

Abstract
AIM
To determine whether and to what extent the gut microbiome is involved in regulating racial disparity in colorectal cancer (CRC).
METHODS
All patients were recruited and experiments were performed in accordance with the relevant guidelines and regulations by the Institutional Review Boards (IRB), committees of the John D. Dingell VAMC and Wayne State University guidelines. African American (AA) and Caucasian American (CA) patients were scheduled for an outpatient screening for colonoscopy, and no active malignancy volunteer patients were doubly consented, initially by the gastroenterologist and later by the study coordinator, for participation in the study. The gut microbial communities in colonic effluents from AAs and CAs were examined using 16sRNA profiling, and bacterial identifications were validated by performing SYBR-based Real Time PCR. For metagenomic analysis to characterize the microbial communities, multiple software/tools were used, including Metastats and R statistical software.
RESULTS
It is generally accepted that the incidence and mortality of CRC is higher in AAs than in CAs. However, the reason for this disparity is not well understood. We hypothesize that the gut microbiome plays a role in regulating this disparity. Indeed, we found significant differences in species richness and diversity between AAs and CAs. Bacteroidetes was more abundant in AAs than in CAs. In particular, the pro-inflammatory bacteria Fusobacterium nucleatum and Enterobacter species were significantly higher in AAs, whereas probiotic Akkermansia muciniphila and Bifidobacterium were higher in CAs. The polyphyletic Clostridia class showed a divergent pattern, with Clostridium XI elevated in AAs, and Clostridium IV, known for its beneficial function, higher in CAs. Lastly, the AA group had decreased microbial diversity overall in comparison to the CA group. In summary, there were significant differences in pro-inflammatory bacteria and microbial diversity between AA and CA, which may help explain the CRC disparity between groups.
CONCLUSION
Our current investigation, for the first time, demonstrates microbial dysbiosis between AAs and CAs, which could contribute to the racial disparity of CRC.
Keywords: Human gut, Microbiome, Colorectal cancer, Fusobacterium nucleatum, African Americans, 16S RNA profiling, Metagenomics
Core tip: Several studies have demonstrated that the incidence of colorectal cancer (CRC) is higher in African Americans than Caucasian Americans. Reasons for this racial disparity are unknown. The current study, for the first time, demonstrated that dysbiosis in the gut microbiome plays a determinant role in the racial disparity of CRC. Determining the influence of the microbiota on the risk of developing CRC will have a major impact on health, since early-stage CRC hinges on the ability to detect early pathological changes. Subsequent translational studies could also be developed to alter microbiota with medications or diet, thus reducing the risk of developing CRC.
INTRODUCTION
Colorectal cancer (CRC) is the second leading cause of cancer death in the United States, and several studies have demonstrated that African Americans (AAs) have the highest rate of CRC in the United States[1-6]. AAs have the highest incidence and death rates for CRC than Caucasian Americans (CAs), Hispanics and Asian/Pacific Islanders[7]. CRC typically correlates with age, reflecting a multistep progression from normal epithelium to carcinoma. However, a significant number of AAs are diagnosed with CRC at a younger age compared to CAs[8-11]. Genomic alterations in oncogenes and tumor suppressor genes drive the epithelial cell transformation to carcinoma - including the Apc/Wnt-β-catenin signaling pathway and the tumor suppressor gene Apc[12-14]. Previous studies have shown that microRNA drivers upregulated in AAs lead to an increased proportion of cancer stem cells in human colonic epithelial cells[15].
The human microbiome is at the interface of intrinsic and environmental factors - and abnormalities in the gut microbiome have been noted in patients with CRC[16-19]. The colonic microbiota is mostly bacteria consisting of approximately 103 different microbial species[20]. Gut microbiota is essential in the maintenance of homeostasis, and it contributes to immune development, inhibits pathogen colonization, processes drug metabolites, metabolizes nutrients from the diet and also modulates their biological activities[10,21]. Dysregulation of gut microbiota and a concomitant state of chronic inflammation and persistent activation of the host immune system have been implicated in the initiation and development of CRC[22-24].
One primary role of gut bacteria is to participate in the biotransformation of products in the gut, which include bile acids secreted from the liver. The gut microbiota may alter cancer susceptibility and has anti-cancer effect through the production of microbial secretory metabolites, such as short chain fatty acids (SCFAs), secondary bile acids (SBA), and trimethylamine N-oxide (TAMO)[25-28]. SBAs, deoxycholic acid (DCA) and lithocholic acid (LCA), are noted in particular for their carcinogenic activity[29-33]. Murine models have demonstrated that DCA shifts the microbiota community to dysbiosis and promotes intestinal tumorigenesis in Apcmin/+ mice when DCA-treated fecal microbiota inoculated in one mouse is transferred to another[34].
Thus, multifactorial reasons underlie the racial disparity of CRC. The current investigation was aimed at studying microbial dysbiosis in the gut between AAs and CAs. In this pilot study, we investigate the diversity and abundance of specific gut microbes in colonic effluents using 16S rRNA gene community profiles in AAs and CAs and their possible role in increased incidence of CRC in AAs.
MATERIALS AND METHODS
Study subjects and collection of samples
In the current pilot investigation, 52 AA and 46 CA patients were recruited. The study was approved by the Institutional Review Boards and Committees of the John D. Dingell-Veterans Affairs Medical Center (JDD-VAMC) and Wayne State University School of Medicine. Patients excluded from the study were those with active malignant disease, inflammatory bowel disease, recent infection and those recently treated with antibiotics. In addition, patients with psychiatric or addictive disorder, hemorrhagic diathesis or on warfarin were excluded. General characteristics of study participants are the same as described in our earlier publication[15]. None of the patients were taking probiotics as supplements. General characteristics of each group of patients are presented in Table 1.
Table 1.
General characteristics of African American and Caucasian American patients
| Race | Gender | Age | Height (inches) | Weight (lbs) | Body mass index | Polyp | Adenoma |
| AA | Male | 65.2 | 70.4 | 181 | 28.9 | 4.6 | 3.8 |
| CA | Male | 62.6 | 69.0 | 194 | 31.0 | 1.43 | 1.0 |
AA: African Americans; CA: Caucasian Americans.
Eligible study subjects were scheduled for an outpatient screening colonoscopy at the JDD-VAMC. All study subjects received standard colonoscopy purgative preparation. Briefly, patients were asked to stay on a clear liquid diet for 24 h and to take a preparation containing 15 mg Bisacodyl the morning prior to their colonoscopy. The patients were also instructed to split the dose (4 L) of poly-ethylene glycol solution (PEG) into a first half (2 L) the evening prior to colonoscopy, and to drink the second half (the remaining 2 L) 5 h prior to the procedure and to finish it 3 h prior to the procedure, regardless of appointment time (morning or afternoon). Colonic effluent was aspirated prior to the colonoscopy through the working channel of the endoscope, as reported earlier[15]. Additionally, 8 forceps biopsies were taken from macroscopically normal appearing colonic mucosa (< 10 cm anal verge), as described previously[15].
DNA extraction for 16S rRNA gene microbial community profiling
Genomic DNA was extracted from colonic effluents using QIAamp DNA Stool Mini Kit (QIAGEN) according to the manufacturer’s instructions. Purified DNA used for analysis of 16S rRNA community profiling was performed by LC Sciences (Houston, Texas). The V3 and V4 regions of the 16S rRNA gene were amplified to generate approximately 469 bp amplicons, automating cluster generation and sequencing on the MiSeq system. For data analysis, the merge paired-end reads from DNA fragments were analyzed using next generation sequencing FLASH (Fast Length Adjustment SHort reads) software, and raw sequencing data quality control was checked using FastQC software. Operational taxonomic units (OTUs) clustering was based on 97% sequence similarity using CD-HIT software. Microbial strain identification software Quantitative Insights Into Microbial Ecology (QIIME) was used for alpha diversity and beta diversity, visualization of high throughput microbial community, and for principal coordinates analysis. Ribosomal Database Project (RDP) classifier, Greengenes, NCBI 16SMicrobial (TUIT tool) and GraPhlAn software were used for taxonomic classification and circular taxonomic phylogenetic trees.
Genomic DNA isolation from colonic effluents and validation
Bacterial genomic DNA was isolated from colonic effluents using QIAamp DNA Stool Mini Kit (QIAGEN) according to the manufacturer’s instructions. For real time PCR, DNA (40-50 ng) and appropriate blank were used for RT-PCR analysis in triplicate using the 2 × Power-Up SYBR Green PCR Master Mix (Applied Biosystems) and the ABI Prism 7500 sequence detection system. PCR consisted of 40 cycles of 95 °C for 10 min and then 95 °C for 15 s, 60 °C for 60 s. The primer sequences were used to evaluate the presence of specific types of bacteria. Ct values were utilized to assess the relative concentration of specific DNA for each sample as described by the manufacturer. Each sample ΔΔCt values was calculated by normalizing to the CT value of total bacteria (Eubacteria). 16S rDNA served as an internal control and each value represented the mean of three replicates. All oligonucleotide primers were synthesized by Integrated DNA technology Inc. (Coralville, IA, United States). The primer set for each gene is listed in Table 2.
Table 2.
List of primers for bacterial genes specific for family, genus and species
| Bacterial gene | Forward primer (5’-3’) | Reverse primer (5’-3’) | Ref. |
| Clostridium cluster XI | TGACGGTACYYNRKGAGGAAGCC | CTACGGTTRAGCCGTAGCCTTT | [63] |
| Clostridium cluster XIVa | GCGGTRCGGCAAGTCTGA | CCTCCGACACTCTAGTMCGAC | [64] |
| Clostridium cluster IV | GCACAAGCAGTGGAGT | CTTCCTCCGTTTTGTCAA | [65] |
| Bifidobacterium genus | GATTCTGGCTCAGGATGAACGC | CTGATAGGACGCGACCCCAT | [66] |
| Lactobacillus spp. | AGCAGTAGGGAATCTTCCA | CACCGCTACACATGGAG | [66] |
| Enterobacter (Family) | CATTGACGTTACCCGCAGAAGAAGC | CTCTACGAGACTCAAGCTTGC | [66] |
| Fusobacterium (genus) | GGATTTATTGGGCGTAAAGC | GGCATTCCTACAAATATCTACGAA | [67] |
| Fusobacterium nucleatum | CAACCATTACTTTAACTCTACCATGTTCA | GTTGACTTTACAGAAGGAGATTATGTAAAAATC | [68] |
| Clostridium sordelli | CTGAGACACGTCCAAACTCTAC | CCTCCTCAAGTACCGTCATTATC | - |
| Total bacteria | CGTGCCAGCAGCCGCGG | TGGACTACCAGGGTATCTAATCCTG |
Real-Time PCR from biopsy
To determine the specific bacterial abundance between serrated and tubular adenomatous patients, total RNA was prepared from patient biopsy samples using TRIzol as recommended by the manufacturer and purified using the Rneasy Mini Kit (QIAGEN). For real time PCR, cDNA was prepared with the SuperScript III First-Strand cDNA synthesis system for RT-PCR (Invitrogen) and analyzed in triplicate using the 2 × Power-Up SYBR Green PCR Master Mix (Applied Biosystems) and the ABI Prism 7500 sequence detection system. Primers and PCR were performed as described in the previous section.
For determination of RT-PCR expression of 7-α-dehydroxylase (BaiCD), primers were as follows, baiCD forward: 5’-GGWTTCAGCCCRCAGATGTTCTTTG-3’; reverse: 5’GAATTCCGGGTTCATGAACATTCTKCKAAG-3’[35].
Statistical analysis
For microbiota data statistical analysis, Metastats software was used for metagenomics sequencing data analyzed from two groups to characterize the microbial communities. CD-HT and R statistical software was used for BIOM-formatted OTU communities clustering and OTU statistics. For examining alpha diversity, QIIME software was used for graphics and statistical purposes. RDP classifier, QIIME, TUIT GraPhlAn, MetaPhlAn, R software/Too were used for taxonomic classification and statistics. For Real Time PCR data, the standard deviation of mean between two groups and t-test were performed to determine the significance level between two groups.
RESULTS
Phylogenetic analysis of microbial communities in colonic effluents
Microbiota composition of colonic effluent was compared by high throughput analysis of 16S small ribosomal subunit gene (16S rRNA) amplicon. Sequencing of the V3 + V4 region was used for OTU clustering based on 97% sequence similarity. We found unique OTUs in AAs (7234) and CAs (5252), with an overlap of 742 OTUs between the two groups (Figure 1A). We found higher species richness and species diversity in CAs using the number of OTUs in Shannon index (Figure 1B). Irrespective of high inter-individual variances, the Principal Coordinates Analysis (PCoA) showed AAs to possess an abundance of common microbiota, compared to dispersed CA counterparts, revealing more microbial homogeneity within AAs than CA patients (Figure 1C).
Figure 1.

Microbial diversity in colonic effluent from African Americans and Caucasian Americans. A: Venn diagram showing the unique Operational taxonomic units (OTUs) in different subsets of African American (AA) and Caucasian Americans (CA) including overlap community; B: Rarefaction curves showing the species richness from the average number of OTUs for AA and CA (alpha diversity); C: Principal coordinates analysis for beta diversity showing the very dissimilar individual in the CA group while closely similar to the group indicated by the yellow circle. CD-HIT and R software were used for BIOM format OTU clustering and OTU statistics. For measurement of alpha diversity of observed species, QIIME software and Shannon index were used and the emperor tool generated the PCoA plot.
The microbiota composition of AAs and CAs showed significant differences in the Bacteroidetes and Proteobacteria phyla (Figure 2A and Table 3). Taxonomic phylum from 11 from AAs and 24 from CAs were identified (Figure 2A). Bacteroidetes was the most abundant bacterial phylum in AAs (70%), whereas Firmicutes occurrence of 36% was higher in CAs (Figure 2A). Phylogenetic analysis further identified 44 classes of microbiota, with CAs showing more diverse population than AAs (Figure 2B). Bacteroidia was significantly higher in AAs, while Clostridia, Bacteria-unclassified, Gammaproteobacteria, Bacilli, Verrucomicrobiae, Acinobacteria, Fusobacteria and Alphaproteobacteria were more abundant in CAs (Figure 2B). Again, the microbial species richness and diversity was higher in CAs compared to AAs (Figure 2B).
Figure 2.

Microbial composition of each group at the phylum and class levels. A: Bar chart shows the relative abundance of phylum; B: Pie chart show the proportion of predominant different classes of microbial community in African Americans and Caucasian Americans. In order to get more comprehensive and accurate taxonomies, multiple databases, Ribosomal Database Project classifier, Greengenes and NCBI 16S Microbial were used for analysis of plot bars and pie charts.
Table 3.
Abundance of microbiota in colonic effluents from African Americans and Caucasian Americans (green, black, blue and red color represent phylum, family, genus and species, respectively)
| Phylum | Family | Genus | Species | AA (%) | CA (%) |
| Bacteroidetes | 70.74 | 43.17 | |||
| Bacteroidaceae | 56.75 | 29.94 | |||
| Bacteroides | 56.8 | 29.9 | |||
| Unclassified | 30.3 | 17.8 | |||
| Caccae | 13.6 | 5.5 | |||
| Massiliensis | 6.7 | 1.7 | |||
| Uniformis | 2.8 | 1.5 | |||
| Fragilis | 1 | 2 | |||
| Rikenellaceae | 6.44 | 5.47 | |||
| Allistipes | Putredinis | 6.4 (3.9) | 5.4 (3.0) | ||
| Porphymonadaceae | 6.4 | 6.95 | |||
| Parabacteroides | Distasonis | 4.9 (3.4) | 3.2 (1.0) | ||
| Barnesiella | Intestinihominis | 0.16 (0.16) | 2.7 (2.6) | ||
| Prevotellaceae | 1.9 | 1.1 | |||
| Paraprevotella | Clara | 1.0 (0.9) | 0.6 (0.6) | ||
| Firmicutes | 26.74 | 36.25 | |||
| Ruminococaceae | 11.35 | 15.7 | |||
| Faecalibacterium | Prausinitzii | 4.5 (4.5) | 3.1 (3.1) | ||
| Unclassified | Unclassified | 2.5 (2.5) | 2.9 (3.0) | ||
| Gemmiger | Formicilis | 1.9 (1.6) | 6.1 (6.0) | ||
| Clostridium IV | Unclassified | 0.3 (0.3) | 1.5 (1.5) | ||
| Lachnospiraceae | 11.19 | 13.7 | |||
| Ruminococcus 2 | Unclassified | 3.4 (3.4) | 4.7 (4.7) | ||
| Clostridium XIVa | Unclassified | 2.05 (2.0) | 1.03 (1.0) | ||
| Unclassified | Unclassified | 1.6 (1.7) | 2.4 (2.4) | ||
| Blautica | Producta | 0.22 (0.06) | 1.2 (0.0) | ||
| Dorea | Unclassified | 1.8 (0.8) | 1.3 (1.0) | ||
| Roseburia | Unclassified | 0.5 (0.3) | 1.3 (0.9) | ||
| Steptococaccae | Steptococcus | Faecium | 0.9 (0.9) (0.8) | 3.2 (3.2) (3.2) | |
| Acidaminococcaceae | Phascolarctobacterium | Unclassified | 1.8 (1.9) (1.4) | 0.9 (0.9) (1.3) | |
| Peptostreptococcaceae | Clostridium XI | Unclassified | 0.02 (0.02) (0.02) | 0.01 (0.008) (0.008) | |
| Veillonellaceae | Veillonella | Atypica | 0.81 (0.03) (0.03) | 1.27 (0.02) (0.02) | |
| Unclassified | 0.6 (0.6) | 0.5 (0.5) | |||
| Dialister | Invisus | 0.002 (0.0) | 0.68 (0.65) | ||
| Lactobacillaceae | Lactobacillus | Sanfrancisecnsis | 0.035 (0.02) (0.004) | 0.01 (0.01) (0.001) | |
| Proteobacteria | 2.02 | 5.9 | |||
| Desulfovibrionaceae | Biophila | Unclassified | 0.16 (0.12) (0.12) | 0.07 (0.07) (0.01) | |
| Sutterrellaceae | Parasuttrella | Excrementihomis | 0.50 (0.76) (0.4) | 0.24 (1.9) (0.1) | |
| Pasteurellaceae | Haemophilus | Parainfluenza | 0.57 (0.9) (0.4) | 1.98 (1.9) (1.9) | |
| Enterobacteiaceae | Escherichia/Shigella | Unclassified | 0.92 (0.8) (0.9) | 3.14 (3.1) (3.1) | |
| Klebsiella | Unclassified | (0.09) (0.02) | 0.003 (0.01) | ||
| Fusobacteria | 0.18 | 0.58 | |||
| Fusobactereaceae | Fusobacterium | Unclassified | 0.18 (0.15) (0.15) | 0.58 (0.6) (0.6) | |
| Verrucomicrobia | 0.04 | 1.9 | |||
| Verrucomicrobiaceae | Akkermansia | Muciniphila | 0.04 (0.04) | 1.9 (1.9) | |
| Bacteria-unclassified | Unclassified | Unclassified | Unclassified | 0 | 5.4 (5.4) (5.4) |
| Unclassified-unclassified | Unclassified | Unclassified | Unclassified | 0 | 4.6 (4.6) (4.6) |
AA: African Americans; CA: Caucasian Americans.
By 16S rRNA gene profiling, there were eight predominant families found between the two racial groups, Bacteroidaceae, Ruminococcaceae, Lachnospiraceae, Rikenallaceae, Porphymonadaceae, streptococaceae, Acidaminococaceae and Veillonellaceae (Table 3). The most predominant genus in AAs and CAs was Bacteroides, comprising 56% and 29%, respectively. Genus abundance of other microbiota was less than 7% with some degree of variation, as observed for Gemmiger, Allistipes, Parabacterroides, Faecalibacterium, Biophila, Ruminococcus2, Escherichia/Shigella (E/S), Streptococcus, Clostridium IV, Clostridium XIVa, Barnesiella, Akkermansia, Phascolartobacterium, Veillonella, Blautica, Roseburia, Haemophilus, Dialister and Fusobacterium (Table 3).
At the species levels, the relative abundance of B. caccae and B. massiliensis, P. distasonis, P. unclassified, Biophila unclassified and Clostridium XI unclassified were noted to be significantly higher in AAs compared to CAs (Table 3). In CA, the abundance of G. formicilis, Clostridium IV, B. intestinihominis, E/S. unclassified, H. parainfluenza, A. muciniphila, D. invisus, S. faecium and F. unclassified abundance was higher than AA patients. However, with respect to F. prausnitzii, Ruminococcus2 unclassified, A. putredinis, and P. clara, the relative abundance was found to be similar in both groups (Table 3). On the other hand, the Fusobacterium genus was higher in CAs compared to AAs (Figure 3A), and the Fusobacterium species level was identified as unclassified by microbial 16sRNA gene profiling.
Figure 3.

Abundance of inflammatory and probiotic bacteria in colonic effluent from two racial groups using RT-PCR. A: Genus, Fusobacterium occurrence higher in Caucasian American (CA); B: The relative abundance of pro-inflammatory Enterobacter occurrence is higher in African Americans (AAs); C and D: Probiotic Bifidobacteria and A. muciniphila is higher in CAs; E: Relative abundance of F. nucleatum is higher in AAs than CAs. Data represent mean ± SD of all samples from each group (bP < 0.001).
To further compare the microbial population between AAs and CAs, we examined the abundance of specific bacterial populations using species-specific primers. The occurrence of the Enterobacter genus was found to be considerably higher in AAs than CAs (Figure 3B), while the Enterobacteriaceae family showed the opposite pattern (Table 3). Taxonomic analysis showed CAs to contain Citrobacter, Klebsiela Escherichia coli, Enterobacter and Shigella sp. (Table 3). In contrast, the relative abundance of the probiotic bacteria genuses Bifidobacterium and Akkermansia muciniphila was considerably lower in AAs compared to CAs (Figures 3C and D). These observations demonstrate that the population of pro-inflammatory bacteria is higher in the gut of AAs than CAs.
Occurrence of Fusobacterium nucleatum and Clostridium genus
The relative abundance of Fusobacterium nucleatum has been associated with the development and progression of CRC[23,36,37]. We found that the relative abundance of F. nucleatum in colonic effluents was significantly higher in AAs than CAs (Figure 3E), indicating a greater risk for the development of CRC in AAs. On the other hand, serrated polyps, which supposedly possess a greater risk of developing CRC, did not exhibit an increased abundancy of F. nucleatum. In fact, we found the relative abundance of F. nucleatum to be higher in tubular adenoma than serrated adenoma (B-Raf proto oncogene, serine/threonine kinase (BRAF) mutation), whereas the probiotic Lactobacillus was lower in both serrated and tubular adenomas than those without adenoma (Figure 4). Likewise, the relative abundance of Bifidobacteria was found to be lower in tubular adenoma than those without adenoma (Figure 4).
Figure 4.

Abundance of probiotic and pro-carcinogenic bacteria in serrated and tubular adenomatous in colonic mucosa. African American and Caucasian American patients were combined. Data represent mean ± SD of all samples from each group (aP < 0.05, bP < 0.001).
Using a clostridium cluster analysis and RT-PCR, we found that the relative abundance of Clostridium IV was higher in CAs (Figure 5A), while Clostridium XI was significantly higher in AAs (Figure 5B). Clostridium IV is known to be mediate anti-inflammatory effects[38-40]. Clostridium sordelli in the Clostridium XI cluster group is known to transform SBA[41]. AA patients also showed higher concentrations of C. sordelli, compared to CAs (Figure 5C). A few species of gut anaerobes in the Clostridium genus promote the biotransformation of primary to SBA. Given the role of SBA (DCA and LCA) in promoting CRC, the expression of 7-α-dehydroxylase, an enzyme that participates in de-conjugation of primary bile acids, was examined. We found the expression of 7-α-dehydroxylase (baiCD) in colonic effluent from AA patients to be markedly higher than CA subjects (Figure 5D).
Figure 5.

Expression of secondary bile acids transforming enzyme 7-α-DH in the colonic effluent from African Americans and Caucasian Americans using RT-PCR. A: Distribution of different Clostridium cluster between African Americans (AA) and Caucasian Americans (CA); B: Clostridium XI expression was higher in AAs; C: Secondary bile acids transforming bacteria Clostridium sordelli was higher in AAs than CAs; D: Increased 7α-DH expression in AAs. Data represent mean ± SD of all samples from each group (aP < 0.05, bP < 0.01).
DISCUSSION
Our pilot study comparing AA and CA gut microbiota from colonic effluents reveal three major differences between the groups: the AA gut microbiota was less diverse overall, AAs had more pro-inflammatory gut bacteria, and AAs had fewer anti-inflammatory gut bacteria. The phylogenetic tree of microbiota between AAs and CAs reveal an abundance of taxon in CA vs AA (Figure 6). Analyzing PCoA of patients, we noted trends found previously in comparing normal colon to adenomas and CRC[36,42,43]. Patients with colonic adenoma typically demonstrate reduced species richness and diversity compared to those without adenomas[44,45]. Furthermore, a fecal microbiota shift occurs in patients with adenomas[46], and they exhibit increased diversity in mucosa than those without adenoma[47].
Figure 6.
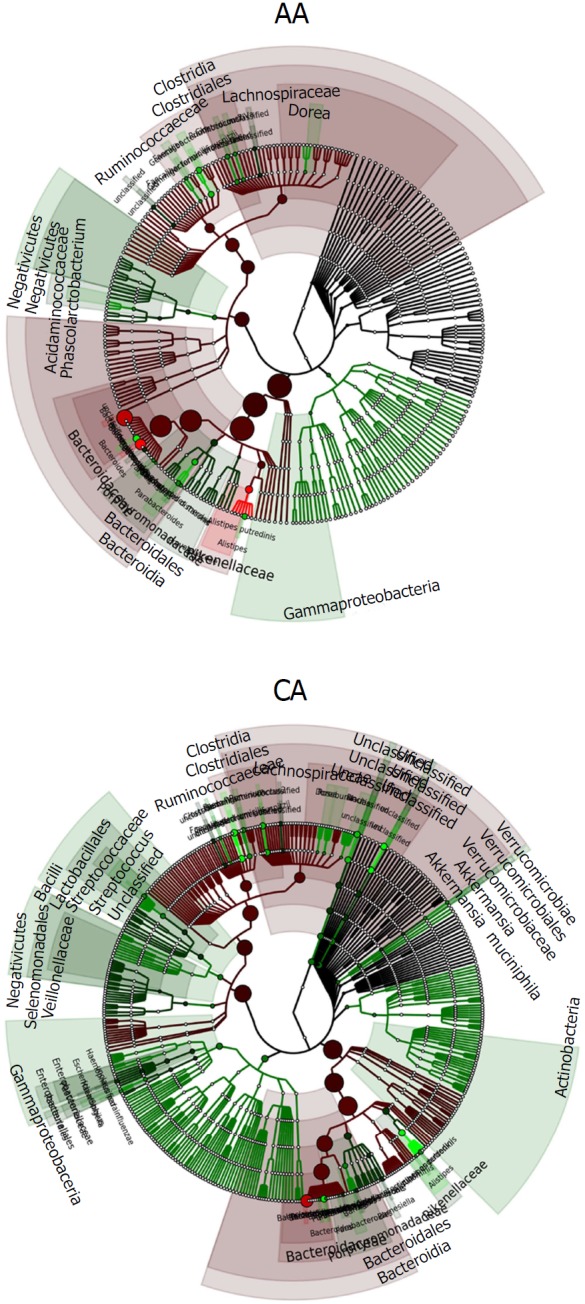
Phylogenetic tree showing the differences and abundance of taxa in African Americans and Caucasian Americans colonic effluents. The taxon size and color indicate the relative abundance of family. GraPhlAn software was used for taxonomic classification and circular taxonomic phylogenetic trees and Ribosomal Database Project classifier, Quantitative Insights into Microbial Ecology, R software/Tool were used for taxonomic data analysis.
The human gut microbiota composition is generally represented by three primary phyla: Firmicutes (30%-50%), Bacteroidetes (20%-40%), and Actinobacteria (1%-10%)[22]. The two predominant bacterial phyla, Bacteroidetes, and Firmicutes, which contribute to 95% of the total GI ecosystem, are associated with adenomas and CRC[48]. The abundance of phylum Bacteroidetes (P. distasonis, Alistipes spp.) in the gut may increase the rate of tumorigenesis[49]. Our current study demonstrates an abundance of Bacteroidetes (70%) and reduction in Firmicute and Actinobacteria in AAs, whereas the microbial balance between predominant groups was unchanged in CAs, as has been demonstrated previously[22]. These observations are similar to what Hester et al[50] noted in their investigation in that the Firmicutes/Bacteroidetes ratio is higher in AAs compared to CAs. There is also a relative abundance of B. massiliensis in AAs compared to CAs. CRC has also been shown to be associated F. nucleatum and pro-inflammatory bacteria, Enterobacter and Clostridium XI-species. F. nucleatum strains have been shown to promote carcinogenesis and invasion of host cells and potentiate tumorigenesis in mouse model of colon cancer[23,37]. CRC patients demonstrate a higher abundance of F. nucleatum and Clostridium difficile, a member of Clostridium XI[51,52]. Others have demonstrated that the relative increase in Clostridium cluster XI and Enterobacteriaceae are associated with intestinal dysbiosis[53].
Previous studies have suggested that a decrease in commensal microbiota in AAs may contribute to the tumorigenic microenvironment and that dysbiosis of gut microbiota may be partially responsible for promoting CRC and colitis-associated CRC[22,42,54]. In line with these observations, we found commensal bacteria B. fragilis to be slightly higher in CAs than AAs. Bacteroides fragilis is an immunomodulatory bacteria, which stimulates anti-inflammatory cytokine IL10 by Foxp3+ regulatory CD+ T (Treg) cells and suppresses mucosal inflammation[55,56]. In contrast, Unclassified-Bacteria and Unclassified-Unclassified micro-organisms were only present in CAs A. municiphila, a member of Verrumicomicrobia, is an intestinal symbiont and is known to induce an anti-inflammatory effect and enhance immune function[57]. Depletion of A. muciniphila is associated with a variety of diseases, including diabetes[58,59]. We found the relative abundance of A. muciniphila to be lower in AAs than their CA counterparts. Collectively, AAs have fewer bacterial populations that are known to suppress inflammation, improve mucus barrier function, and diminish permeability[60].
Human genetic variants can modulate the effects of the microbiome composition, and both are associated with many human complex diseases[61]. Microbiota changes with diet and stimulatory agents and can modulate disease development and progression[61,62]. Microbial dysbiosis in AAs may serve as a point for prevention and ultimately treatment of CRC. Identifying a microbial signature associated with CRC is complicated by many factors. This study was limited by focusing on a specific population with limited sample size and did not investigate dietary differences. However, there were significant differences between the colonic effluent microbiota of the AA and CA study groups - with less diversity of bacteria, greater abundance of pro-inflammatory bacteria, and reduced anti-inflammatory bacteria. Mechanisms for tumorigenesis may include bacteria that promote SBA transformation, as suggested by higher 7-α-dehydroxylase in the AA vs CA group. Further study is needed to evaluate the role of decreased diversity and structural imbalance in the colon microbial communities and the development of CRC.
ARTICLE HIGHLIGHTS
Research background
The incidence of colorectal cancer (CRC), the third most common malignancy, is not only higher among African Americans (AAs), but is also associated with higher mortality. In addition, AAs tend to be diagnosed with CRC at a younger age than Caucasian Americans (CAs) and exhibit worse prognoses than their CA counterparts. Despite this grim outlook, neither the extrinsic/intrinsic factor(s) nor the underlying molecular and/or biochemical mechanisms are fully understood. We hypothesize that imbalance in the gut microbiome between AAs and CAs results in alterations of metabolites, which changes symbiotic relationships and enhance gastrointestinal diseases, including CRC. A number of bacteria are known to promote carcinogenesis in the colon by altering gut microbial composition, which may play a major role in colorectal carcinogenesis.
Research motivation
CRC is the third leading malignancy world-wide, affecting both males and females equally. It represents one of the most common cancers in the United States and is estimated to be the second and third leading cause of cancer-related deaths in men and women, respectively, in the United States. Several studies have also demonstrated that AAs have the highest rate of CRC than any other racial group in the USA, and also AA men are even more likely to die from CRC than AA women. With these grim statistics, it is important to gain a better understanding of the underlying mechanism(s), particularly the role of gut microbiota, in regulating racial disparity in colorectal carcinogenesis.
Research objectives
The current investigation was aimed at studying microbial dysbiosis in the gut between AAs and CAs. The primary endpoint of this investigation was to determine whether the increased incidence of CRC in AAs could be attributed to alterations in gut microbiota. In this pilot study, we investigated the diversity and abundance of specific gut microbial communities in colonic effluents using 16S rRNA gene profiling in AAs and CAs and their possible role in the increased incidence of CRC in AAs.
Research methods
Male and female AA and CA patients, aged between 40 and 80 years, undergoing routine colonoscopy at the John D. Dingell VA Medical Center in Detroit were asked to participate. To determine the microbial diversity and the microbial richness in AAs and CAs, colonic effluent from each patient was used for DNA extraction and 16s RNA gene-based microbial community profiling which was performed and analyzed by LC Sciences (Houston, Texas, United States). The composition of OTU and alpha diversity was measured by Venn diagram and Rarefaction measurement method. The relative abundance of phylum and classes was depicted by bar and pie chart. The phylogenic tree was plotted for AA and CA to determine the relative abundance of family in microbial community. Several inflammatory and probiotic bacterial marker candidates such as Enterobacteria, Bifidobacteria, Lactobacillus, Fusobacterium and/or Clostridium genus and species-specific bacteria were identified by real-time qPCR using specific primers designed on the basis of conserved and variable region in bacterial 16S rRNA genes according to our standard protocol. Statistical analysis was performed for each experiment accordingly.
Research results
The relative abundance of Fusobacterium nucleatum, which has been associated with the development and progression of CRC, was found to be significantly higher in AAs than CAs, indicating a greater risk for the development of CRC in AAs. Clostridium IV, a known mediator of anti-inflammatory effects, was found to be higher in CAs than AAs.
Research conclusion
The human colon harbors a complex microbial flora. Bacterial density in the human colon is among the highest found in nature, approaching 1012 bacteria/gm wet weight of feces. These bacteria are in a symbiotic relationship with the intestine, utilizing undigested nutrients as substrates and in return, produce various vitamins, amino acids, transform bile salts and assist in the maintenance of the intestinal barrier, and the appropriate immune response against pathogens. This homeostasis is altered in a state of dysbiosis, which is overgrowth of pathogenic bacteria that are normally inhibited by commensal bacteria. Our current investigation, for the first time, demonstrates microbial dysbiosis between AAs and CAs. This imbalance, we believe, is partially responsible for the racial disparity in CRC observed between AAs and CAs.
Research perspective
Although numerous studies have demonstrated that the incidence of CRC is higher in AAs than CAs, the reasons for this racial disparity are not fully understood. Data generated from this investigation reveal a role for the gut microbiome in racial disparity. The precise mechanisms by which changes in gut microbiota would lead to an increase CRC in AAs remain unexplored. However, it is tempting to speculate that this dysbiosis or overgrowth of certain bacteria in the gut of AAs resulting in alterations in microbial metabolites, specifically deoxycholic acid and lithocholic acid, which are known for their co-carcinogenic activity, could induce the process(es) of carcinogenesis in the colon of AAs. However, the levels of microbial metabolites, including bile acids in AAs and CAs with and without adenomas have not determined. Moreover, no information is available whether the observed dysbiosis in AAs is due to changes in diet and/or lifestyle. Undoubtedly, further investigations are needed to gain a better and fuller understanding of the intrinsic and extrinsic factors that are critically involved in regulating racial disparity in CRC.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C, C
Grade D (Fair): D
Grade E (Poor): 0
Institutional review board statement: The study followed a Full Board Review by the Wayne State University Institutional Review Board.
Informed consent statement: All routine colonoscopy, collection of colonic effluents and biopsy specimens from the patients were performed after obtaining informed consent and ethical permission.
Conflict-of-interest statement: To the best of our knowledge, no conflict of interest exists.
ARRIVE guidelines statement: The ARRIVE Guidelines had been adopted.
Peer-review started: May 11, 2018
First decision: July 10, 2018
Article in press: August 26, 2018
P- Reviewer: Huerta-Franco M, Mann O, Maric I, McMillin MA S- Editor: Cui LJ L- Editor: Filipodia E- Editor: Yin SY
Contributor Information
Lulu Farhana, Department of Internal Medicine, John D Dingell Veterans Affairs Medical Center, Detroit, MI 48201, United States; Department of Internal Medicine, Wayne State University School of Medicine, Detroit, MI 48201, United States.
Fadi Antaki, Department of Internal Medicine, Wayne State University School of Medicine, Detroit, MI 48201, United States; Division of Gastroenterology, John D Dingell VA Medical Center, Detroit, MI 48201, United States.
Farhan Murshed, Department of Internal Medicine, John D Dingell Veterans Affairs Medical Center, Detroit, MI 48201, United States.
Hamidah Mahmud, Department of Internal Medicine, John D Dingell Veterans Affairs Medical Center, Detroit, MI 48201, United States.
Stephanie L Judd, Division of Gastroenterology, John D Dingell VA Medical Center, Detroit, MI 48201, United States.
Pratima Nangia-Makker, Department of Internal Medicine, John D Dingell Veterans Affairs Medical Center, Detroit, MI 48201, United States; Department of Internal Medicine, Wayne State University School of Medicine, Detroit, MI 48201, United States; Department of Medicine, Karmanos Cancer Institute, Detroit, MI 48201, United States.
Edi Levi, Department of Pathology Service, John D Dingell VA Medical Center, Detroit, MI 48201, United States.
Yingjie Yu, Department of Internal Medicine, John D Dingell Veterans Affairs Medical Center, Detroit, MI 48201, United States; Department of Internal Medicine, Wayne State University School of Medicine, Detroit, MI 48201, United States.
Adhip PN Majumdar, Department of Internal Medicine, John D Dingell Veterans Affairs Medical Center, Detroit, MI 48201, United States. a.majumdar@wayne.edu; Department of Internal Medicine, Wayne State University School of Medicine, Detroit, MI 48201, United States; Department of Medicine, Karmanos Cancer Institute, Detroit, MI 48201, United States.
References
- 1.Dimou A, Syrigos KN, Saif MW. Disparities in colorectal cancer in African-Americans vs Whites: before and after diagnosis. World J Gastroenterol. 2009;15:3734–3743. doi: 10.3748/wjg.15.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Lieberman DA, Holub JL, Moravec MD, Eisen GM, Peters D, Morris CD. Prevalence of colon polyps detected by colonoscopy screening in asymptomatic black and white patients. JAMA. 2008;300:1417–1422. doi: 10.1001/jama.300.12.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 5.Wallace K, Hill EG, Lewin DN, Williamson G, Oppenheimer S, Ford ME, Wargovich MJ, Berger FG, Bolick SW, Thomas MB, et al. Racial disparities in advanced-stage colorectal cancer survival. Cancer Causes Control. 2013;24:463–471. doi: 10.1007/s10552-012-0133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallace K, Sterba KR, Gore E, Lewin DN, Ford ME, Thomas MB, Alberg AJ. Prognostic factors in relation to racial disparity in advanced colorectal cancer survival. Clin Colorectal Cancer. 2013;12:287–293. doi: 10.1016/j.clcc.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashktorab H, Kupfer SS, Brim H, Carethers JM. Racial Disparity in Gastrointestinal Cancer Risk. Gastroenterology. 2017;153:910–923. doi: 10.1053/j.gastro.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amri R, Bordeianou LG, Berger DL. The conundrum of the young colon cancer patient. Surgery. 2015;158:1696–1703. doi: 10.1016/j.surg.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 9.Ashktorab H, Vilmenay K, Brim H, Laiyemo AO, Kibreab A, Nouraie M. Colorectal Cancer in Young African Americans: Is It Time to Revisit Guidelines and Prevention? Dig Dis Sci. 2016;61:3026–3030. doi: 10.1007/s10620-016-4207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carethers JM. Screening for colorectal cancer in African Americans: determinants and rationale for an earlier age to commence screening. Dig Dis Sci. 2015;60:711–721. doi: 10.1007/s10620-014-3443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laryea JA, Siegel E, Klimberg S. Racial disparity in colorectal cancer: the role of equal treatment. Dis Colon Rectum. 2014;57:295–302. doi: 10.1097/DCR.0000000000000056. [DOI] [PubMed] [Google Scholar]
- 12.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 13.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Todaro M, Iovino F, Eterno V, Cammareri P, Gambara G, Espina V, Gulotta G, Dieli F, Giordano S, De Maria R, et al. Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res. 2010;70:8874–8885. doi: 10.1158/0008-5472.CAN-10-1994. [DOI] [PubMed] [Google Scholar]
- 15.Farhana L, Antaki F, Anees MR, Nangia-Makker P, Judd S, Hadden T, Levi E, Murshed F, Yu Y, Van Buren E, et al. Role of cancer stem cells in racial disparity in colorectal cancer. Cancer Med. 2016;5:1268–1278. doi: 10.1002/cam4.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexander DD, Cushing CA, Lowe KA, Sceurman B, Roberts MA. Meta-analysis of animal fat or animal protein intake and colorectal cancer. Am J Clin Nutr. 2009;89:1402–1409. doi: 10.3945/ajcn.2008.26838. [DOI] [PubMed] [Google Scholar]
- 17.Carethers JM. Racial and ethnic factors in the genetic pathogenesis of colorectal cancer. J Assoc Acad Minor Phys. 1999;10:59–67. [PubMed] [Google Scholar]
- 18.Huxley RR, Ansary-Moghaddam A, Clifton P, Czernichow S, Parr CL, Woodward M. The impact of dietary and lifestyle risk factors on risk of colorectal cancer: a quantitative overview of the epidemiological evidence. Int J Cancer. 2009;125:171–180. doi: 10.1002/ijc.24343. [DOI] [PubMed] [Google Scholar]
- 19.Sandler RS. Aspirin and other nonsteroidal anti-inflammatory agents in the prevention of colorectal cancer. Important Adv Oncol. 1996:123–137. [PubMed] [Google Scholar]
- 20.Dethlefsen L, Eckburg PB, Bik EM, Relman DA. Assembly of the human intestinal microbiota. Trends Ecol Evol. 2006;21:517–523. doi: 10.1016/j.tree.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Abreu MT, Peek RM Jr. Gastrointestinal malignancy and the microbiome. Gastroenterology. 2014;146:1534–1546.e3. doi: 10.1053/j.gastro.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gagnière J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, Bringer MA, Pezet D, Bonnet M. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol. 2016;22:501–518. doi: 10.3748/wjg.v22.i2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tözün N, Vardareli E. Gut Microbiome and Gastrointestinal Cancer: Les liaisons Dangereuses. J Clin Gastroenterol. 2016;2015:S191–S196. doi: 10.1097/MCG.0000000000000714. [DOI] [PubMed] [Google Scholar]
- 25.Boutagy NE, Neilson AP, Osterberg KL, Smithson AT, Englund TR, Davy BM, Hulver MW, Davy KP. Short-term high-fat diet increases postprandial trimethylamine-N-oxide in humans. Nutr Res. 2015;35:858–864. doi: 10.1016/j.nutres.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Fleming SE, Fitch MD, Chansler MW. High-fiber diets: influence on characteristics of cecal digesta including short-chain fatty acid concentrations and pH. Am J Clin Nutr. 1989;50:93–99. doi: 10.1093/ajcn/50.1.93. [DOI] [PubMed] [Google Scholar]
- 27.Northfield TC, McColl I. Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut. 1973;14:513–518. doi: 10.1136/gut.14.7.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zitvogel L, Pietrocola F, Kroemer G. Nutrition, inflammation and cancer. Nat Immunol. 2017;18:843–850. doi: 10.1038/ni.3754. [DOI] [PubMed] [Google Scholar]
- 29.Ajouz H, Mukherji D, Shamseddine A. Secondary bile acids: an underrecognized cause of colon cancer. World J Surg Oncol. 2014;12:164. doi: 10.1186/1477-7819-12-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narisawa T, Magadia NE, Weisburger JH, Wynder EL. Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-methyl-N’-nitro-N-nitrosoguanidine in rats. J Natl Cancer Inst. 1974;53:1093–1097. doi: 10.1093/jnci/53.4.1093. [DOI] [PubMed] [Google Scholar]
- 31.Ou J, DeLany JP, Zhang M, Sharma S, O’Keefe SJ. Association between low colonic short-chain fatty acids and high bile acids in high colon cancer risk populations. Nutr Cancer. 2012;64:34–40. doi: 10.1080/01635581.2012.630164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Payne CM, Crowley-Skillicorn C, Bernstein C, Holubec H, Moyer MP, Bernstein H. Hydrophobic bile acid-induced micronuclei formation, mitotic perturbations, and decreases in spindle checkpoint proteins: relevance to genomic instability in colon carcinogenesis. Nutr Cancer. 2010;62:825–840. doi: 10.1080/01635581003695756. [DOI] [PubMed] [Google Scholar]
- 33.Reddy BS, Martin CW, Wynder EL. Fecal bile acids and cholesterol metabolites of patients with ulcerative colitis, a high-risk group for development of colon cancer. Cancer Res. 1977;37:1697–1701. [PubMed] [Google Scholar]
- 34.Cao H, Xu M, Dong W, Deng B, Wang S, Zhang Y, Wang S, Luo S, Wang W, Qi Y, et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int J Cancer. 2017;140:2545–2556. doi: 10.1002/ijc.30643. [DOI] [PubMed] [Google Scholar]
- 35.Wells JE, Williams KB, Whitehead TR, Heuman DM, Hylemon PB. Development and application of a polymerase chain reaction assay for the detection and enumeration of bile acid 7alpha-dehydroxylating bacteria in human feces. Clin Chim Acta. 2003;331:127–134. doi: 10.1016/s0009-8981(03)00115-3. [DOI] [PubMed] [Google Scholar]
- 36.Mira-Pascual L, Cabrera-Rubio R, Ocon S, Costales P, Parra A, Suarez A, Moris F, Rodrigo L, Mira A, Collado MC. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J Gastroenterol. 2015;50:167–179. doi: 10.1007/s00535-014-0963-x. [DOI] [PubMed] [Google Scholar]
- 37.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 40.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30:332–338. doi: 10.1097/MOG.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WK, Ng SC, Tsoi H, Dong Y, Zhang N, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. 2015;6:8727. doi: 10.1038/ncomms9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, Randall TA, Galanko J, Benson A, Sandler RS, et al. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J. 2012;6:1858–1868. doi: 10.1038/ismej.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, Yang L. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013;105:1907–1911. doi: 10.1093/jnci/djt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peters BA, Dominianni C, Shapiro JA, Church TR, Wu J, Miller G, Yuen E, Freiman H, Lustbader I, Salik J, et al. The gut microbiota in conventional and serrated precursors of colorectal cancer. Microbiome. 2016;4:69. doi: 10.1186/s40168-016-0218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hale VL, Chen J, Johnson S, Harrington SC, Yab TC, Smyrk TC, Nelson H, Boardman LA, Druliner BR, Levin TR, et al. Shifts in the Fecal Microbiota Associated with Adenomatous Polyps. Cancer Epidemiol Biomarkers Prev. 2017;26:85–94. doi: 10.1158/1055-9965.EPI-16-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N, Jovov B, Abdo Z, Sandler RS, Keku TO. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1:138–147. doi: 10.4161/gmic.1.3.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allen-Vercoe E, Jobin C. Fusobacterium and Enterobacteriaceae: important players for CRC? Immunol Lett. 2014;162:54–61. doi: 10.1016/j.imlet.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baxter NT, Zackular JP, Chen GY, Schloss PD. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome. 2014;2:20. doi: 10.1186/2049-2618-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hester CM, Jala VR, Langille MG, Umar S, Greiner KA, Haribabu B. Fecal microbes, short chain fatty acids, and colorectal cancer across racial/ethnic groups. World J Gastroenterol. 2015;21:2759–2769. doi: 10.3748/wjg.v21.i9.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Franks AH, Harmsen HJ, Raangs GC, Jansen GJ, Schut F, Welling GW. Variations of bacterial populations in human feces measured by fluorescent in situ hybridization with group-specific 16S rRNA-targeted oligonucleotide probes. Appl Environ Microbiol. 1998;64:3336–3345. doi: 10.1128/aem.64.9.3336-3345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukugaiti MH, Ignacio A, Fernandes MR, Ribeiro Júnior U, Nakano V, Avila-Campos MJ. High occurrence of Fusobacterium nucleatum and Clostridium difficile in the intestinal microbiota of colorectal carcinoma patients. Braz J Microbiol. 2015;46:1135–1140. doi: 10.1590/S1517-838246420140665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marteau P, Chaput U. Bacteria as trigger for chronic gastrointestinal disorders. Dig Dis. 2011;29:166–171. doi: 10.1159/000323879. [DOI] [PubMed] [Google Scholar]
- 54.Sobhani I, Amiot A, Le Baleur Y, Levy M, Auriault ML, Van Nhieu JT, Delchier JC. Microbial dysbiosis and colon carcinogenesis: could colon cancer be considered a bacteria-related disease? Therap Adv Gastroenterol. 2013;6:215–229. doi: 10.1177/1756283X12473674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. 2016;14:20–32. doi: 10.1038/nrmicro3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zitvogel L, Daillère R, Roberti MP, Routy B, Kroemer G. Anticancer effects of the microbiome and its products. Nat Rev Microbiol. 2017;15:465–478. doi: 10.1038/nrmicro.2017.44. [DOI] [PubMed] [Google Scholar]
- 57.de Vos WM. Microbe Profile: Akkermansia muciniphila: a conserved intestinal symbiont that acts as the gatekeeper of our mucosa. Microbiology. 2017;163:646–648. doi: 10.1099/mic.0.000444. [DOI] [PubMed] [Google Scholar]
- 58.Belzer C, de Vos WM. Microbes inside--from diversity to function: the case of Akkermansia. ISME J. 2012;6:1449–1458. doi: 10.1038/ismej.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, Chilloux J, Ottman N, Duparc T, Lichtenstein L, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med. 2017;23:107–113. doi: 10.1038/nm.4236. [DOI] [PubMed] [Google Scholar]
- 60.Plotnikoff GA. Three measurable and modifiable enteric microbial biotransformations relevant to cancer prevention and treatment. Glob Adv Health Med. 2014;3:33–43. doi: 10.7453/gahmj.2014.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luca F, Kupfer SS, Knights D, Khoruts A, Blekhman R. Functional Genomics of Host-Microbiome Interactions in Humans. Trends Genet. 2018;34:30–40. doi: 10.1016/j.tig.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ou J, Carbonero F, Zoetendal EG, DeLany JP, Wang M, Newton K, Gaskins HR, O’Keefe SJ. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr. 2013;98:111–120. doi: 10.3945/ajcn.112.056689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 64.Matsuki T, Watanabe K, Fujimoto J, Miyamoto Y, Takada T, Matsumoto K, Oyaizu H, Tanaka R. Development of 16S rRNA-gene-targeted group-specific primers for the detection and identification of predominant bacteria in human feces. Appl Environ Microbiol. 2002;68:5445–5451. doi: 10.1128/AEM.68.11.5445-5451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matsuki T, Watanabe K, Fujimoto J, Takada T, Tanaka R. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl Environ Microbiol. 2004;70:7220–7228. doi: 10.1128/AEM.70.12.7220-7228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Collado MC, Isolauri E, Laitinen K, Salminen S. Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr. 2008;88:894–899. doi: 10.1093/ajcn/88.4.894. [DOI] [PubMed] [Google Scholar]
- 67.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–298. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]