).
RNA isolation and qRT-PCR
Total RNA was isolated from BMVEC (1 ×106) by Trizol (Life Technologies, Carlsbad, CA) extraction according to the manufacturer’s instructions and quantified by Nanodrop (Thermo Scientific, Wilmington, DE). cDNA synthesis was performed using SuperScript VILO cDNA synthesis kit (Life Technologies). Relative mRNA expression of PrPc, 18S, and GAPDH was determined using a Taqman Gene Expression Assay (Life Technologies). Results are represented as relative expression of PrPc normalized to 18S and/or GAPDH as housekeeping genes using 2-ΔCt. The ΔCt value was determined by subtracting the average Ct of the housekeeping gene from the average Ct of PrPc.
PrPc knockdown with shRNA
shRNAs corresponding to the human PRNP gene were synthesized by the shRNA Core at Albert Einstein College of Medicine. These shRNA clones were built on the pGIPZ vector53 that encodes GFP and a puromycin selection marker making it possible to monitor and select transfected cells. To generate PrPc knockdown cell lines, BMVEC were infected with three lentiviruses carrying three different shRNA’s, with an MOI of 80 in the presence of polybrene (5ug/ml) (Sigma) for 48h. Cells were grown to confluence in fresh media for an additional 48h. Transfected cells were selected with puromycin (0.5ug/ml) (Sigma) and maintained in puromycin containing media. Three different cell lines with different shRNA sequences were generated. The specific shRNA sequences used were 5’ TGCATGTTCTTGTTTTGTT 3’ for PrPcKD1, 5’GACATATTCACAGTGAACA 3’ for PrPc KD2, and 5’ TGCGTCAATATCACAATCA 3’ for PrPcKD3. These three shRNAs target different regions in the 3’UTR of the PRNP gene. A lentivirus with a pGIPZ vector with no PRNP shRNA was used as a control. To assay for knockdown of PrPc, we determined the level of PrPc mRNA in the different cell lines and found mRNA reduction of 90%, 25% and 80% in PrPc KD1, PrPc KD2, and PrPc KD3, respectively. We also assayed for reduction in surface PrPc by flow cytometry and found 90%, 40% and 86% reduction in surface PrPc in PrPc KD1, PrPc KD2, and PrPc KD3, respectively.
Permeability measurement
Transfected BMVEC were plated on tissue culture inserts with 3μm pores (Corning, Corning, NY) at 4 ×104 cells per insert and grown to confluence for 3–4 days. Cultures were maintained in puromycin containing media. Inserts were washed with phenol red-free DMEM (Thermo Fisher Scientific) and placed in 24-well tissue culture plates containing 400µL of phenol red-free DMEM/10% FBS in each well. Dextran-FITC (125 μg/mL, 70kDa, Sigma) (200µL) was added to the top of the insert, and after 5 min at 37°C, media was collected from the lower chamber and fluorescence was analyzed with a fluorescence-detecting plate reader (excitation λ 488 nm; emission λ 510 nm).
Immunofluorescence
Primary BMVEC were grown to confluence on 0.2% gelatin (Fisher Scientific) coated 35 mm ibiTreat dishes (ibidi USA, Madison, WI). Cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for 30 min and permeablized in 0.01% Triton X-100 for 1 min. Blocking was performed for 2 h at room temperature with 5 mM EDTA (Thermo Fisher Scientific), 1% fish gelatin (Sigma), 1% essentially immunoglobulin-free BSA (Sigma), 1% heat-inactivated human serum type AB (Lonza), and 1% goat serum (Vector, Burlingame, CA, USA) in deionized water. Cells were incubated overnight at 4°C in primary antibody anti-occludin, (Abcam, Cambridge, MA), and anti-claudin-5 (Santa Cruz Biotechnology, Dallas, TX) diluted 1:500 and 1:300, respectively. Cells were washed 3 times with PBS at room temperature and incubated with the appropriate secondary antibodies conjugated to Cy3, and labeled with phalloidin to identify the shape of the cells. ibiTreat dishes were then mounted using antifade reagent with DAPI to identify nuclei and the cells were then examined by confocal microscopy using a Nikon A1 confocal microscope with spectrum detection (Nikon, Tokyo, Japan). Due to the nature of the distribution of occludin and claudin-5 in BMVEC, we identified XY areas that were positive for PrPc siRNA (areas that were GFP positive) and stained as described above. After identifying the cells that were positive for PrPc siRNA, we characterized the intracellular and membrane expression by positivity for actin filament and membrane expression by lack of positivity for actin. Using the previous acquired XY coordinates with GFP from the PrPc siRNA, we compared the expression in areas with or without knockdown using NIS elements by quantifying the numbers of positive pixels for each location.
Results
TNF-α and VEGF decrease surface PrPc from BMVEC
BMVEC monolayers are characterized by the presence of intercellular TJ and adhesion proteins, and are a major component of the BBB. The BBB contributes to cerebral homeostasis and controls leukocyte migration, as well as pathogen invasion, into the brain.19–21 Immunofluorescence, as detected by confocal microscopy, showed that PrPc was localized to junctions between adjacent endothelial cells similar to the adhesion protein platelet endothelial cell adhesion molecule-1 (PECAM-1), suggesting that PrPc may be part of endothelial adhesion complexes that maintain integrity of the endothelial monolayer.9 In intestinal epithelial cells, PrPc localizes to adhesion complexes as surface PrPc has been shown to regulate intestinal barrier functions.26
TNF-α increases BBB permeability directly by downregulating TJ proteins occludin, claudin-5, and ZO-1 and indirectly by inducing the production of chemokines and cytokines that regulate TJ and AJ proteins and affect barrier integrity.54, 55 VEGF decreases endothelial barrier permeability by downregulating claudin-5 and occludin.46 We hypothesized that another mechanism by which these inflammatory factors disrupt TJ formation and BBB integrity is by changing the surface expression of PrPc.
To determine the effect of TNF-α and VEGF on cell surface PrPc, human BMVEC were cultured to confluence on 0.2% gelatin coated tissue cultures plates. Cultures were treated with VEGF (100ng/ml) or TNF-α (10ng/ml) for 18h and 24h. Earlier time points, 30 min, 6h and 12h were examined and we determined that significant decreases in PrPc expression occur starting at 18h post treatment. Cells were dissociated from plates using TrypLE and stained with PrPc antibody or isotype matched control antibody and analyzed by flow cytometry.
Fold changes in PrPc mean fluorescent intensity after TNF-α or VEGF treatments were compared with control untreated cells, which was set to one. Changes in PrPc mean fluorescence were determined by subtracting the mean fluorescent intensity of the isotype matched negative control antibody from the mean fluorescent intensity of the PrPc specific antibody. TNF-α treatment decreased surface PrPc on human BMVEC at 18h and 24h as shown by a representative histogram (Figure 1A). Quantification of 3 independent experiments shows that TNF-α decreases PrPc by 0.5 and 0.4 fold at 18h and 24h, respectively (Figure 1B, **p < 0.01). Similarly, VEGF decreased surface PrPc on human BMVEC at 18h and 24h as shown by a representative histogram (Figure 1C). Quantification of 3 independent experiments shows that VEGF decreases PrPc by 0.4 fold at 18h and by 0.5 fold at 24h (Figure 1D, **p < 0.01). For both mediators, this is a 40–50% reduction, that are highly significant.
Figure 1.
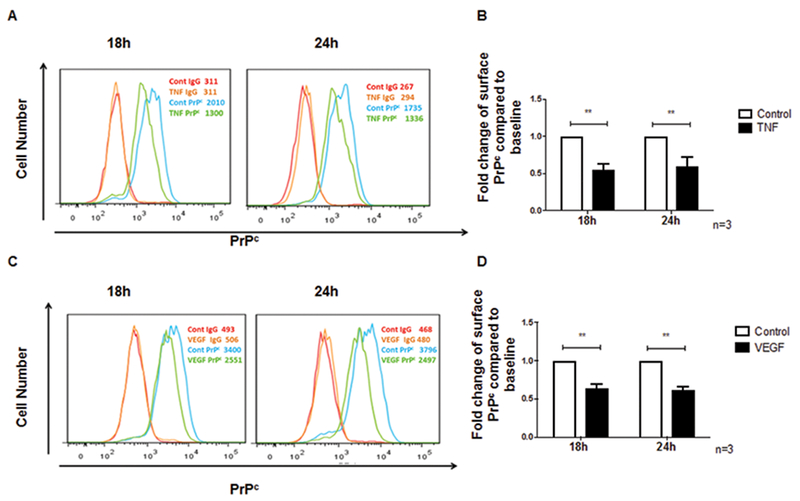
TNF-α and VEGF decrease PrPc surface expression on endothelial cells. Surface PrPc was analyzed by flow cytometry after treatment with TNF-α (10ng/ml) or VEGF (100ng/ml) (A) Representative plot showing the change in surface PrPc determined by FACS analysis after treatment with TNF-α for 18h and 24h. Three independent experiments were quantified (B) The fold change in mean fluorescence intensity (MFI) of PrPc on BMVEC after TNF-α treatment as compared to control was calculated after subtracting the contribution of the isotype matched negative control antibody. Surface PrPc after 18h and 24h of TNF-α treatment decreased by 0.5 fold (50%) and 0.4 fold (40%), respectively. (C) Representative plot showing the change in surface PrPc determined by FACS analysis after treatment with VEGF for 18h and 24h (D). The fold change in MFI of PrPc on BMVEC after VEGF treatment as compared to control was calculated after subtracting the contribution of the isotype matched negative control antibody. Surface PrPc after 18h and 24h of VEGF treatment decreased by 0.4 fold (40%) and 0.5 fold (50%), respectively. Significance was determined using a two-tailed paired Student t test. *p < 0.05, **p < 0.01.
TNF-α and VEGF decrease total PrPc in BMVEC
To determine whether TNF-α and VEGF decrease surface PrPc by changing the localization of this protein or by decreasing total protein levels of PrPc, we treated BMVEC with these factors and studied changes in total PrPc protein by Western blotting. Cells were grown to confluence and treated with TNF-α and VEGF for 18h or 24h, after which cell lysates were prepared and changes in total PrPc were quantified. Densitometry was performed in which total PrPc was compared to a loading control, GAPDH, and these values were compared between control and TNF-α treated cells, and control and VEGF treated cells. Results are reported as fold change relative to control. We found that TNF-α decreased total PrPc by 0.4 fold (40%) at 18h and that PrPc protein levels returned to baseline by 24h (Figure 2A and 2B, *p < 0.05). VEGF decreased total PrPc in BMVEC by 0.2 fold (20%) at 18h and by 0.6 fold (60%) at 24h (Figure 2C and2D,*p < 0.05). These results suggest that during neuroinflammation, total PrPc in BMVEC is downregulated by inflammatory factors. We propose that this downregulation in turn may negatively impact endothelial function by decreasing barrier integrity.
Figure 2.
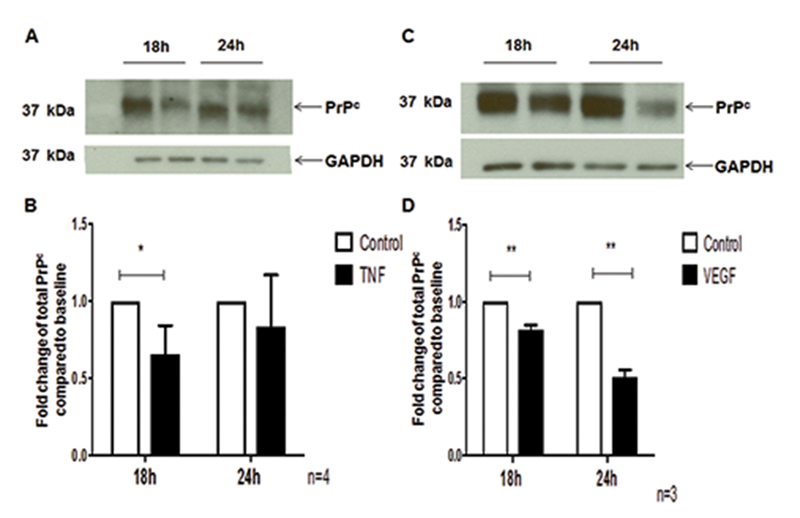
TNF-α and VEGF decrease total PrPc in endothelial cells. (A). Representative blot showing PrPc decrease in BMVEC treated with TNF-α (10ng/ml) for either 18h or 24h and lysates prepared to examine total PrPc by Western blotting. GAPDH was used as a loading control (B) Four independent experiments were quantified. TNF-α decreased total PrPc by 0.4-fold (40%) at 18h with PrPc returning to baseline at 24h. (C) Representative blot showing PrPc decrease in BMVEC treated with VEGF (100 ng/ml) for either 18h or 24h and lysates prepared to examine total PrPc by Western blotting. GAPDH was used as a loading control (D) Three independent experiments were quantified. VEGF decreased total PrPc by 0.2 fold (20%) at 18h and by 0.6 fold (60%) at 24h. Significance was determined using a two-tailed paired Student’s t test. *p < 0.05, **p < 0.01.
TNF-α and VEGF decrease PrPc gene expression in BMVEC
To examine the mechanism by which TNF-α and VEGF regulate PrPc, we treated BMVEC with these mediators and analyzed PrPc mRNA by qRT-PCR. We found that TNF-α decreased PrPc mRNA at 12h and 18h by 0.2 fold (20%) and 0.4 fold (40%), respectively. By 24h, PrPc message returned to baseline (Figure 3A, *p < 0.05). VEGF decreased PrPc mRNA significantly at 12h, 18h, and 24h in BMVEC by 0.4 fold (40%), 0.5 fold (50%) and 0.6 fold (60%), respectively, (Figure 3B, *p < 0.05 and ***p< 0.0005). These results indicate that TNF-α and VEGF decrease de novo synthesis of PrPc.
Figure 3.
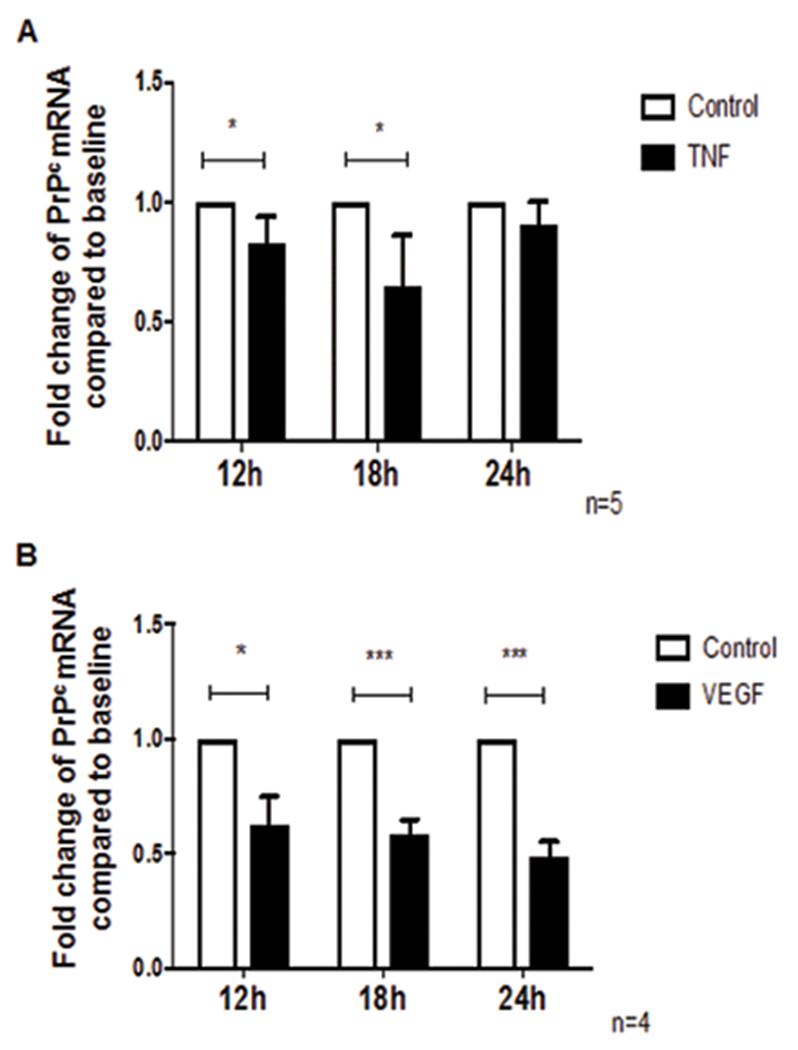
TNF-α and VEGF decrease PrPc mRNA in endothelial cells. (A) BMVEC were treated with TNF-α (10ng/ml) for 12h, 18h, or 24h and mRNA levels of PrPc were evaluated using qRT-PCR. TNF-α decreased PrPc mRNA at 12h and 18h by 0.2 fold (20%) and 0.4 fold (40%), respectively, and PrPc mRNA returned to baseline at 24h, n=5. (B) BMVEC were treated with VEGF (100ng/ml) for 12h, 18h, or 24h and mRNA levels of PrPc were evaluated using qRT-PCR. VEGF decreased PrPc mRNA by 0.4 fold (40%), 0.5 fold (50%), and 0.6 fold (60%) at 12h, 18h, and 24h respectively, n=3. Significance was determined using a two-tailed paired Student’s t test. *p < 0.05, **p < 0.01, ***p< 0.0005.
shRNA interference decreases surface PrPc on BMVEC
To examine the specific role of PrPc in endothelial barrier integrity and function and the consequences of its downregulation, we generated 3 different PrPc knockdown BMVEC cell lines using shRNA interference. Cells were infected with lentivirus carrying shRNA against PRNP (the PrPc gene) and GFP and puromycin selection markers (as described in Materials and Methods). Control cells were infected with a lentivirus vector that did not contain PRNP shRNA. Cells were grown to confluence in puromycin containing media. Infections were performed 2 independent times with the 3 shRNAs described.
To confirm PrPc mRNA was reduced in the BMVEC, we analyzed PrPc mRNA in the 3 different cell lines by qRT-PCR. All cells lines were generated using the same donor of BMVEC, but unique shRNA sequences that target different regions of the 3’UTR of the PrPc gene. We found that shRNA knockdown of the PRNP gene resulted in a 90% reduction of PrPc mRNA in one cell line (PrPcKD1) as compared to the control cell line. In two other cell lines, shRNA knockdown resulted in 25% and 80% reduction of PrPc mRNA. These cell lines were termed PrPcKD2 and PrPcKD3, respectively (Figure 4A, *p < 0.05 and **p < 0.01).
Figure 4.
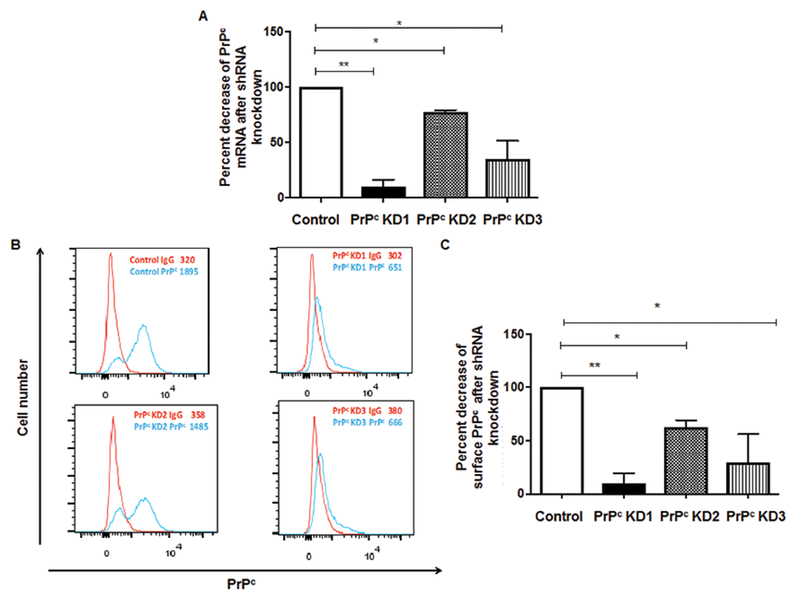
Significant knockdown of PrPc in BMVEC with shRNA. (A) mRNA levels of PrPc were analyzed by qRT-PCR in control cells and 3 lines infected with PRNP shRNA. One cell line (PrPc KD1) showed 90% decrease in PrPc mRNA. The second cell line (PrPc KD2) and the third cell line (PrPc KD3) showed 25% and 65% fold decrease in PrPc mRNA, respectively (B) Surface PrPc was analyzed by flow cytometry in control cell lines and 3 cell lines that were infected with lentivirus containing PRNP shRNA. (C) Change in the mean fluorescence intensity (MFI) of PrPc on BMVEC cell lines infected with PRNP shRNA as compared to control was calculated after subtracting the contribution of the isotype matched negative control antibody. PrPc KD1 showed a 90% decrease in surface PrPc while PrPc KD2 showed a 40% decrease in surface PrPc and PrPc KD3 showed a 70% decrease in PrPc.
We also analyzed changes in surface PrPc levels in the shRNA knockdown BMVEC lines by flow cytometry. BMVEC were dissociated from plates using TrypLE and stained with PrPc antibody or isotype matched control antibody and analyzed by flow cytometry. Changes in PrPc mean fluorescence were determined by subtracting the mean fluorescent intensity of the isotype matched negative control antibody from the mean fluorescent intensity of the PrPc specific antibody. We found that shRNA knockdown of the PRNP gene resulted in 90% reduction of cell surface PrPc in the cell line PrPc KD1. PrPcKD2 and PrPcKD3 showed 40% and 70% reduction of cell surface PrPc, respectively (Figure 4B and 4C, *p < 0.05 and **p < 0.01). Although all PrPcKD2 cells were GFP positive, indicating that they had lentivirus incorporated into their genome, only a percentage of these cells exhibited a decrease in PrPc as shown on the FACS histograms (Figure 4B). PrPc knockdown did not affect cell proliferation or viability as evidenced by counting of cells with trypan blue staining.
PrPc contributes to barrier integrity in BMVEC
To examine whether PrPc knockdown results in changes in permeability, control and PRNP shRNA transfected BMVEC were cultured on gelatin coated 3μm pore culture inserts placed in 24 well tissue culture plates. Changes in permeability of BMVEC monolayers of PrPc knockdown cell lines were assessed by the movement of dextran-FITC (70kDA) across the monolayers. Dextran-FITC (125 μg/mL) was added to the top of the insert, and after 5 min at 37°C, media was collected from the lower chamber and fluorescence determined. In cell lines that exhibited 90% knockdown of PrPc (PrPcKD1), permeability increased by 2 fold as compared to control cell lines. The cell line with a 25% decrease in PrPc mRNA (PrPcKD2) did not show any change in permeability, while PrPcKD3 showed a 1.5 fold increase in permeability (Figure 5, *p < 0.05 and***p< 0.0005). In previous studies, we have used other measurements of barrier integrity including transendothelial electrical resistance (TEER) and flux of albumin conjugated to Evans blue dye across monolayers.56, 57 Measurement of movement of dextran-FITC across BMVEC monolayers is comparable to measurement with Evans blue dye conjugated to albumin, and therefore we used dextran-FITC to assess permeability in this study.
Figure 5.
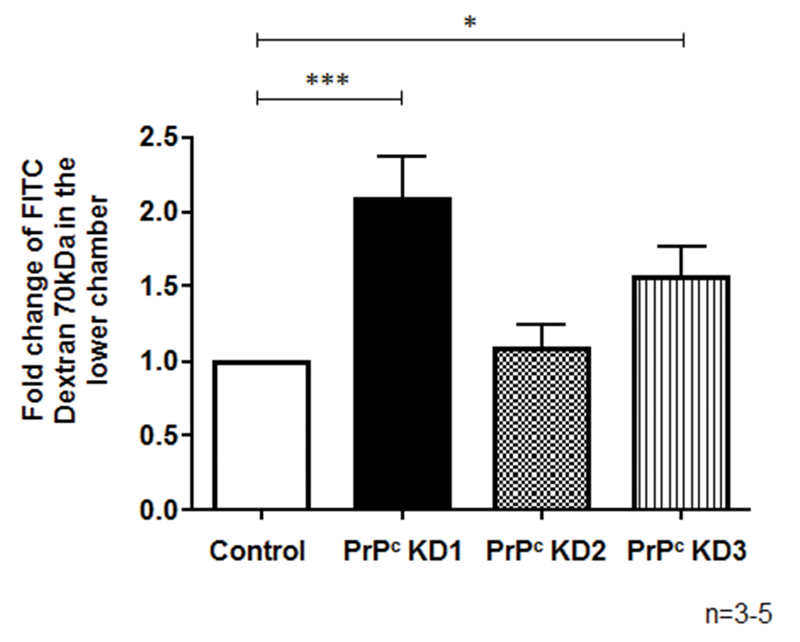
PrPc knock down results in increased BMVEC monolayer permeability. Permeability was measured by the passage of dextran-FITC (70kDA) across BMVEC monolayer. Transfected BMVECs were cultured on tissue culture inserts with 3 μm pores placed in 24-well tissue culture plates. Permeability of the barrier was measured by passage of dextran-FITC (70kDA) (125ug/ml) as described in the Materials and Methods section. The cell line with a 90% knock down of PrPc (PrPc KD1) showed a 2.0-fold increase in permeability as compared to control cell lines. PrPc KD2, which has a 40% knock down of PrPc, showed no increase in permeability while PrPc KD3, which has a 70% knock down of PrPc, showed a 1.6 fold increase in permeability. Significance was determined using a two-tailed paired Student’s t test. *p < 0.01, ***p< 0.0005.
PrPc knock down downregulates total and membrane associated expression of occludin and claudin-5.
To examine the mechanism by which PrPc silencing leads to increased permeability of BMVEC monolayers, we studied the expression of occludin and claudin-5 in BMVEC which have decreased PrPc. In human Caco-2/TC7 enterocytes, downregulation of PrPc changes the localization and expression of tight junction proteins.26 We hypothesized that the absence of PrPc on BMVEC will lead to downregulation and redistribution of these TJ proteins on the cell surface which leads to a leaky barrier. To examine whether PrPc is required for expression and localization of occludin and claudin-5 in BMVEC, total expression and localization of these tight junction proteins in our KD lines was assessed by confocal microscopy and subsequent imaging analysis. We used actin as an intercellular marker. Proteins that did not localize with, but that were close to actin were considered membrane proteins. Control cells had 40–50% of their total occludin as membrane protein (Figure 6A, 6B). PrPc KD1 and PrPc KD3 had reduced total occludin expression ((Figure 6B, black bars, * p≤0.001 as compared to control) as well as reduced membrane occludin (Figure 6B, red bars, # p≤0.0002 as compared to membrane control). Using the same approach described for occludin, we performed analysis for claudin-5. PrPc KD1 and PrPc KD3 had reduced total expression of claudin-5 as well as cell to cell membrane localization (Figure 6C and 6D). This change was not as pronounced as for occludin but the reduction was significant. Negative controls using IgGs and control sera did not show positive staining (data not shown). Thus, PrPc knockdown decreased expression and membrane localization of occludin and claudin-5.
Figure 6.
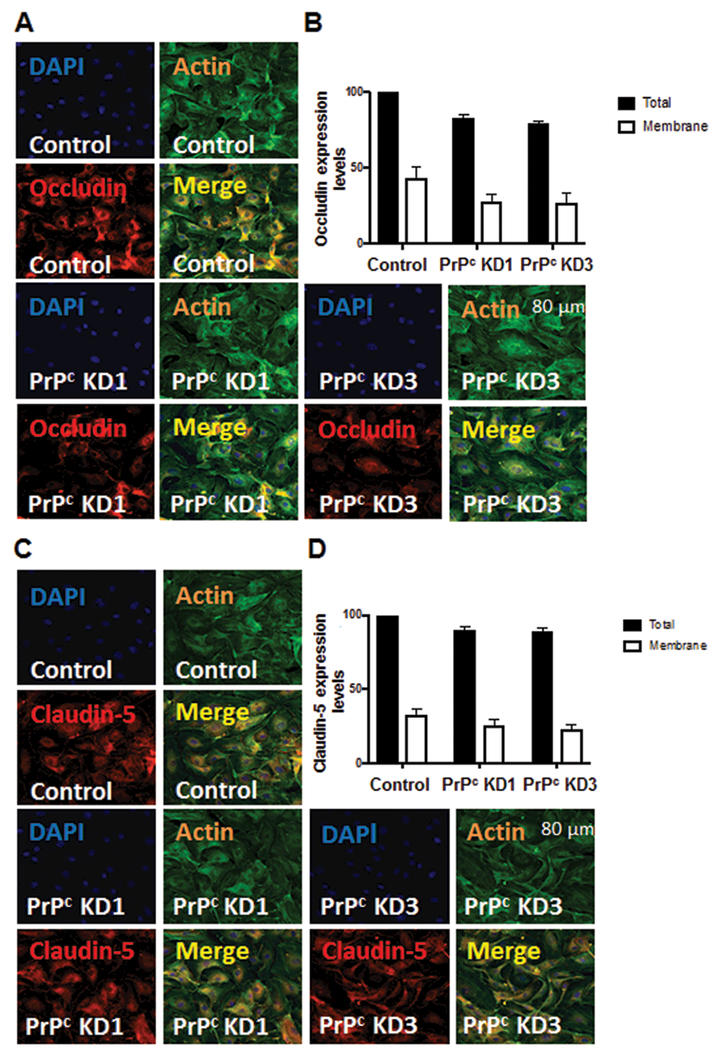
PrPc knock down downregulates total and membrane associated expression of occludin and claudin-5. Occludin and claudin-5 expression was measured by confocal microscopy and analyzed. Control and PrPc knock down cells were plated on ibdi dishes and fixed with PFA. Cells were stained using occludin and claudin-5 antibodies and analyzed for changes in expression and localization. (A) Cells were stained with DAPI to identify cell nuclei and labeled with phalloidin to identify the shape of the cells. PrPc KD1 and PrPc KD3 show reduced total occludin and cell membrane occludin. (B) To compare conditions, similar number of cells (300 to 800 cells) and total area was analyzed. Control cells had 43% of their total occludin as membrane protein. PrPc KD1 had reduced membrane occludin (28%) while PrPc KD3 had 27% of their total occludin as membrane occludin. (C) Cells were stained with DAPI to identify cell nuclei and labeled with phalloidin to identify the shape of the cells. PrPc KD1 and PrPc KD3 show reduced total claudin-5 and cell membrane claudin-5. (D) To compare conditions, similar number of cells (300 to 800 cells) and total area was analyzed. Control cells had 32.64% of their total claudin-5 as membrane protein. PrPc KD1 had reduced membrane claudin-5 (25%) while PrPc KD3 had 22.65% of their total claudin-5 as membrane claudin-5. Significance was determined using ANOVA test. * p≤0.001, # p≤0.0002.
Discussion
The blood brain barrier, consisting primarily of BMVEC and astrocyte foot processes, separates the brain from the periphery.58 TJ proteins, including occludin and claudin-5, connect adjacent BMVEC and restrict paracellular transmigration, rendering the barrier semi permeable.59, 60
PrPc is an adhesion molecule expressed at cell junctions in endothelial and epithelial cells.7 In brain endothelial cells isolated from mice, rats, and humans, this protein was localized at intercellular junctions between two adjacent cells.9 In the same study, anti-PrPc antibodies, blocked the migration of U937 human monocytic cell line and freshly isolated human monocytes across a human endothelial monolayer, suggesting PrPc is a junctional protein in BMVEC and that it is essential for monocyte migration across the BBB.9 PrPc is also localized at cell junctions in human enterocytes and keratinocytes.7, 61 This junctional localization of PrPc is dependent on the expression of PrPc on two adjacent cells. In co-cultures of PrPc expressing brain endothelial cells (WT) and PrPc KO cells, PrPc was expressed only between adjacent WT cells, suggesting that this protein is involved in homotypic interactions and contributes to intercellular adhesion by that mechanism.9
In this study we examined the effect of neuroinflammation on cell associated PrPc in the context of HIV infection of the CNS. HIV enters the CNS and induces neuroinflammation, BBB dysfunction, and production of toxic factors that can collectively lead to neuronal injury.62, 63 This process results in neurocognitive impairment in 40–60 % of HIV infected individuals, despite antiretroviral therapy (ART).64, 65 Adhesion molecules such as PrPc are expressed on cells of the CNS and the BBB, as well as infiltrating leukocytes, and regulate CNS homeostasis. They act as membrane receptors, transduce intracellular signals, and promote cellular communication and adhesion.66, 67 During HIV infection of the brain, adhesion molecule expression is dysregulated and many physiological processes regulated by these proteins are disrupted, contributing to CNS pathology.68–70
Previously, we showed that, shed (sPrPc) is increased specifically in the CSF of HIV positive people with cognitive impairment as compared to HIV positive people with no cognitive impairment.71 This increase in sPrPc correlated with CSF CCL2 levels, indicating that PrPc shedding is increased during neuroinflammation. In addition, we also demonstrated that PrPc shedding from cultured human astrocytes is increased in the presence of inflammatory factors CCL2 and TNF-α. We characterized the mechanism by which PrPc is cleaved, which is by the A disintegrin and metalloproteinase domain-containing protein 10 (ADAM10).72 Shed PrPc in turn, increased the release of cytokines and chemokines from astrocytes while decreasing glutamate uptake,72 suggesting an additional mechanism by which shed PrPc may play a role in neuroinflammation.
In cerebellar granule cells, PrPc has been shown to be localized in lipid rafts in a specific detergent resistant domain known as Prion domain.73, 74 These cholesterol and sphingolipid enriched areas of the cell membrane are suggested to be important to stabilize PrPc in its native (α-helical) conformation but don’t seem to affect its cleavage.75 In addition, the conversion of PrPc to the scrapie form (PrPsc ) seems to occur in lipid rafts.75 Studies implicate ADAM10 and ADAM17 in the shedding of PrPc in the brain and specifically from neurons.76 However, no studies have been conducted in astrocytes to study the role of lipid rafts in cleavage or conformational stability of PrPc.
In this current study we examined the effect of inflammation on BMVEC cell surface PrPc expression by treating cells with TNF-α and VEGF, and identifying potential consequences of changes in this expression. TNF-α and VEGF are increased during neuroinflammation and have been shown to decrease BBB integrity.31, 39, 46, 47 We found that both TNF-α and VEGF decrease surface PrPc. To determine the mechanisms for this decrease, we examined changes in PrPc protein and mRNA expression in response to these inflammatory factors. Both TNF-α and VEGF decrease PrPc total protein in BMVEC as well as PRNP expression, suggesting that these factors regulate the de novo synthesis of PrPc.
In our experiments we used TNF-α at 10 ng/ml, which is the concentration used in many in vitro studies. This has been shown to elicit responses in cultured cells that often reflect what can be detected in vivo. Although this value is higher than the physiological TNF-α levels of serum, plasma, and tissue, in pathological conditions serum concentrations of TNF-α of up to 50 ng/ml have been quantified.77, 78 Similarly, serum VEGF levels in normal physiological conditions are lower than the concentration used in our study (100ng/ml). However, serum VEGF values as high as 1208 ng/ml have been shown in pathological states.79
Previously PRNP, the gene encoding PrPc, was thought to be a housekeeping gene because of the absence of a TATA box. However, later evidence showed that this gene has binding sites for several transcription factors including NFAT, AP1, AP2, MEF2, and p53, indicating that PrPc expression may be regulated by various cellular factors.80–83 In human neuroblastoma cells, TNF-α and IL-β cause increased PrPc transcripts while IFN-γ decreased PrPc mRNA.84 In addition to cytokines, heat shock and oxidative stress have also been shown to increase PrPc expression.85, 86
The role of cell associated PrPc in intercellular adhesion and changes in the expression of this protein in response to inflammation have also been studied in other diseases such as irritable bowel syndrome (IBS).26 In intestinal epithelium PrPc localizes to cell junctions and in colonic epithelium of individuals with Crohn’s disease (CD) and ulcerative colitis (UC), hypothesized to be caused, in part, by disrupted barriers, levels of PrPc at cell junctions were decreased. These results suggest that PrPc is essential for barrier integrity and function of the intestine.26
While the presence of PrPc at endothelial cell junctions was shown, its specific contribution to BMVEC monolayer integrity has not been determined. In the present study, we used lentivirus shRNA delivery to knockdown PrPc in BMVEC and examined the effect of PrPc loss on endothelial monolayer permeability. We found that cell lines that had low level surface PrPc formed monolayers that exhibited increased permeability as determined by a FITC-dextran permeability assay. In human Caco-2/TC7 enterocytes, PrPc silencing led to decreased barrier properties of this cell type by changing the localization and expression of junctional and adhesion proteins E-cadherin, claudin-5, and occludin.26 One potential mechanism by which PrPc knockdown increases BMVEC monolayer permeability is by changing the distribution and expression of tight junction and adhesion proteins. Both VEGF and TNF-α downregulate TJ proteins claudin-5, and occludin and increase endothelial barrier permeability.39, 46, 47 In this study we showed that VEGF and TNF-α decrease PrPc on BMVEC and that the absence of PrPc from these cells increases endothelial monolayer permeability. We propose that the downregulation of PrPc induced by VEGF and TNF-α will contribute directly to changes in TJ proteins affecting endothelial monolayer integrity.
Interestingly, in our studies, we found that only nearly complete knockdown of PrPc has an effect on the permeability of endothelial monolayers. These results suggest that during partial knockdown of this protein, the function of PrPc might be compensated by another redundant protein, possibly an adhesion or junction protein. However, when PrPc is completely absent, this compensatory mechanism might not be enough to compensate for the functional role of PrPc in barrier impermeability. Another possible mechanism is that there is excess PrPc on the cell surface and the amount of PrPc in cell lines that showed partial knockdown may be enough to maintain barrier integrity.
In intestinal epithelium, PrPc is important in maintaining the intestinal barrier by activating Src- signaling and regulating expression of TJ and adherence junctions (AJ) proteins including occludin, ZO-1 and E-cadherin.26 In epithelial cell lines including A431, MCF7, and Hela cells, PrPc regulates expression of E-Cadherin and B-catenin through a reggie-1 and EGF-R mediated pathway affecting cell-cell adhesion.7 However, in BMVEC, adhesion or tight junction proteins that are regulated by PrPc, or signaling molecules that are activated or modulated by this protein to affect endothelial barrier integrity have not yet been determined. Therefore, we examined whether knockdown of surface PrPc affects the expression and distribution of TJ proteins that will contribute to a more permeable barrier. We found that PrPc knockdown results in decreased expression of occludin and claudin-5. Since PrPc is not a transmembrane protein, it is possible that it causes these changes in expression of proteins that are essential for membrane integrity by coupling with other adaptor proteins. In this study, we examined the effect of PrPc loss on the expression and localization of occludin and claudin-5. However, since both TNF-α and VEGF regulate the expression of ZO-1,39, 87 we plan on expanding our studies to include the effects of PrPc loss on the regulation of ZO-1 expression.
An intact BBB is essential for maintaining brain homeostasis and highly selective endothelial permeability, and its impairment has been shown to lead to neuroinflammation. Tight junction and adhesion molecules including claudin-5, occludin, and PrPc, are essential for regulating endothelial permeability. Thus, proper localization and expression of these proteins are critical for BBB integrity.19, 20, 22, 23 Our study shows that TNF-α and VEGF, factors associated with BBB disruption, decrease PrPc in BMVEC. PrPc on the cell surface of BMVEC is essential for monolayer integrity and its loss increases permeability. PrPc also regulates the expression and localization of TJ proteins claudin-5 and occludin which are important in maintaining a proper barrier. Therefore, a possible mechanism by which TNF-α and VEGF increase BBB permeability is by downregulating PrPc, which in turn will affect TJ protein expression and localization. This may result in imbalance of ions and transmitters, less efficient prevention of toxin and pathogen infiltration, as well as increased leukocyte diapedesis across the barrier, which can subsequently lead to neuroinflammation.88 In an in vivo setting, a complex interaction of several cell types including astrocytes, microglia and macrophages leads to the neuropathogenesis that is subsequent to HIV infection of the CNS. Our in vitro system in this study enables us to elucidate individual mechanisms which lead to transient permeability of the endothelial layer and blood brain barrier integrity loss. Future studies will examine the effect of inflammation on BBB permeability and cognitive function in PrPc knockout mice in the context of HIV infection.
Supplementary Material
Acknowledgments
We thank Dr. Tina M. Calderon, Dr. Loreto-Torres Carvallo, Dr. Mike Veenstra, Dr. Matias Jaureguiberry, and Lillie Lopez for their valuable contributions to this project. We also thank the shRNA Core and the Flow Cytometry Core at Albert Einstein College of Medicine. This work was supported by U.S. National Institutes of Health Grants R01MH075679 (to JWB); R01DA025567–04 (to JWB); R21MH102113–01A1 (to JWB); R01MH090958 (to JWB and BWM); R01MH07754208 (to JWB and BWM); MH096625 and PHRI Funding (to EAE).
Footnotes
Disclosure/Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Watts JC, Bourkas MEC, Arshad H. The function of the cellular prion protein in health and disease. Acta Neuropathol 2018;135(2):159–178. [DOI] [PubMed] [Google Scholar]
- 2.Sarnataro D, Pepe A, Zurzolo C. Cell Biology of Prion Protein. Prog Mol Biol Transl Sci 2017;150:57–82. [DOI] [PubMed] [Google Scholar]
- 3.Wulf MA, Senatore A, Aguzzi A. The biological function of the cellular prion protein: an update. BMC Biol 2017;15(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linden R The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front Mol Neurosci 2017;10:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castle AR, Gill AC. Physiological Functions of the Cellular Prion Protein. Front Mol Biosci 2017;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirsch TZ, Hernandez-Rapp J, Martin-Lannerée S, et al. PrP C signalling in neurons: From basics to clinical challenges. Biochimie 2014;104:2–11. [DOI] [PubMed] [Google Scholar]
- 7.Petit CS, Besnier L, Morel E, et al. Roles of the cellular prion protein in the regulation of cell-cell junctions and barrier function. Tissue Barriers 2013;1(2):e24377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeon JW, Park BC, Jung JG, et al. The Soluble Form of the Cellular Prion Protein Enhances Phagocytic Activity and Cytokine Production by Human Monocytes Via Activation of ERK and NF-kappaB. Immune Netw 2013;13(4):148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viegas P, Chaverot N, Enslen H, et al. Junctional expression of the prion protein PrPC by brain endothelial cells: a role in trans-endothelial migration of human monocytes. J Cell Sci 2006;119(22):4634–4643. [DOI] [PubMed] [Google Scholar]
- 10.Rousset M, Leturque A, Thenet S. The nucleo-junctional interplay of the cellular prion protein: A new partner in cancer-related signaling pathways? Prion 2016;10(2):143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis V, Hooper NM. The role of lipid rafts in prion protein biology. Front Biosci (Landmark Ed) 2010;16:151–168. [DOI] [PubMed] [Google Scholar]
- 12.Sarnataro D, Campana V, Paladino S, et al. PrPC association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol Biol Cell 2004;15(9):4031–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mattei V, Garofalo T, Misasi R, et al. Association of cellular prion protein with gangliosides in plasma membrane microdomains of neural and lymphocytic cells. Neurochem Res 2002;27(7–8):743–749. [DOI] [PubMed] [Google Scholar]
- 14.Rudd PM, Merry AH, Wormald MR, et al. Glycosylation and prion protein. Curr Opin Struct Biol 2002;12(5):578–586. [DOI] [PubMed] [Google Scholar]
- 15.Rudd PM, Wormald MR, Wing DR, et al. Prion glycoprotein: structure, dynamics, and roles for the sugars. Biochemistry 2001;40(13):3759–3766. [DOI] [PubMed] [Google Scholar]
- 16.Lima FR, Arantes CP, Muras AG, et al. Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J Neurochem 2007;103(6):2164–2176. [DOI] [PubMed] [Google Scholar]
- 17.Brown DR, Besinger A, Herms JW, et al. Microglial expression of the prion protein. Neuroreport 1998;9(7):1425–1429. [DOI] [PubMed] [Google Scholar]
- 18.Richardson DD, Tol S, Valle-Encinas E, et al. The prion protein inhibits monocytic cell migration by stimulating beta1 integrin adhesion and uropod formation. J Cell Sci 2015;128(16):3018–3029. [DOI] [PubMed] [Google Scholar]
- 19.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008;57(2):178–201. [DOI] [PubMed] [Google Scholar]
- 20.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 2006;7(1):41–53. [DOI] [PubMed] [Google Scholar]
- 21.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 2004;84(3):869–901. [DOI] [PubMed] [Google Scholar]
- 22.Theodorakis PE, Muller EA, Craster RV, et al. Physical insights into the blood-brain barrier translocation mechanisms. Phys Biol 2017;14(4):041001. [DOI] [PubMed] [Google Scholar]
- 23.Begley DJ, Brightman MW. Structural and functional aspects of the blood-brain barrier. Prog Drug Res 2003;61:39–78. [DOI] [PubMed] [Google Scholar]
- 24.Stamatovic SM, Keep RF, Wang MM, et al. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem 2009;284(28):19053–19066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schenk GJ, de Vries HE. Altered blood-brain barrier transport in neuro-inflammatory disorders. Drug Discov Today Technol 2016;20:5–11. [DOI] [PubMed] [Google Scholar]
- 26.Petit CS, Barreau F, Besnier L, et al. Requirement of cellular prion protein for intestinal barrier function and mislocalization in patients with inflammatory bowel disease. Gastroenterology 2012;143(1):122–132. e115. [DOI] [PubMed] [Google Scholar]
- 27.Pan W, Kastin AJ. Tumor necrosis factor and stroke: role of the blood–brain barrier. Prog Neurobiol 2007;83(6):363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lv S, Song HL, Zhou Y, et al. Tumour necrosis factor‐α affects blood–brain barrier permeability and tight junction‐associated occludin in acute liver failure. Liver Int 2010;30(8):1198–1210. [DOI] [PubMed] [Google Scholar]
- 29.Friedl J, Puhlmann M, Bartlett DL, et al. Induction of permeability across endothelial cell monolayers by tumor necrosis factor (TNF) occurs via a tissue factor–dependent mechanism: relationship between the procoagulant and permeability effects of TNF. Blood 2002;100(4):1334–1339. [PubMed] [Google Scholar]
- 30.Scutari R, Alteri C, Perno CF, et al. The Role of HIV Infection in Neurologic Injury. Brain Sci 2017;7(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montgomery SL, Bowers WJ. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J Neuroimmune Pharmacol 2012;7(1):42–59. [DOI] [PubMed] [Google Scholar]
- 32.John CC, Panoskaltsis-Mortari A, Opoka RO, et al. Cerebrospinal fluid cytokine levels and cognitive impairment in cerebral malaria. Am J Trop Med Hyg 2008;78(2):198–205. [PMC free article] [PubMed] [Google Scholar]
- 33.Persidsky Y, Buttini M, Limoges J, et al. An analysis of HIV-1-associated inflammatory products in brain tissue of humans and SCID mice with HIV-1 encephalitis. J Neurovirol 1997;3(6):401–416. [DOI] [PubMed] [Google Scholar]
- 34.Feuerstein G, Liu T, Barone F. Cytokines, inflammation, and brain injury: role of tumor necrosis factor-alpha. Cerebrovasc Brain Metab Rev 1993;6(4):341–360. [PubMed] [Google Scholar]
- 35.Tyor WR, Glass JD, Griffin JW, et al. Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol 1992;31(4):349–360. [DOI] [PubMed] [Google Scholar]
- 36.Benveniste EN, Sparacio SM, Norris JG, et al. Induction and regulation of interleukin-6 gene expression in rat astrocytes. J Neuroimmunol 1990;30(2):201–212. [DOI] [PubMed] [Google Scholar]
- 37.Eugenin EA, Osiecki K, Lopez L, et al. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood–brain barrier: a potential mechanism of HIV–CNS invasion and NeuroAIDS. J Neurosci 2006;26(4):1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ehrlich LC, Hu S, Sheng WS, et al. Cytokine regulation of human microglial cell IL-8 production. J Immunol 1998;160(4):1944–1948. [PubMed] [Google Scholar]
- 39.Aveleira CA, Lin C-M, Abcouwer SF, et al. TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 2010;59(11):2872–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobrogowska D, Lossinsky A, Tarnawski M, et al. Increased blood–brain barrier permeability and endothelial abnormalities induced by vascular endothelial growth factor. J Neurocytol 1998;27(3):163–173. [DOI] [PubMed] [Google Scholar]
- 41.Proescholdt MA, Jacobson S, Tresser N, et al. Vascular endothelial growth factor is expressed in multiple sclerosis plaques and can induce inflammatory lesions in experimental allergic encephalomyelitis rats. J Neuropathol Exp Neurol 2002;61(10):914–925. [DOI] [PubMed] [Google Scholar]
- 42.Greenberg DA, Jin K. From angiogenesis to neuropathology. Nature 2005;438(7070):954–959. [DOI] [PubMed] [Google Scholar]
- 43.Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci USA 2000;97(18):10242–10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Storkebaum E, Lambrechts D, Dewerchin M, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci 2005;8(1):85–92. [DOI] [PubMed] [Google Scholar]
- 45.Lange C, Storkebaum E, de Almodovar CR, et al. Vascular endothelial growth factor: a neurovascular target in neurological diseases. Nat Rev Neurol 2016;12(8):439–454. [DOI] [PubMed] [Google Scholar]
- 46.Argaw AT, Gurfein BT, Zhang Y, et al. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA 2009;106(6):1977–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Argaw AT, Zhang Y, Snyder BJ, et al. IL-1β regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol 2006;177(8):5574–5584. [DOI] [PubMed] [Google Scholar]
- 48.Esser S, Lampugnani MG, Corada M, et al. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 1998;111(13):1853–1865. [DOI] [PubMed] [Google Scholar]
- 49.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the β-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 2006;8(11):1223–1234. [DOI] [PubMed] [Google Scholar]
- 50.Grant I, Atkinson JH, Hesselink JR, et al. Evidence for early central nervous system involvement in the acquired immunodeficiency syndrome (AIDS) and other human immunodeficiency virus (HIV) infections: Studies with neuropsychologic testing and magnetic resonance imaging. Ann Int Med 1987;107(6):828–836. [DOI] [PubMed] [Google Scholar]
- 51.Price RW, Brew B, Sidtis J, et al. The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science 1988;239(4840):586–592. [DOI] [PubMed] [Google Scholar]
- 52.Ton H, Xiong H. Astrocyte Dysfunctions and HIV-1 Neurotoxicity. J AIDS Clin Res 2013;4(11):255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ansaloni S, Lelkes N, Snyder J, et al. A streamlined sub-cloning procedure to transfer shRNA from a pSM2 vector to a pGIPZ lentiviral vector. J RNAi Gene Silencing 2010;6(2):411–415. [PMC free article] [PubMed] [Google Scholar]
- 54.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001;410(6831):988–994. [DOI] [PubMed] [Google Scholar]
- 55.Saha RN, Pahan K. Tumor necrosis factor‐α at the crossroads of neuronal life and death during HIV‐associated dementia. J Neurochem 2003;86(5):1057–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eugenin EA, Berman JW. Chemokine-dependent mechanisms of leukocyte trafficking across a model of the blood-brain barrier. Methods 2003;29(4):351–361. [DOI] [PubMed] [Google Scholar]
- 57.Eugenin EA, Clements JE, Zink MC, et al. Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J Neurosci 2011;31(26):9456–9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ballabh P, Braun A, Nedergaard M. The blood–brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis 2004;16(1):1–13. [DOI] [PubMed] [Google Scholar]
- 59.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 2005;57(2):173–185. [DOI] [PubMed] [Google Scholar]
- 60.Luissint A-C, Artus C, Glacial F, et al. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012;9(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morel E, Fouquet S, Chateau D, et al. The cellular prion protein PrPc is expressed in human enterocytes in cell-cell junctional domains. J Biol Chem 2004;279(2):1499–1505. [DOI] [PubMed] [Google Scholar]
- 62.Saylor D, Dickens AM, Sacktor N, et al. HIV-associated neurocognitive disorder--pathogenesis and prospects for treatment. Nat Rev Neurol 2016;12(4):234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spudich S, Gonzalez-Scarano F. HIV-1-related central nervous system disease: current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb Perspect Med 2012;2(6):a007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan P, Hellmuth J, Spudich S, et al. Cognitive Impairment and Persistent CNS Injury in Treated HIV. Curr HIV/AIDS Rep 2016;13(4):209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heaton RK, Clifford DB, Franklin DR Jr., et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010;75(23):2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dietrich J-B. The adhesion molecule ICAM-1 and its regulation in relation with the blood–brain barrier. J Neuroimmunol 2002;128(1):58–68. [DOI] [PubMed] [Google Scholar]
- 67.Aplin AE, Howe AK, Juliano R. Cell adhesion molecules, signal transduction and cell growth. Curr Opin Cell Biol 1999;11(6):737–744. [DOI] [PubMed] [Google Scholar]
- 68.Williams DW, Eugenin EA, Calderon TM, et al. Monocyte maturation, HIV susceptibility, and transmigration across the blood brain barrier are critical in HIV neuropathogenesis. J Leukoc Biol 2012;91(3):401–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang W, Eum SY, Andras IE, et al. PPARalpha and PPARgamma attenuate HIV-induced dysregulation of tight junction proteins by modulations of matrix metalloproteinase and proteasome activities. FASEB J 2009;23(5):1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seilhean D, Dzia-Lepfoundzou A, Sazdovitch V, et al. Astrocytic adhesion molecules are increased in HIV-1-associated cognitive/motor complex. Neuropathol Appl Neurobiol 1997;23(2):83–92. [PubMed] [Google Scholar]
- 71.Roberts TK, Eugenin EA, Morgello S, et al. PrP C, the cellular isoform of the human prion protein, is a novel biomarker of HIV-associated neurocognitive impairment and mediates neuroinflammation. Am J Pathol 2010;177(4):1848–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Megra BW, Eugenin EA, Berman JW. The Role of Shed PrP(c) in the Neuropathogenesis of HIV Infection. J Immunol 2017;199(1):224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Botto L, Masserini M, Cassetti A, et al. Immunoseparation of Prion protein-enriched domains from other detergent-resistant membrane fractions, isolated from neuronal cells. FEBS Lett 2004;557(1–3):143–147. [DOI] [PubMed] [Google Scholar]
- 74.Farina F, Botto L, Chinello C, et al. Characterization of prion protein-enriched domains, isolated from rat cerebellar granule cells in culture. J Neurochem 2009;110(3):1038–1048. [DOI] [PubMed] [Google Scholar]
- 75.Botto L, Cunati D, Coco S, et al. Role of lipid rafts and GM1 in the segregation and processing of prion protein. PLoS One 2014;9(5):e98344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Linsenmeier L, Altmeppen HC, Wetzel S, et al. Diverse functions of the prion protein - Does proteolytic processing hold the key? Biochim Biophys Acta 2017;1864(11 Pt B):2128–2137. [DOI] [PubMed] [Google Scholar]
- 77.Andrade DR Junior, Santos SA, Castro I, et al. Correlation between serum tumor necrosis factor alpha levels and clinical severity of tuberculosis. Braz J Infect Dis 2008;12(3):226–233. [DOI] [PubMed] [Google Scholar]
- 78.Damas P, Reuter A, Gysen P, et al. Tumor necrosis factor and interleukin-1 serum levels during severe sepsis in humans. Crit Care Med 1989;17(10):975–978. [DOI] [PubMed] [Google Scholar]
- 79.Di Raimondo F, Azzaro MP, Palumbo GA, et al. Elevated vascular endothelial growth factor (VEGF) serum levels in idiopathic myelofibrosis. Leukemia 2001;15(6):976–980. [DOI] [PubMed] [Google Scholar]
- 80.Inoue S, Tanaka M, Horiuchi M, et al. Characterization of the bovine prion protein gene: the expression requires interaction between the promoter and intron. J Vet Med Sci 1997;59(3):175–183. [DOI] [PubMed] [Google Scholar]
- 81.Mahal SP, Asante EA, Antoniou M, et al. Isolation and functional characterisation of the promoter region of the human prion protein gene. Gene 2001;268(1):105–114. [DOI] [PubMed] [Google Scholar]
- 82.Varela-Nallar L, Toledo EM, Larrondo LF, et al. Induction of cellular prion protein gene expression by copper in neurons. Am J Physiol Cell Physiol 2006;290(1):C271–C281. [DOI] [PubMed] [Google Scholar]
- 83.Sander P, Hamann H, Drögemüller C, et al. Bovine prion protein gene (PRNP) promoter polymorphisms modulate PRNP expression and may be responsible for differences in bovine spongiform encephalopathy susceptibility. J Biol Chem 2005;280(45):37408–37414. [DOI] [PubMed] [Google Scholar]
- 84.Satoh J-i, Kurohara K, Yukitake M, et al. Constitutive and cytokine-inducible expression of prion protein gene in human neural cell lines. J Neuropathol Exp Neurol 1998;57(2):131–139. [DOI] [PubMed] [Google Scholar]
- 85.Sauer H, Wefer K, Vetrugno V, et al. Regulation of intrinsic prion protein by growth factors and TNF-alpha: the role of intracellular reactive oxygen species. Free Radic Biol Med 2003;35(6):586–594. [DOI] [PubMed] [Google Scholar]
- 86.Shyu WC, Harn HJ, Saeki K, et al. Molecular modulation of expression of prion protein by heat shock. Mol Neurobiol 2002;26(1):1–12. [DOI] [PubMed] [Google Scholar]
- 87.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 2008;32(2):200–219. [DOI] [PubMed] [Google Scholar]
- 88.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med 2013;19(12):1584–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.