

Abstract
Polymorphic forms of nucleic acids provide platforms for new nanomaterials, and transition metal cations give access to alternative arrangements of nucleobases by coordinating with electron-rich functional groups. Interaction of Ag+ with 5’-guanosine monophosphate (5’-GMP) is considered in this work. Ag+ promotes nucleotide stacking and aggregation, as indicated by the increased viscosity of 5’-GMP solutions with Ag+, magnification of the circular dichroism response of guanine by Ag+, and exothermic reactions between Ag+ and guanine derivatives. Isothermal titration calorimetry studies show that the reaction is favored starting at 10 μM 5’-GMP. Utilizing the exothermic heat change associated with reaction of Ag+ with 5’-GMP, local structure within the aggregate was assessed. Based on salt dependence and comparison with the corresponding nucleoside, the dianionic phosphate of 5’-GMP is one binding site for Ag+, although this electrostatic interaction is not a dominant contribution to the overall heat change. Another binding site is the N7 on the nucleobase, as determined via studies with 7-deazaguanosine. Besides this binding site, Ag+ also associates with the O6, as earlier studies deduced from the shift in the carbonyl stretching frequency associated with adduct formation. With these two binding sites on the nucleobase, the empirical stoichiometry of ~1 Ag+:nucleobase derived from the calorimetry studies indicates that Ag+ coordinates two nucleobases. The proposed structural model is a Ag+-mediated guanine dimer within a base stacked aggregate.
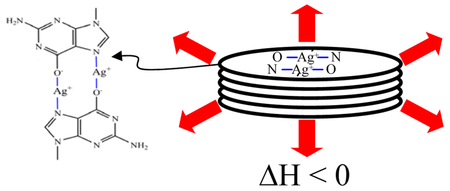
Graphical Abstarct
Introduction
DNA hybridization has enabled creation of novel nanomaterials, such as self-assembled biomolecular electronic components whose small size could greatly expand the information density of electronic devices.1 Nucleobase assembly is dictated by the directionality, strength, and interplay of noncovalent interactions such as base stacking and hydrogen bonding.2,3 By promoting alternative base pairing arrangements through coordination with the electron-rich groups of nucleobases, transition metal cations broaden the repertoire of base interactions while also conferring magnetic and electronic functionality to DNA-based nanostructures.4,5,6 Indirectly, these cations can change patterns of hydrogen bond donor and acceptors by favoring particular tautomers or by altering ionization constants of acidic groups on the nucleobase. In addition, transition metal cations can directly chelate electron-rich nitrogen and oxygen groups to produce both canonical and noncanonical base pairs such as coordination of adenine and thymine by Pt2+, adenine and cytosine by Ag+, and two thymines by Hg2+.7,8,9
A new class of nanomaterial that provided the motivation for the present studies is fluorescent silver clusters that form on DNA templates.10,11,12 By reducing Ag+ that is bound with DNA, clusters with ~10 atoms are formed. These chromophores are distinguished by their spectral tunability that depends on base sequence, high fluorescence quantum yield, high photostability, and limited blinking.11,13 Our goals are to better understand how Ag+ is organized within a DNA matrix and how this cation influences DNA structure. This paper considers interaction of Ag+ with isolated guanines and the resulting nucleobase organization in dilute solutions (10 – 500 ¼M). Ag+ favors σ bond formation with the electron-rich binding sites of aromatic rings, as indicated by the 30 fold greater affinity for pyridine relative to benzene, and site specific adducts have been characterized by infrared spectroscopy and x-ray crystallography studies. 14,15,16 Beyond isolated 1:1 adducts, Ag+ facilitates higher order interactions such as base pairing due to it’s propensity for linear coordination.16,17,18,19,20 This paper evaluates interaction with guanine derivatives, and three potential binding sites on the nucleobase have been considered. Spectroscopic and crystallographic studies show association with N7, which has been attributed to the Lewis basicity at this site.21,22 Association with N1 is possibly connected with the decrease in pH that accompanies Ag+ addition to guanine in slightly basic solutions.15 Alternatively, association could occur via an enolate-like tautomer that concentrates electron density on the O6, as supported by infrared spectroscopic studies.15,21 Consistent with linear coordination, Ag+-mediated chelation between guanines has been proposed to produce specific dimeric and tetrameric nucleobase arrangments.15,18 Further higher order organization via stacking is considered in this work.
This paper examines chelation and aggregation of 5’-guanosine monophosphate (5-GMP) and related derivatives by Ag+ in pH = 8 buffers (Fig. 1). This choice of ribonucleoside and pH were motivated by extensive studies of 5’-GMP aggregation in basic solutions.23 For example, NMR studies have used the combination of chemical shifts and translational diffusion coefficients to discern aggregates of 5’-GMP.24 The quartet based aggregate represents a distinctive nucleic acid form with alternating C2’-endo and C3’-endo sugar puckers within the helical base arrangement. With Ag+, three observations indicate that aggregation of 5’-GMP depends on this transition metal cation. First, viscosities of solutions of the nucleotide increase with Ag+. Second, circular dichroism associated with the guanine base greatly increases in the presence of Ag+. These changes are accompanied by a reduction in the extinction coefficient of guanine. Third, exothermic heat changes result from association of Ag+ with the nucleobase, and the magnitude of these changes is in the range expected for base stacking. Using the calorimetric signature for the reaction, the local aggregate structure was investigated using 5’-GMP and structural variants. Exothermic heat changes are driven by association of ~ Ag+ with the guanine base. When the potential binding sites for Ag+ on the nucleobase are considered, a dimeric base arrangement within the aggregate is considered to be most reasonable.
Figure 1.

Structures of the nucleotides and nucleosides used for these studies. For 5’-guanosine monophosphate, the functional groups that were considered as binding sites for Ag+ are emphasized in red. The proposed structure for the dimer of guanine with Ag+-coordination between N7 and O6 is shown in (F).
Experimental Section
Silver nitrate (99.9995%, Alfa Aesar), 5’-guanosine monophosphate (Sigma-Aldrich, 99+%), guanosine (Acros Organics, 99%), inosine monophosphate (Acros Organics, 99+%), 5’-cytosine monophosphate (Sigma-Aldrich, 99%), and 7-deazaguanosine (Berry & Associates, ≥97%) were used as received. Buffers were prepared in deionized water (Elix 10 Water Purification System, Millipore) and the desired pH was obtained using a total concentration of 10 mM of the acid/conjugate base. A concentration of 10 mM H3BO3/H2BO3− was used to maintain pH = 8 in the solutions, and supporting electrolytes were NaClO4 and LiClO4. Nucleotide and nucleoside concentrations were determined using extinction coefficients of 14,230 M−1cm−1 for 5’-GMP at 252 nm, 8,860 M−1cm−1 for 5’-cytosine monophosphate at 271 nm, 11,700 M−1cm−1 for inosine monophosphate at 246 nm, 12,300 M−1cm−1 for 7-deazaguanosine at 260 nm, and 12,500 M−1 cm−1 for guanosine at 260 nm.25,26,27 For the latter compound, its solubility at the 100 ¼M concentrations used for the calorimetry studies was experimentally verified.
Using previously described protocols, calorimetry studies were conducted using a Microcal VP-ITC (Northhampton, MA) controlled by Origin 7.0 software.28 Following degassing, a solution of AgNO3 was titrated into a nucleotide or nucleoside solution. The solutions were degassed prior to loading. Heat changes associated with the titration were determined by integrating the power required to maintain reference and sample cells at the same temperature. Heat changes associated with dilution of AgNO3 were subtracted after saturation of the binding sites. The single site binding model in the manufacture software was appropriate for fitting the binding isotherms to determine the enthalpy and free energy changes and the adduct stoichiometry. Entropy changes were derived from the free energy and enthalpy changes. Uncertainties were derived from a minimum of three experiments on separate samples. The highest 5’-GMP concentration at which a full binding isotherm could be acquired was 500 ¼M. Absorption (Cary 50, Varian) and circular dichroism (J-710, Jasco) spectra were acquired using quartz cuvettes with 1 or 0.2 cm pathlengths. Viscosity studies followed the protocol from prior experiments.29 Measurements were made with a Cannon-Ubbelohde semi-micro viscometer with a capillary diameter of 0.54 mm (Model 75, Cannon Instrument Company), and the viscometer was submerged in a water bath to maintain a constant temperature of 25.00 ± 0.05 °C. Flow times between the viscometer timing marks were 117.92 ± 0.03 s for the buffer and 118.41 ± 0.02 s for a 1 mM solution of 5’-GMP. Flow times were measured at least in triplicate, error was propagated using standard procedures to give uncertainties of ±1σ.30 Low nucleotide concentrations allow intrinsic viscosities to be calculated from differences between the flow times for the DNA solutions and for the buffer using:31,32
where [η] and [η]o are intrinsic viscosities of the 5’-GMP solutions with and without Ag+, respectively. Flow times of the buffer, 5’-GMP, and 5’-GMP:Ag+ solutions are tb, to, and t, respectively. Nucleoside solubility at the high concentrations needed for viscosity precluded studies with other derivatives.
Results
Base Aggregation and Stacking
Addition of Ag+ to solutions with 5’-GMP results in solutions with higher viscosities, while control experiments without 5’-GMP give no significant viscosity changes (Fig. 2). These results indicate that Ag+ induces aggregation of 5’-GMP. After increasing significantly over the range of 1 – 3 Ag+:GMP, the relative viscosity ratio reaches a plateau of ~400. This limiting ratio is maintained with higher concentrations of Ag+, suggesting that available binding sites are saturated and no further reactions such as precipitation occur. Approximate size differences between 5’-GMP and its Ag+ adduct are extracted by treating both as spherical solutes with identical shape factors of 2.5.33 Using the limiting relative viscosity, the aggregate is ~7 times larger than monomeric 5’-GMP. The concentration used for these studies allowed reliable measurements in both the absence and presence of Ag+ yet is higher than the 100 ¼M concentrations used for most of the studies is this work. Calorimetry measurements up to 500 μM suggest that the same reaction occurs at 100 μM, as indicated by the similarities of the thermodynamic parameters and relative stoichiometry.
Figure 2.

Graph of the relative viscosity (η/η0) of a 1 mM 5’-GMP solution with increasing relative amounts of Ag+ (closed circles). For control experiments, Ag+ was added to the buffer without 5’-GMP (open circles). Trend lines are provided to connect the data, and the error bars are ± 1σ.
Spectroscopy studies indicate that stacked bases provide the structural basis of the aggregate. For a 100 μM solution of 5’-GMP, the circular dichroism response of the nucleobase is magnified by Ag+, as shown by development of two bands with negative ellipticity at 220 and 275 nm (Fig. 3). Given the importance of base stacking in the circular dichroism response of DNA, the spectral changes that are induced by Ag+ are consistent with a chiral aggregate that is comprised of stacked bases.32 Similar to the viscosity studies, the circular dichroism response reaches a plateau at ~2 Ag+:5’-GMP. Ag+ also alters the absorption spectrum of guanine by suppressing the primary band with λmax = 252 nm while a shoulder develops at ~300 nm (Fig. 3). Up to 2 Ag+:5’-GMP, addition of Ag+ results in isosbestic points, which supports conversion of 5’-GMP from unbound to to a single type of bound form. Earlier studies attributed these changes to adduct formation, but temperature dependent changes suggest that base stacking and aggregation contribute.15,22 For a solution of 100 μM 5’-GMP, spectra without Ag+ are comparable at 25 °C and 90 °C, which is expected as the monomeric form of 5’-GMP is favored at this low concentration.23 Addition of 2 equivalents of Ag+ at 90 °C has a minor influence, which suggests that monomeric 5’-GMP is still favored at this high temperature. Upon cooling this solution to 20 °, the absorbance diminishes from 240 – 290 nm and a shoulder develops at ≈300 nm. These spectral changes do not vary with complete temperature cycling, suggesting that noncovalent interactions in the complex are disrupted at higher temperatures (Fig. 4). A two-state transition between an aggregate at low temperature and a monomer at high temperature forms is supported by isosbestic points.
Figure 3.

Circular dichroism (top) and absorbance (bottom) spectra of 100 μM 5’-GMP solutions in a 100 mM NaClO4 solution at pH = 8 with 0.2 molar increments of Ag+. The spectra of 5’-GMP solution alone are represented by dashed lines. The arrows indicate the development of the bands, and the inset summarizes the stoichiometry dependence of the spectroscopic changes.
Figure 4.

(Top) Absorption spectra of 100 μM solutions of 5’-GMP without and with Ag+. Without Ag+, spectra were collected at 25 ° (dashed line) and 90 °C (dashed – dotted line). With 2 equivalents of Ag+, spectra were collected at 25 ° (dotted line) and 90 °C (solid line). (Bottom) Absorption spectra of a solution of 100 μM 5’-GMP with 200 μM Ag+ in 100 mM NaClO4. The temperature was increased from 25 °C to 95 °C. At the completion of the experiment, the temperature was returned to 25 °C, and the two spectra at 25 °C overlap.
Association of Ag+ with 5’-GMP is further characterized by a negative enthalpy change of −9.8 (± 0.1) kcal/(mole Ag+) and a negative entropy change of −11 (± 2) cal/(K mole Ag+), with the former dominating the free energy change (Fig. 5 and Table 1). Base specificity is indicated by comparison with 5’-cytosine monophosphate. Despite identical phosphate and ribose groups for both ribonucleotides, reaction of Ag+ with 5’-cytosine monophosphate has an affinity that is 70 times lower and an enthalpy change that is 3.4 times lower relative to the reaction with the guanine analog (Table 1). Because Ag+ displaces a proton from 5’-GMP, the intrinsic enthalpy change for the reaction is −6.6 kcal/(mole Ag+) when accounting for protonation of H2BO3− in the buffer.15,34 This heat change is similar to values associated with the stacking of purine nucleobases in aqueous solution, which suggests that Ag+ promotes that stacking of 5’-GMP.35,36 This reaction has a threshold concentration for calorimetric detection of ~10 μM 5’-GMP, and enthalpy changes and stoichiometries are consistent from 100 – 500 μM (Fig. 5).
Figure 5.

Binding isotherm from the titration of 10 mM Ag+ into 100 μM 5’-GMP at 25 °C. The inset shows the enthalpy changes for the reaction of Ag+ with 5’-GMP as a function of the concentration of 5’-GMP.
Table I.
Thermodynamic Parameters for Association of Ag+ with Nucleotides and Nucleosidesa
| Ligand | ΔH (kcal/mole) |
N (Ag+:Ligand) |
K (M−1) | ΔG (kcal/mole) |
ΔS (cal/(mole K)) |
|---|---|---|---|---|---|
| 5’-guanosine monophosphate | −9.8 ± 0.5 | 2.3 ± 0.1 | 9.6 (± 1.9) × 104 | −6.8 ± 0.1 | −10 ± 2 |
| 5’-cytosine monophosphate | −3.0 ± 0.8 | 1.1 ± 0.3 | 1.3 (± 0.3) × 103 | −4.3 ± 0.1 | −4 ± 3 |
| guanosine | −14.7 ± 1.0 | 1.33 ± 0.05 | 1.8 (± 0.2) × 105 | −7.2 ± 0.1 | −25 ± 3 |
| 5’-guanosine monophosphate in 1 M NaClO4 | −12.1 ± 0.8 | 1.5 ± 0.1 | 6.6 (± 1.2) × 104 | −6.6 ± 0.1 | −19 ± 3 |
| 5’-guanosine monophosphate in 100 mM LiClO4 | −11.0 ± 1.0 | 2.1 ± 0.2 | 6.0 (± 0.6) × 104 | −6.5 ± 0.1 | −15 ± 3 |
| 7-deazaguanosine | −1.4± 0.3 | 1.15 ± 0.25 | 1.6 (± 0.7) × 104 | −5.7 ± 0.3 | 14 ± 1 |
| 5’-inosine monophosphate | −9.7 ± 0.2 | 2.3 ± 0.1 | 9.2 (± 0.2) × 104 | −6.8 ± 0.1 | −10. ± 1 |
Nucleotide and nucleoside concentrations were 100 μM
Local Structure of the Ag+-Guanosine Complex
Viscometry and spectroscopic studies indicate that Ag+ induces aggregation of 5’-GMP. The binding sites on 5’-GMP for Ag+ are considered using pH and thermodynamic studies. A decrease in pH accompanies reaction of Ag+ and 5’-GMP.15 In an unbuffered solution with 100 mM NaClO4, a 100 μM solution of the dianionic form of 5’-GMP has a pH = 7.6, which is similar to the pH = 8 conditions used for these studies. Addition of 5 mole equivalents of Ag+ completes complex formation and decreases the pH to 4.4, whereas no significant pH change occurs without 5’-GMP. Thus, Ag+ drives protons from the nucleotides. Because the phosphate is doubly ionized prior to addition of Ag+, the most labile proton is associated with the N1 position of guanine, which has a pKa = 9.2.37 Stoichiometric information on the Ag+-GMP complex in basic solution was derived from a pH stat titration.15,38 With addition of 1.2 equivalents of OH−, the displaced H+ is neutralized to recover the initial pH of 7.6. Besides Ag+ complexation with N1, another possible adduct is with O6, as electron density can be localized on either site in tautomeric forms.15
By changing reaction conditions and by using structural variants of 5’-GMP, other binding sites for Ag+ were evaluated via calorimetry studies. For 5’-GMP, a relative stoichiometry of 2.3 ± 0.1 Ag+:GMP was measured, and two observations suggest that the phosphate group is one binding site (Fig. 1, Table 1). First, the stoichiometry decreases to 1.3 ± 0.1 Ag+ when guanosine is used. Both the nucleoside and nucleotide have similar free energy changes for their reaction with Ag+, so the stoichiometry difference suggests that 1 Ag+ binds with the phosphate group. Second, increasing the salt concentration from 100 mM to 1 M Na+ does not influence the free energy changes but decreases the empirical stoichiometry to 1.5 ± 0.1 Ag+:5’-GMP. This change suggests that higher concentrations of Na+ screen the negatively charged phosphates to inhibit Ag+ coordination.39 The salt dependence was also examined using Li+. Relative to solutions with 100 mM Na+, stoichiometries and thermodynamic parameters for reaction of Ag+ with 5’-GMP are similar with Li+, thus further suggesting that cations in the supporting electrolyte predominantly act to electrostatically screen assembled phosphates (Table 1). While these experiments indicate that Ag+ associates with the phosphate in the nucleotide, both removing the phosphate using guanosine and reducing the involvement of phosphate using higher salt concentrations makes the reaction more enthalpically favored. These trends suggest that electrostatic interactions are not a dominant contribution to the enthalpy changes, which is consistent with a more favored reaction of Ag+ with nucleobases relative to phosphates (Table 1).17
Two other binding sites were considered using structural variations in the guanine base (Table 1). A ten-fold lower enthalpy change is observed for reaction of Ag+ with 7-deazaguanosine relative to guanosine, and this significant change indicates that adduct formation with N7 is necessary for guanosine aggregation. Another potential binding site is the exocyclic amine. However, absence of N2 in inosine monophosphate does not influence the thermodynamic parameters, which suggests that this amine is not complexed with Ag+. This finding is consistent with structural studies that indicate that this group is not a favorable binding site for transition metal cations.21
Discussion
A key finding from these studies is that adduct formation between Ag+ and guanosine induces aggregation of the nucleobase in the 102 μM range. In support, viscosities of solutions of 5’-GMP increase with Ag+, and a spherical model for the solutes predicts that the aggregate is limited to be ~7 times larger than the 5’-GMP monomer. This size determination is qualitative, as aggregate shape cannot be deduced from these experiments and aggregates could be polydisperse. The nucleobases are stacked in this aggregate, as indicated by a chiral structure that develops with addition of Ag+ to 5’-GMP. Because exciton coupling of electronic states associated with the bases is the largest contribution to the circular dichroism spectra of nucleic acids, this observation suggests that Ag+-GMP complexes are stacked, possibly with a helical arrangment.32 Furthermore, the circular dichroism spectrum is distinct from other assembled forms of guanosine, which suggests that Ag+ alters electronic coupling between assembled bases in this polymorphic form of 5’-GMP. 40 A reduction in the extinction coefficient of 5’-GMP with the addition of Ag+ is also consistent with base stacking.33 Furthermore, enthalpy changes associated with reaction of Ag+ with 5’-GMP are similar to prior measurements of purine association in aqueous solution.
With respect to binding sites for Ag+, favored association with the N7, N1, and O6 of guanine is well-established from solid-state and solution studies. Ten-fold lower enthalpy changes with 7-deazaguanosine relative to guanosine indicate that Ag+ coordinates with the N7 position. This binding site was identified earlier by changes in infrared linear dichroism spectra that accompany the methylation of the N7 position.22 For the present studies, 7-deazaguanosine was chosen because the overall charge of the molecule is not altered as with methylation, thus reducing concerns regarding how electrostatics could influence aggregation. A decrease in pH accompanies complexation of Ag+ with guanine. This correlation could be due to adduct formation with N1, as this nitrogen is largely protonated in slightly basic solutions. Alternatively, the pKa may be reduced when Ag+ binds with N7, thereby changing the electronic distribution in the nucleobase. This altered electronic distribution may shift electron density to the tautomeric form of the anion to favor Ag+ association with O6. In support, infrared spectra show a significant change in the carbonyl stretching frequency for Ag+ adducts with 5’-GMP.15 An additional binding site on 5’-GMP is the phosphate group, as demonstrated by an ~ Ag+ reduction in stoichiometry when the nucleoside is used in place of the nucleotide and when higher concentrations of Na+ are used with 5’-GMP. Coordination of Ag+ with the phosphate suggests that compensation of electrostatic repulsion could be an important factor for nucleotide assembly.24,41 It is difficult to discern the mechanism of aggregation from our studies, as chelation and aggregation may not be coupled.
These studies utilized exothermic heat changes as a signature of Ag+-induced aggregation of 5’-GMP. By using calorimetry to monitor reaction progress, a relative stoichiometry of ~1 Ag+:guanine nucleobase was derived (Table 1). Yet, two binding sites at O6 and N7 were identified. This difference between the empirical stoichiometry and the number of binding sites suggests Ag+ mediates coordination between guanines. Crystallographic studies demonstrate that Ag+ favors linear coordination geometries, so the following possibilities utilize this tendency. Calorimetry studies with 7-deazaguanosine demonstrate that aggregation is dependent on Ag+ complexation with N7, which is consistent with favored association of many transition metals with this site.42 One model is based on guanine dimers in which two Ag+ are coordinated by O6 and N7 groups from opposing guanines (Fig. 1F).15 This structure is supported by the loss or dramatic shift of the carbonyl stretching frequency when Ag+ complexes with guanine and by the proton displacement if Ag+ coordinates with the enolate-like tautomer. Although the O6 position is usually considered to be a less favorable binding site for Ag+, crystallographic studies have demonstrated that formation of such adducts.43 Another general type of coordinated structure involves four bases, and a well-studied form is the G-quartet. The N1-H and N2-H are hydrogen bonded with O6 and N7, respectively, of an adjacent guanine, and the planar macrocycle forms an electronegative cavity for a variety of cations, including Group I and II cations, Tl+, Pb2+, and NH4+.44,45,46,47,48 Three observations argue against this base arrangement with Ag+ mediated chelation. First, quartets of 5’-GMP are most favored in the concentration range 0.1 to 1 M in basic solution, whereas Ag+-induced aggregation is favored in the 10−4 M range. Second, G-quartets are stabilized by hydrogen bonding interactions between N7 and N2, but the studies with inosine show that the free energy change is not significantly influenced by the absence of the exocyclic amine. Third, the centrally arranged carbonyls of G-quartets coordinate cations, and their stabilizing effect depends on the size and hydration of the cation. However, the reaction of Ag+ with 5’-GMP is not influenced by Li+ vs. Na+. Other quartet models have been proposed, but these involve no coordination of Ag+ with N7 or steric interactions between the exocyclic amines. 49,50 Experiments are underway to distinguish these possibilities and to determine the local structure of the aggregate.
These studies provide two general observations. First, Ag+ induced base stacking suggests that aggregates with a high aspect ratio can be produced, which is significant for efforts to develop biomolecular based electronics. Furthermore, the relatively high concentration of transition metal cations could have an important impact on the electronic properties of the aggregates. Thus, efforts to determine the global shape of the aggregate are focusing on the aspect ratio of the possible rod-like structure using light scattering methods. To supplement information from circular dichroism studies, other techniques such as NMR spectroscopy would provide valuable insight into the relative arrangement of the bases in the aggregate.51 Furthermore, Ag+ could produce a helically organized aggregate.24 Specific oligonucleotides may provide more precise control of the aggregate length, but altered pKa’s of guanine’s functional groups relative to the isolated nucleotides might alter interaction with Ag+.37 Second, coordination of multiple bases by Ag+ has implications for understanding how fluorescent silver clusters form with DNA. For example, we have recently shown that a red emissive silver cluster is stabilized within a four-stranded i-motif structure, and cluster stabilization via coordination with multiple bases is a possibility. The question of interest is whether these interactions reflect the original base arrangement prior to reduction of the DNA bound Ag+.
Conclusion
Aggregation results when Ag+ forms adducts with guanosine. The following evidence supports a base stacked aggregate: the viscosity of a 5’-guanosine monophosphate solution increases in the presence of Ag+, the circular dichroism response and the extinction coefficient of guanine are significantly influenced by Ag+, and the exothermic heat changes are base dependent and their magnitude is similar to that of base stacking for purines. The empirical stoichiometry is ~1 Ag+:guanine, but the nucleobase possesses two binding sites for Ag+. A decrease in pH when Ag+ is added to 5’-GMP and a reduced enthalpy change with 7-deazaguanosine support binding association with the O6 and N7 positions, respectively. These groups are arranged such that Ag+ could adopt its favored linear coordination geometry within a Ag+-linked dimer. The general conclusions from these studies are that Ag+ favors self-association of guanine and derivatives in dilute solution and also promotes formation of a specific type of specific structure within the aggregate. This alternative base arrangement with a relatively high concentration of Ag+ could influence the electronic properties of this aggregated form of 5’-GMP, thereby expanding functionality. These studies also provide important insight in to the role of base coordination in the synthesis of fluorescent silver nanomaterials.
ACKNOWLEDGMENT:
We thank the National Science Foundation (CHE-0718588 and CBET-0853692), the National Institutes of Health (R15GM071370 and P20 RR-016461 (from the National Center for Research Resource)), and the Henry Dreyfus Teacher-Scholar Awards Program for support. We are grateful for the assistance of G. Hendrickson and A. Lyon in the initial stages of the work.
References
- 1.Davis JT; Spada GP, Supramolecular architectures generated by self-assembly of guanosine derivatives. Chemical Society Reviews 2007, 36, (2), 296–313. [DOI] [PubMed] [Google Scholar]
- 2.Simmel FC, Three-Dimensional Nanoconstruction with DNA. Angewandte Chemie International Edition 2008, 47, (32), 5884–5887. [DOI] [PubMed] [Google Scholar]
- 3.Sivakova S; Rowan SJ, Nucleobases as supramolecular motifs. Chemical Society Reviews 2005, 34, (1), 9–21. [DOI] [PubMed] [Google Scholar]
- 4.Muller J, Metal-Ion-Mediated Base Pairs in Nucleic Acids. European Journal of Inorganic Chemistry 2008, 2008, (24), 3749–3763. [Google Scholar]
- 5.Lippert B, Effects of Metal-Ion Binding on Nucleobase Pairing: Stabilization, Prevention, and Mismatch Formation. Journal of the Chemical Society-Dalton Transactions 1997, 3971–3976. [Google Scholar]
- 6.Lippert B, Multiplicity of metal ion binding patterns to nucleobases. Coordination Chemistry Reviews 2000, 200-202, 487–516. [Google Scholar]
- 7.Krizanovic O; Sabat M; Beyerle-Pfnuer R; Lippert B, Metal-modified nucleobase pairs: mixed adenine, thymine complexes of trans-a2platinum(II) (a = ammonia, methylamine) with Watson-Crick and Hoogsteen orientations of the bases. Journal of the American Chemical Society 1993, 115, (13), 5538–5548. [Google Scholar]
- 8.Menzer S; Sabat M; Lippert B, Ag(I) Modified Base-Pairs Involving Complementary (G, C) and Noncomplementary (A, C) Nucleobases - on the Possible Structural Role of Aqua Ligands in Metal-Modified Nucleobase Pairs. Journal of the American Chemical Society 1992, 114, (12), 4644–4649. [Google Scholar]
- 9.Tanaka Y; Oda S; Yamaguchi H; Kondo Y; Kojima C; Ono A, 15N-15N J-Coupling Across HgII: Direct Observation of HgII-Mediated T-T Base Pairs in a DNA Duplex. Journal of the American Chemical Society 2007, 129, (2), 244–245. [DOI] [PubMed] [Google Scholar]
- 10.Petty JT; Zheng J; Hud NV; Dickson RM, DNA-templated Ag nanocluster formation. Journal of the American Chemical Society 2004, 126, (16), 5207–12. [DOI] [PubMed] [Google Scholar]
- 11.Richards CI; Choi S; Hsiang J-C; Antoku Y; Vosch T; Bongiorno A; Tzeng Y-L; Dickson RM, Oligonucleotide-Stabilized Ag Nanocluster Fluorophores. Journal of the American Chemical Society 2008, 130, (15), 5038–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Neill PR; Velazquez LR; Dunn DG; Gwinn EG; Fygenson DK, Hairpins with Poly-C Loops Stabilize Four Types of Fluorescent Agn:DNA. The Journal of Physical Chemistry C 2009, 113, (11), 4229–4233. [Google Scholar]
- 13.Vosch T; Antoku Y; Hsiang J-C; Richards CI; Gonzalez JI; Dickson RM, Strongly emissive individual DNA-encapsulated Ag nanoclusters as single-molecule fluorophores. Proceedings of the National Academy of Sciences of the United States of America 2007, 104, (31), 12616–12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Andrews LJ; Keefer RM, Cation Complexes of Compounds Containing Carbon-Carbon Double Bonds. VI. The Argentation of Substituted Benzenes. Journal of the American Chemical Society 1950, 72, (7), 3113–3116. [Google Scholar]; (b) Fyfe WS, Relationship of Electron Distribution in Amines to Ammine Stability. Nature 1952, 169, (4289), 69–70. [Google Scholar]
- 15.Tu AT; Reinosa JA, The Interaction of Silver Ion with Guanosine, Guanosine Monophosphate, and Related Compounds. Determination of Possible Sites of Complexing. Biochemistry 1966, 5, (10), 3375–3383. [Google Scholar]
- 16.Sabat M; Lippert B, Metal ions in multiple-stranded DNA. Metal Ions in Biological Systems 1996, 33, 143–76. [PubMed] [Google Scholar]
- 17.Jensen RH; Davidson N, Spectrophotometric, potentiometric, and density gradient ultracentrifugation studies of the binding of silver ion by DNA. Biopolymers 1966, 4, (1), 17–32. [Google Scholar]
- 18.Belanger-Gariepy F; Beauchamp AL, Crystal structure of bis(9-methylhypoxanthine)silver(I) perchlorate monohydrate, a model complex for the silver-poly(I) system. Journal of the American Chemical Society 1980, 102, (10), 3461–3464. [Google Scholar]
- 19.Fox BS; Beyer MK; Bondybey VE, Coordination Chemistry of Silver Cations. Journal of the American Chemical Society 2002, 124, (45), 13613–13623. [DOI] [PubMed] [Google Scholar]
- 20.(a) Menzer S; Sabat M; Lippert B, Ag(I) Modified Base-Pairs Involving Complementary (G, C) and Noncomplementary (A, C) Nucleobases - on the Possible Structural Role of Aqua Ligands in Metal-Modified Nucleobase Pairs. Journal of the American Chemical Society 1992, 114, (12), 4644–4649. [Google Scholar]; (b) Purohit CS; Mishra AK; Verma S, Four-Stranded Coordination Helices Containing Silver-Adenine (Purine) Metallaquartets. Inorganic Chemistry 2007, 46, (21), 8493–8495. [DOI] [PubMed] [Google Scholar]
- 21.Aoki K, General Conclusions form Solid State Studies of Nucleotide-Metal Ion Complexes. Metal Ions in Biological Systems 1996, 32, 91–134. [Google Scholar]
- 22.Matsuoka Y, Norden B, and Kurucsev T (1984) Nucleic acid-metal interactions. 2. Complexes of silver(i) with guanosine and 7-methylguanine from studies of isotropic and dichroic spectra, The Journal of Physical Chemistry 88, 971–976. [Google Scholar]
- 23.Davis JT, G-Quartets 40 Years Later: From 5-GMP to Molecular Biology and Supramolecular Chemistry. Angewandte Chemie-International Edition 2004, 43, (6), 668–698. [DOI] [PubMed] [Google Scholar]
- 24.(a) Wong A; Ida R; Spindler L; Wu G, Disodium Guanosine 5’-Monophosphate Self-Associates into Nanoscale Cylinders at pH 8: A Combined Diffusion NMR Spectroscopy and Dynamic Light Scattering Study. Journal of the American Chemical Society 2005, 127, (19), 6990–6998. [DOI] [PubMed] [Google Scholar]; (b) Wu G; Kwan ICM, Helical Structure of Disodium-Guanosine Monophosphate Self-Assembly in Neutral Solution. Journal of the American Chemical Society 2009, 131, (9), 3180–3182. [DOI] [PubMed] [Google Scholar]
- 25.Fasman GD, CRC Handbook of Biochemistry and Molecular Biology. ed.; CRC Press: Cleveland, 1975. [Google Scholar]
- 26.Cavaluzzi MJ; Borer PN, Revised UV extinction coefficients for nucleoside-5'-monophosphates and unpaired DNA and RNA. Nucleic Acids Research 2004, 32, (1), e13–e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganguly M; Wang F; Kaushik M; Stone MP; Marky LA; Gold B, A study of 7-deaza-2'-deoxyguanosine 2'-deoxycytidine base pairing in DNA. Nucleic Acids Research 2007, 35, (18), 6181–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Kiser JR; Monk RW; Smalls RL; Petty JT, Hydration Changes in the Association of Hoechst 33258 with DNA. Biochemistry 2005, 44, (51), 16988–16997. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Degtyareva NN; Fresia MJ; Petty JT, DNA Conformational Effects on the Interaction of Netropsin with A-tract Sequences. Biochemistry 2007, 46, (51), 15136–15143. [DOI] [PubMed] [Google Scholar]
- 29.Bordelon JA; Feierabend KJ; Siddiqui SA; Wright LL; Petty JT, Viscometry and Atomic Force Microscopy Studies of the Interactions of a Dimeric Cyanine Dye with DNA. The Journal of Physical Chemistry B 2002, 106, (18), 4838–4843. [Google Scholar]
- 30.Bevington PR; Robinson DK Data Reduction and Error Analysis for the Physical Sciences. McGraw-Hill: NY, 1992. [Google Scholar]
- 31.Wakelin LPG; Waring MJ, The binding of echinomycin to deoxyribonucleic acid. Biochemical Journal 1976, 157, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cantor CR; Schimmel PR Biophysical Chemistry. W. H. Freeman: NY, 1980. [Google Scholar]
- 33.van Holde KE; Johnson WC; Ho PS, Principles of Physical Biochemistry ed.; Pearson/Prentice Hall: Upper Saddle River, NJ, 2006; p 710. [Google Scholar]
- 34.Goldberg RN; Kishore N; Lennen RM, Thermodynamic quantities for the ionization reactions of buffers. Journal of Physical and Chemical Reference Data 2002, 31, (2), 231–370. [Google Scholar]
- 35.Marenchic MG; Sturtevant JM, Calorimetric investigation of the association of various purine bases in aqueous media. Journal of Physical Chemistry 1975, 77, (4), 544–548. [DOI] [PubMed] [Google Scholar]
- 36.Tso PO, Bases, Nucleosides, and Nucleotides ed.; Academic Press: New York, 1974; Vol. I, p.453–584. [Google Scholar]
- 37.Bloomfield VA; Crothers DM; Tinoco J, Ignacio, Nucleic Acids: Structures, Properties, and Functions ed.; University Science Books: Sausaltio, CA, 2000. [Google Scholar]
- 38.Yamane T; Davidson N, On the Complexing of Deoxyribonucleic Acid (DNA) by Mercuric Ion. Journal of the American Chemical Society 1961, 83, (12), 2599–2607. [Google Scholar]
- 39.Manning GS, The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Quarterly Reviews of Biophysics 1978, 11, (2), 179–246. [DOI] [PubMed] [Google Scholar]
- 40.(a) Gottarelli G; Lena S; Masiero S; Pieraccini S; Spada GP, The use of circular dichroism spectroscopy for studying the chiral molecular self-assembly: An overview. Chirality 2008, 20, (3-4), 471–485. [DOI] [PubMed] [Google Scholar]; (b) Yu Y; Nakamura D; DeBoyace K; Neisius AW; McGown LB, Tunable Thermoassociation of Binary Guanosine Gels. Journal of Physical Chemistry B 2008, 112, (4), 1130–1134. [DOI] [PubMed] [Google Scholar]
- 41.Chantot JF, and Guschlbauer W (1972) Mechanism of gel formation by guanine nucleosides, Jerusalem Symposium on Quantum Chemistry and Biochemistry 4, 205–216. [Google Scholar]
- 42.Martin RB, Dichotomy of Metal Ion Binding to N1 and N7 of Purines. Metal Ions in Biological Systems: Interactions of Metal Ions with Nucleotides, Nucleic Acids, and their Constituents 1996, 32, 61–89. [Google Scholar]
- 43.Marzilli LG; Kistenmacher TJ; Rossi M, An extension of the role of O(2) of cytosine residues in the binding of metal ions. Synthesis and structure of an unusual polymeric silver(I) complex of 1-methylcytosine. Journal of the American Chemical Society 1977, 99, 2797–2798. [DOI] [PubMed] [Google Scholar]
- 44.Davis JT, G-Quartets 40 Years Later: From 5-GMP to Molecular Biology and Supramolecular Chemistry. Angewandte Chemie-International Edition 2004, 43, (6), 668–698. [DOI] [PubMed] [Google Scholar]
- 45.Hud NV; Smith FW; Anet FAL; Feigon J, The Selectivity for K+ versus Na+ in DNA Quadruplexes Is Dominated by Relative Free Energies of Hydration: A Thermodynamic Analysis by 1H NMR. Biochemistry 1996, 35, (48), 15383–15390. [DOI] [PubMed] [Google Scholar]
- 46.Gill ML; Strobel SA; Loria JP, Crystallization and characterization of the thallium form of the Oxytricha nova G-quadruplex. Nucleic Acids Research 2006, 34, (16), 4506–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mondragon-Sanchez JA; Liquier J; Shafer RH; Taillandier E, Tetraplex structure formation in the thrombin-binding DNA aptamer by metal cations measured by vibrational spectroscopy. Journal of Biomolecular Structure and Dynamics 2004, 22, (3), 365–73. [DOI] [PubMed] [Google Scholar]
- 48.Podbevsek P; Hud NV; Plavec J, NMR evaluation of ammonium ion movement within a unimolecular G-quadruplex in solution. Nucleic Acids Research 2007, 35, (8), 2554–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shin YA; Eichhorn GL, Induction of Helicity in Polyuridylic Acid and Polyinosinic Acid by Silver Ions. Biopolymers 1980, 19, (3), 539–556. [DOI] [PubMed] [Google Scholar]
- 50.Belanger-Gariepy F; Beauchamp AL, Crystal Structure of Bis(9-methylhypoxanthine)silver(I) perchlorate Monohydrate, A Model Complex for the Silver-Poly(I) system. Journal of the American Chemical Society 1980, 102, (10), 3461–3464. [Google Scholar]
- 51.Gottarelli G; Lena S; Masiero S; Pieraccini S; Spada GP, The use of circular dichroism spectroscopy for studying the chiral molecular self-assembly: An overview. Chirality 2008, 20, (3-4), 471–485. [DOI] [PubMed] [Google Scholar]