

Abstract
Trichomonas vaginalis infects approximately 300 million people worldwide annually. Infected individuals have a higher susceptibility to more serious conditions such as cervical and prostate cancer. The parasite has developed increasing resistance to current drug therapies, with an estimated 5% of clinical cases resulting from resistant strains, creating the need for new therapeutic strategies with novel mechanisms of action. Nucleoside salvage pathway enzymes represent novel drug targets since these pathways are essential for the parasite’s survival. The guanosine/adenosine/cytidine nucleoside hydrolase (GACNH) may be particularly important as its expression is upregulated under glucose-limiting conditions mimicking those that occur during infection establishment. GACNH was screened against the NIH Clinical Collection to explore its druggability. Seven compounds were identified with IC50 values less than 20 μM. Extensive overlap was found between inhibitors of GACNH and the adenosine/guanosine nucleoside hydrolase (AGNH), but no overlap was found with inhibitors of the uridine nucleoside hydrolase (UNH). The guanosine analog ribavirin was the only compound found to be specific for GACNH. Compounds that inhibit both AGNH and GACNH purine salvage pathway enzymes may prove critical given the role that GACNH appears to play in the early stages of infection.
Keywords: NIH Clinical Compound Collection, NMR, Nucleoside hydrolase, ribavirin, Trichomonas vaginalis
Graphical Abstract
Trichomonas vaginalis guanosine/adenosine/cytidine nucleoside hydrolase (GACNH) was screened against the NIH Clinical Collection to explore its druggability. Seven compounds were identified with IC50 values less than 20 μM. Extensive overlap was found between inhibitors of GACNH and the adenosine/guanosine nucleoside hydrolase (AGNH), but no overlap was found with inhibitors of the uridine nucleoside hydrolase (UNH). The guanosine analog ribavirin was the only compound found to be specific for GACNH.
The parasitic protozoan Trichomonas vaginalis causes the sexually transmitted infection trichomoniasis. It affects approximately 300 million people worldwide annually with the majority of cases occurring in developing nations.[1–3] The Centers for Disease Control and Prevention recognizes trichomoniasis as a neglected parasitic infection, with an infection prevalence close to 4 million in the United States.[4] Individuals with trichomonal infections have a higher susceptibility to more serious conditions such as cervical cancer, HIV-1, and pelvic inflammatory disease.[5,6] More recently, trichomonal infection has also been associated with prostate cancer and benign prostatic hyperplasia.[7–9]
Trichomoniasis is typically treated with 5-nitroimidazole drugs such as metronidazole and tinidazole, with metronidazole being the first-line treatment used in the United States since the 1960s.[5,10] The compounds themselves are inactive, but are anaerobically reduced by redox enzymes in the parasitic hydrogenosome forming toxic nitro radical anions which target thymine and adenine residues in the pathogen’s DNA.[5,10] Resistance to metronidazole is increasing, with an estimated 5% of trichomoniasis clinical cases resulting from T. vaginalis strains with some resistance.[10] The need to combat the rising resistant strains of T. vaginalis has created a demand for new therapeutic strategies with novel mechanisms of action.
T. vaginalis is incapable of the de novo synthesis of purines[11] and pyrimidines[12] and therefore relies on salvage pathway enzymes to scavenge nucleosides from host cells to obtain its necessary nucleobases. RNA interference and gene knock out experiments in the closely related parasite Trypanosoma brucei have demonstrated the essential nature of these salvage pathways.[13,14] The key enzymes used in these pathways are nucleoside hydrolases (NHs). NHs catalyze the hydrolysis of nucleosides obtained from the host at the N-glycosidic bond, yielding a molecule of ribose and a free nucleic base. The parasite in turn uses the free nucleic bases to synthesize its own DNA to carry out its metabolic processes. NHs are specific for ribonucleosides, but display variability in their preference for the nucleic base.[15] The identification of inhibitors of NHs that block this pathway can lead to the development of antiparasitic drugs that are mechanistically distinct from current treatment strategies.
The consensus amino acid sequence DXDXXXDD[15] was used to probe the T. vaginalis genome[16] and identify at least three NHs: adenosine/guanosine nucleoside hydrolase (TVAG_213720),[17] guanosine/adenosine/cytidine nucleoside hydrolase (TVAG_305790),[18] and uridine nucleoside hydrolase (TVAG_092730).[19] Kinetic characterizations with reducing sugar assays were used to determine their respective substrate specificities based on kcat/Km catalytic efficiency values.[17,18] AGNH efficiently hydrolyzes adenosine and guanosine but has only barely detectable activity toward cytidine or uridine. GACNH has broad activity toward guanosine, adenosine, and cytidine but does not hydrolyze uridine. UNH is highly specific for uridine, with only marginal activity toward cytidine and no measurable activity for the other nucleosides. AGNH and GACNH likely have larger active sites compared to UNH, enabling them to accommodate the larger purine rings. A fourth putative NH was also identified, TVAG_424130, but it has not yet been characterized to confirm its function. These four nucleoside hydrolases share a high degree of sequence homology with a T-Coffee score of 86,[17] and their putative function was independently annotated in the T. vaginalis genome database.[20]
The druggability of AGNH and UNH have been tested previously by developing NMR-based activity assays to screen the National Institutes of Health Clinical Compound Collection for inhibitors. Flavonoids were identified as strong inhibitors of AGNH,[21] while the proton pump inhibitors, omeprazole, pantoprazole, and rabeprazole were identified as potent inhibitors of UNH.[22] Both enzymes are now the basis of fragment-based drug discovery projects. This paper describes 1H NMR-based activity assays to delineate the druggability of GACNH. As in our previous work, NMR was used for screening in order to minimize assay interference at the higher concentrations of compounds tested. Inhibition of GACNH may be particularly important as its expression is upregulated under glucose-limiting conditions mimicking those that occur during infection establishment.[23] Novel drugs that target purine NHs will likely need to be effective against both AGNH and GACNH.
1H NMR data sets were collected on a Bruker AvanceIII 500 MHz NMR spectrometer equipped with a BBFO probe and a SampleXpress. The acquisition and relaxation delay times of 2.045 s and 1.0 s, respectively, resulted in a total acquisition time of 13 min per spectrum with 256 scans. Compounds screened were a high solubility subset of the NIH Clinical Collection and NIH Clinical Collection 2 and provided as curated 10 mM DMSO solutions.[24] To increase throughput, compounds were screened in mixtures of three. For each reaction sample, 12 μL of 5 mM adenosine and 3 μL of each test compound were added to microfuge tubes. The reaction was then initiated with 585 μL of a stock solution containing 50 mM potassium phosphate buffer with 0.3 M KCl at pH 6.5, 10% 2H2O, and 12 nM GACNH. The final concentrations of adenosine and each compound were 100 μM and 50 μM, respectively. After 40 min, reactions were quenched with 10 μL of 1.5 M HCl. In total, 573 of the 727 compounds from the NIH Collection were screened; compounds with predicted water solubilities less than 50 μM were excluded from screening.[22] To determine IC50 values, compounds were serially diluted with DMSO to concentrations ranging from 250 μM to 0.08 μM. Since compounds screened at such high concentrations can be false positives because of aggregation, IC50 values were also determined in the presence of 0.01% Triton X-100. IC50 curves and values were obtained using GraphPad Prism. Activity assays for AGNH and UNH were carried out using 1H NMR and 19F NMR, respectively, as described previously.[21,22]
Figure 1 illustrates an example of a screened NIH Clinical Collection plate NGP-202-04. Ten assay mixtures were screened, totaling 30 test compounds. The spectra for control samples without test compounds for time 0 and 40 min and for 10 assayed mixtures are shown. Despite the presence of 1H resonances from the test compounds, substrate adenosine signals (6.10 and 8.48 ppm) and product adenine signals (8.33 ppm) are clearly resolved in all spectra. In comparison to the 40 min control spectrum, mixture 1 exhibited significant inhibition evidenced by its lack of product signal at 8.33 ppm. Mixture 1 was subsequently deconvoluted by retesting compounds individually to determine the compound responsible for inhibition. This deconvolution is shown in Figure 2. In comparison to the control spectra, the spectrum obtained with ribavirin displayed significant inhibition because a product signal at 8.33 ppm was not observed.
Figure 1.

Control (0 min and 40 min) and selected mixture (M1–M10) 1H NMR data sets for NIH Clinical Collection plate NGP-202-04.
Figure 2.

Control (0 min and 40 min) and deconvolution (A2, B2, C2) 1H NMR data sets for mixture 1 from NIH Clinical Collection plate NGP-202-04. Wells A2, B2, and C2 contain the compounds propranolol hydrochloride, ribavirin, and terazosin, respectively.
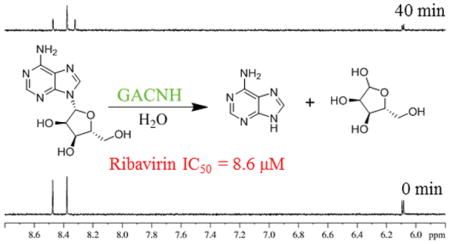
The 16 compounds displaying > 40% inhibition (Supplementary Table 1) were obtained commercially as pure solids and then tested to determine their IC50 values. To determine IC50 values, the hits were assayed in a concentration-dependent manner. Figure 3 shows the dose-dependent spectra for ribavirin. Spectra for higher concentrations (250 and 50 μM) closely resemble the 0 min control, indicating strong inhibition. As the concentration becomes increasingly dilute, significant product formation is observed. The IC50 value determined for ribavirin from these data was 8.6 ± 4.4 μM. Table 1 summarizes the IC50 values for the top seven GACNH inhibitors identified.
Figure 3.

Control (0 min and 40 min) and variable concentration (250 μM to 0.04 μM) 1H NMR data sets for ribavirin.
Table 1.
IC50 values for GACNH inhibitors in the presence and absence of Triton X-100.
| Compound | IC50 (μM) no Triton | IC50 (μM) with Triton |
|---|---|---|
| Benzbromarone | 20.6 ± 19.9 | 133.7 ± 32.7 |
| CCPAa | 14.3 ± 1.5 | 14.0 ± 5.8 |
| Ribavirin | 8.6 ± 4.4 | 9.0 ± 3.3 |
| Hyperoside | 5.5 ± 4.4 | 22.6 ± 7.6 |
| Icariin | 0.2 ± 0.1 | 5.0 ± 1.7 |
| Isoquercitrin | 0.5 ± 0.3 | 2.2 ± 0.6 |
| (±)-Taxifolin | 20.4 ± 11.7 | 41.6 ± 20.4 |
2-chloro-N(6)-cyclopentyladenosine.
Screens run at high concentrations often identify pan assay interference compounds which nonspecifically bind to biological targets rather than specifically binding within the active site.[25] Since nonspecific inhibition can be caused by compounds aggregating in solution, counter screens run in the presence of Triton X-100, a detergent that breaks up aggregates, are useful.[26] Thus IC50 values were determined in the presence of 0.01% TX-100 to weed out false positives resulting from nonspecific inhibition. If the IC50 curve shifted in the presence of detergent, then it was suspected that the inhibitor was aggregating in solution and causing nonspecific inhibition.[27] IC50 values in the presence of TX-100 are included in Table 1. Further evidence for the lack of aggregation was observed in the dose dependent NMR spectra. For example, as the concentration of ribavirin increased from 50 μM to 250 μM, its 1H resonance at 8.7 ppm remained sharp and did not shift (Figure 3). Thus, inhibition by ribavirin is not aggregation-based. Similar results were obtained for CCPA and (±)-taxifolin ruling out nonspecific inhibition for these two compounds as well. However, modest shifts were observed in the IC50 values of hyperoside, icariin, and isoquercitrin in the presence of TX-100. These results suggest that the glycoside moieties are increasing the self-attractive properties of these compounds and thereby causing some aggregation. The data also suggest that benzbromarone is a nonspecific inhibitor.
Three structural classes of GACNH inhibitors were identified: flavonoids, adenosine analogs, and ribavirin. Counter screens in the presence of detergent along with the SAR described below for the flavonoids and ribavirin analogs validate these screening hits according to recently published ACS standards.[28] Flavonoids have a wide range of biological and pharmacological properties. More specifically, they are known to have anti-microbial, anticancer, and anti-inflammatory activities.[29] Isoquercitrin, hyperoside, icariin, (±)-taxifolin, and (+)-taxifolin are classified as flavonoids. These inhibitors have a common catechol moiety that is speculated to be important for inhibition because it resembles the hydroxyl groups of ribose found in the other classes of inhibitors as shown in Figure 4. The 2′- and 3′ hydroxyl groups likely coordinate the Ca2+ in the active site to position the substrate for hydrolysis.[15] As in our previous work on AGNH, (±)-taxifolin was found to be more potent than (+)-taxifolin (IC50 value 81.5 ± 38.6 μM), indicating that (−)-taxifolin is also the more potent dihydroflavonol stereoisomer for GACNH.
Figure 4.

Chemical structures of CCPA, ribavirin, and (−)-taxifolin.
2-Chloro-N6-cyclopentyladenosine (CCPA) is from a second structural class, namely adenosine analogs. CCPA is a receptor agonist for the adenosine A1 receptor.[30] CCPA resembles adenosine in that it contains a pyrimidine ring connected by an N-glycosidic bond to ribose. It is interesting that this bond is not hydrolyzed by GACNH since NHs are responsible for cleaving N-glycosidic bonds. Studies have revealed that the NH catalyzed hydrolysis of β-ribonucleosides proceeds via an SN1 mechanism with an oxocarbenium-ion-like transition state.[15] It is hypothesized that the chlorine substitution at the 2 position on the adenine ring may preclude formation of this transition state, preventing GACNH from hydrolyzing the N-glycosidic bond.
The flavonoid and adenosine compounds found to inhibit GACNH were previously found to inhibit AGNH. This finding is not so surprising given the fact that both enzymes catalyze the hydrolysis of adenosine and guanosine with near equal catalytic efficiency. The guanosine analog, ribavirin, however, was identified as a unique inhibitor of GACNH. IC50 values for ribavirin against AGNH and UNH could not be determined as complete inhibition was not observed at the highest concentrations tested. However, the % inhibition at 250 μM was determined to be 83% and 58% for AGNH and UNH, respectively. Since GACNH also catalyzes the hydrolysis of cytidine, the amino group of ribavirin could also be forming similar interactions in the active site as the cytosine base. This might explain ribavirin’s lack of UNH inhibition, with the UNH active site being optimized for the uracil ring containing a carbonyl group in place of the cytosine amino group.
To further explore the SAR of ribavirin, several analogs were obtained commercially and tested for activity against the three enzymes. Structures and selectivity data are shown in Figure 5 and Table 2, respectively. Addition of polar amino or hydroxyl groups to the nucleobase five-membered ring resulted in diminished activity against all three enzymes, suggesting a conserved structural feature that disfavors hydrogen bonding involving this part of the inhibitor. Replacement of the carboxamide group with a carboxamidine group enhances the potency against GACNH by about 10-fold and, interestingly, against AGNH by more than 100-fold. This compound, viramidine, a prodrug of ribavirin, has a resulting 10-fold selectivity toward AGNH over GACNH. The surprise finding that viramidine is a potent AGNH inhibitor may provide insights into the design of AGNH inhibitors. We are attempting to build homology models based on nucleoside hydrolase structures from related species in order to further understand the structural basis for this selectivity.
Figure 5.

Chemical structures of ribavirin nucleobase analogs.
Table 2.
IC50 values (μM) or % inhibition at 250 μM for ribavirin analogs against AGNH, GACNH, and UNH.
| Compound | AGNH | GACNH | UNH |
|---|---|---|---|
| Acadesine | NMa | 70% | 51% |
| Mizoribine | NM | NM | NM |
| Ribavirin | 83% | 8.6 ± 4.4 | 58% |
| Viramidine | 0.12 ± 0.03 | 1.1 ± 0.6 | 55% |
Not measurable (percent inhibition < 10%).
There was no overlap between the 16 GACNH inhibitors and the proton pump inhibitors previously identified for UNH. This finding is also not surprising, and furthers supports the notion that the pyrimidine-specific UNH is a separately druggable target from the purine-specific AGNH and GACNH. The substantial crossover activity between AGNH and GACNH is important mainly for two reasons. First, our fragment-based drug discovery efforts focusing on AGNH will likely identify compounds that inhibit both AGNH and GACNH. It will be critical to include GACNH assays in the discovery funnel to verify this. Novel drugs that target purine NHs will likely need to be effective against both AGNH and GACNH so that one enzyme does not compensate for selective inhibition of the other. Second, a recent transcript profiling study[23] found that GACNH is upregulated 10-fold under conditions of glucose restriction that enhance cell survival. Neither AGNH nor UNH is upregulated under these conditions. GACNH may play a unique role that is necessary to establish an infection, such as providing ribose as an alternative energy source.
Supplementary Material
Supplementary Table 1. Structures of the 16 compounds displaying > 40% GACNH inhibition.
Acknowledgments
Research reported in this publication was supported by the National Institute Of Allergy And Infectious Diseases of the National Institutes of Health under Award Number R15AI128585. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest
The authors declare no commercial or financial conflicts of interest
References
- 1.Kissinger P. BMC Infect Dis. 2015;15:307. doi: 10.1186/s12879-015-1055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bradic M, Warring SD, Tooley GE, Scheid P, Secor WE, Land KM, Huang PJ, Chen TW, Lee CC, Tang P, Sullivan SA, Carlton JM. Genome Biol Evol. 2017;9:1658. doi: 10.1093/gbe/evx110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouchemal K, Bories C, Loiseau PM. Clin Microbiol Rev. 2017;30:811. doi: 10.1128/CMR.00109-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. [Accessed 1 March 2018]; http://www.cdc.gov/std/trichomonas/stats.htm.
- 5.Secor WE. Expert Rev Anti Infect Ther. 2012;10:107. doi: 10.1586/eri.11.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirt RP, Sherrard J. Curr Opin Infect Dis. 2015;28:72. doi: 10.1097/QCO.0000000000000128. [DOI] [PubMed] [Google Scholar]
- 7.Stark JR, Judson G, Alderete JF, Mundodi V, Kucknoor AS, Giovannucci EL, Platz EA, Sutcliffe S, Fall K, Kurth T, Ma J, Stampfer MJ, Mucci LA. J Natl Cancer Inst. 2009;101:1. doi: 10.1093/jnci/djp306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitteregger D, Aberle SW, Makristathis A, Walochnik J, Brozek W, Marberger M, Kramer G. Med Microbiol Immunol. 2012;201:113. doi: 10.1007/s00430-011-0205-2. [DOI] [PubMed] [Google Scholar]
- 9.Twu O, Dessí D, Vu A, Mercer F, Stevens GC, de Miguel N, Rappelli P, Cocco AR, Clubb RT, Fiori PL, Johnson PJ. Proc Natl Acad Sci USA. 2014;111:8179. doi: 10.1073/pnas.1321884111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cudmore SL, Delgaty KL, Hayward-McClelland SF, Petrin DP, Garber GE. Clin Microbiol Rev. 2004;17:783. doi: 10.1128/CMR.17.4.783-793.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heyworth PG, Gutteridge WE, Ginger CD. FEBS Lett. 1982;141:106. doi: 10.1016/0014-5793(82)80026-4. [DOI] [PubMed] [Google Scholar]
- 12.Heyworth PG, Gutteridge WE, Ginger CD. FEBS Lett. 1984;176:55. doi: 10.1016/0014-5793(84)80910-2. [DOI] [PubMed] [Google Scholar]
- 13.Berg M, Kohl L, Van der Veken P, Joossens J, Al-Salabi MI, Castagna V, Giannese F, Cos P, Versées W, Steyaert J, Grellier P, Haemers A, Degano M, Maes L, de Koning HP, Augustyns K. Antimicrob Agents Chemother. 2010;54:1900. doi: 10.1128/AAC.01787-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ali JAM, Tagoe DNA, Munday JC, Donachie A, Morrison LJ, de Koning HP. PLoS One. 2013;8:e58034. doi: 10.1371/journal.pone.0058034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Versées W, Steyaert J. Curr Opin Struct Biol. 2003;13:731. doi: 10.1016/j.sbi.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Carlton JM, Hirt RP, Silva JC, Delcher AL, Schatz M, Zhao Q, Wortman JR, Bidwell SL, Alsmark UCM, Besteiro S, Sicheritz-Ponten T, Noel CJ, Dacks JB, Foster PG, Simillion C, Van de Peer Y, Miranda-Saavedra D, Barton GJ, Westrop GD, Muller S, Dessi D, Fiori PL, Ren Q, Paulsen I, Zhang H, Bastida-Corcuera FD, Simoes-Barbosa A, Brown MT, Hayes RD, Mukherjee M, Okumura CY, Schneider R, Smith AJ, Vanacova S, Villalvazo M, Haas BJ, Pertea M, Feldblyum TV, Utterback TR, Shu CL, Osoegawa K, de Jong PJ, Hrdy I, Horvathova L, Zubacova Z, Dolezal P, Malik SB, Logsdon JM, Jr, Henze K, Gupta A, Wang CC, Dunne RL, Upcroft JA, Upcroft P, White O, Salzberg SL, Tang P, Chiu CH, Lee YS, Embley TM, Coombs GH, Mottram JC, Tachezy J, Fraser-Liggett CM, Johnson PJ. Science. 2007;315:207. doi: 10.1126/science.1132894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benzie AL. Honors Thesis. Adelphi University; May, 2011. http://libraries.adelphi.edu. [Google Scholar]
- 18.Barskaya A. Honors Thesis. Adelphi University; May, 2011. http://libraries.adelphi.edu. [Google Scholar]
- 19.Rosario I. Honors Thesis. Adelphi University; May, 2011. http://libraries.adelphi.edu. [Google Scholar]
- 20. [Accessed 27 April 2018]; http://trichdb.org/trichdb/
- 21.Beck S, Muellers SN, Benzie AL, Parkin DW, Stockman BJ. Bioorg Med Chem Lett. 2015;25:5036. doi: 10.1016/j.bmcl.2015.10.030. [DOI] [PubMed] [Google Scholar]
- 22.Shea TA, Burburan PJ, Matubia VN, Ramcharan SS, Rosario I, Jr, Parkin DW, Stockman BJ. Bioorg Med Chem Lett. 2014;24:1080. doi: 10.1016/j.bmcl.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Huang KY, MChen YY, Fang YK, Cheng WH, Cheng CC, Chen YC, Wub TE, Ku FM, Chen SC, Lin R, Tang PP. Biochim Biophys Acta. 2014;1840:53. doi: 10.1016/j.bbagen.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 24. [Accessed 5 May 2016]; http://www.nihclinicalcollection.com.
- 25.Baell JB, Holloway GA. J Med Chem. 2010;53:2719. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 26.Feng BY, Shoichet BK. Nat Protoc. 2006;1:550. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LaPlante SR, Carson R, Gillard J, Aubry N, Coulombe R, Bordeleau S, Bonneau P, Little M, O’Meara J, Beaulieu PL. J Med Chem. 2013;56:5142. doi: 10.1021/jm400535b. [DOI] [PubMed] [Google Scholar]
- 28.Aldrich C, Bertozzi C, Georg GI, Kiessling L, Lindsley C, Liotta D, Merz KM, Jr, Schepartz A, Wang S. ACS Cent Sci. 2017;3:143. doi: 10.1021/acscentsci.7b00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benavente-García O, Castillo J. J Agric Food Chem. 2008;56:6185. doi: 10.1021/jf8006568. [DOI] [PubMed] [Google Scholar]
- 30.Lohse MJ, Klotz KN, Schwabe U, Cristalli G, Vittori S, Grifantini M. Naunyn-Schmiedeberg’s Arch Pharmacol. 1988;337:687. doi: 10.1007/BF00175797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Structures of the 16 compounds displaying > 40% GACNH inhibition.