

Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disorder characterized by episodically exuberant heterotopic ossification (HO), whereby skeletal muscle is abnormally converted into misplaced, but histologically normal bone. This HO leads to progressive immobility with catastrophic consequences, including death by asphyxiation. FOP results from mutations in the intracellular domain of the type I BMP (bone morphogenetic protein) receptor ACVR1; the most common mutation alters arginine 206 to histidine (ACVR1R206H) and has been thought to drive inappropriate bone formation as a result of receptor hyperactivity. We unexpectedly found that this mutation rendered ACVR1 responsive to the activin family of ligands, which generally antagonize BMP signaling through ACVR1 but cannot normally induce bone formation. To test the implications of this finding in vivo, we engineered mice to carry the Acvr1R206H mutation. Because mice that constitutively express Acvr1[R206H] die perinatally, we generated a genetically humanized conditional-on knock-in model for this mutation. When Acvr1[R206H] expression was induced, mice developed HO resembling that of FOP; HO could also be triggered by activin A administration in this mouse model of FOP but not in wild-type controls. Finally, HO was blocked by broad-acting BMP blockers, as well as by a fully human antibody specific to activin A. Our results suggest that ACVR1R206H causes FOP by gaining responsiveness to the normally antagonistic ligand activin A, demonstrating that this ligand is necessary and sufficient for driving HO in a genetically accurate model of FOP; hence, our human antibody to activin A represents a potential therapeutic approach for FOP.
One-sentence summary:
In patients with fibrodysplasia ossificans progressiva, the disease-causing mutation changes the ligand response profile of the ACVR1 receptor so that it is activated by the normally antagonistic ligand activin A.
Editor’s Summary:
Explaining bone overgrowth
Fibrodysplasia ossificans progressiva (FOP) is a rare, but deadly, genetic condition that causes growth of bony structures in place of normally soft tissues such as muscle and ligaments. The causal mutation, in the bone morphogenetic protein receptor ACVR1, has been thought to boost the receptor’s activity, triggering inappropriate bone formation. Hatsell et al. suggest that it works instead by a different mode of action—acquiring the ability to respond to the injury-related factor activin, which may explain some of the puzzling aspects of the disease.
The mutated ACVR1 receptor, expressed in cultured cells, responded to activin as well as to its natural ligand, bone morphogenetic protein. When the mutated gene was engineered to be expressed in adult mice (to avoid its perinatal lethal effects), the animals developed heterotopic ossification, as in FOP. Unexpectedly, for heterotypic ossification, the receptor required stimulation by the endogenous ligand activin. Small sponges soaked with activin ossified after they were implanted into the animals. Normally, activin blocks binding of the ACVR1 receptor by its natural ligand bone morphogenetic protein-2.
Finally, the authors confirmed that activin A is a potential therapeutic target for the treatment of FOP: Animals carrying the mutated receptor that were also treated with a monoclonal antibody to activin A did not show heterotopic ossification, even as long as 6 weeks after introduction of the mutation. A mutation-induced ligand specificity change is an unusual cause of disease, but this mechanism may explain why the ossification in FOP patients is triggered by injury or trauma to tissues—a situation that induces high concentrations of activin.
INTRODUCTION
Fibrodysplasia ossificans progressiva (FOP) [Mendelian Inheritance in Man (MIM) #135100] is a rare autosomal-dominant disorder that is characterized chiefly by episodic heterotopic ossification (HO) of muscle, fascia, ligaments, and tendons; because of the serious consequences of this disease, it is generally seen as a result of de novo mutations. Surgery to remove the heterotopic bone in FOP patients often results in more aggressive episodes of HO so that such interventions are counterproductive and contraindicated. Hence, HO is cumulative and results in progressive immobility with catastrophic comorbidities (1). FOP is driven by mutations in the intracellular domain of ACVR1, a bone morphogenetic protein (BMP) type I receptor; the most common (accounting for about 97% of cases) results in an Arg206His (R206H) amino acid change within the glycine-serine (GS) subdomain of ACVR1 (1, 2).
Previous studies have explored the molecular mechanism whereby ACVR1[R206H] brings about HO (3–8), generally concluding that ACVR1[R206H] is a hyperactive receptor, yet with conflicting results regarding requirement for ligand. Several studies have investigated the in vivo properties of ACVR1[R206H]-expressing cells, but these experiments have been performed using cell implants (9,10), a setting that does not mirror the situation in FOP patients. Moreover, a previous attempt at generating a knock-in model of ACVR1R206H-Acvr1tm1Emsh resulted in chimeric animals (11). Although these chimeric F0 mice develop HO that resembles FOP, they cannot be established as a line because F1 Acvr1tm1Emsh/+ mice exhibit perinatal lethality (12).
To address these technical issues and to provide a more versatile model of FOP, we generated a conditional-on knock-in model of ACVR1R206H in mice. Using both the corresponding targeted mouse embryonic stem (ES) cells and carefully matched overexpressing human cell lines, we explored the signaling properties of ACVR1[R206H] in comparison to wild-type ACVR1. In parallel, after demonstrating the faithfulness and utility of our genetically humanized mouse model, we used it to confirm the mechanism by which ACVR1[R206H] drives HO in vivo and to translate our findings into a potential therapy for FOP.
RESULTS
ACVR1R206H is an activin-responsive receptor
ACVR1 transduces signals through Smad1/5/8. To explore how the R206H change might alter the signaling properties of ACVR1, we generated HEK293/BRE-Luc reporter lines stably expressing nearly identical levels of either ACVR1 or ACVR1[R206H] (fig. S1) and tested their responses to a panel of ligands belonging to the BMP and transforming growth factor-β (TGF-β) families. ACVR1[R206H] displayed increased signaling in response to some, but not all, of its canonical ligands. Specifically, the response to BMP2, BMP4, BMP7, BMP9, and BMP10 was enhanced, whereas the response to BMP2/7, BMP4/7, BMP5, and BMP6 remained unchanged (Fig. 1A, Table 1, and fig. S2). These experiments uncovered an unexpected property of ACVR1[R206H]: it was responsive to activin A, AB, AC, and B and, to lesser degree, BMP15, a set of ligands to which wild-type ACVR1shows no measurable response (Fig. 1B and Table 1).
Fig. 1. Acvr1 [R206H] signaling to Smad 1/5/8 is stimulated by activin A.
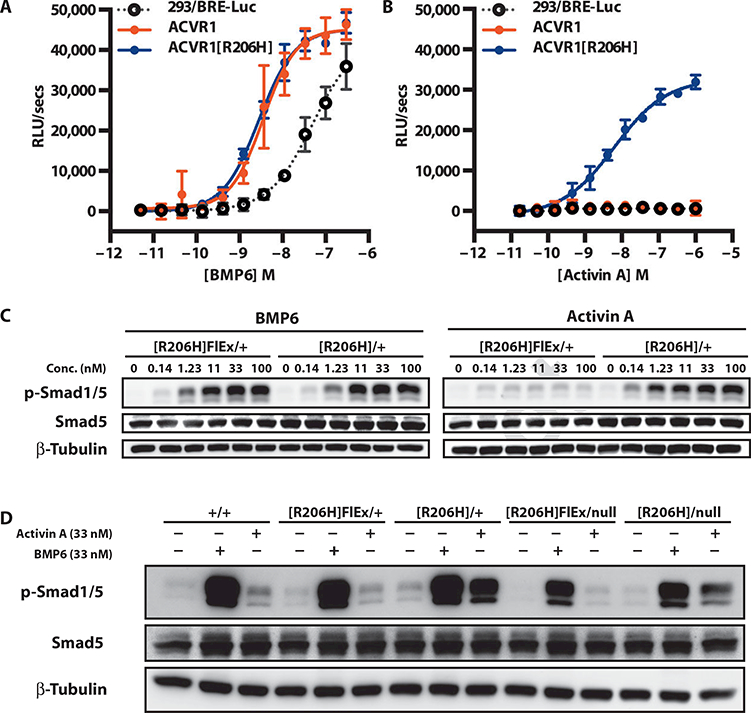
(A and B) Stable pools of HEK293/BRE-Luc reporter cells overexpressing human ACVR1 (solid red circles) or ACVR1[R206H] (solid blue circles) were treated overnight with BMP6 (A) or activin A (B), and activation of canonical BMP signaling was visualized using the Smad 1/5/8 reporter BRE-Luc. The response of the parental line is shown for comparison (293/BRE-Luc; open gray circles). (C) Mouse ES cells Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ and its post-Cre counterpart, Acvr1[R206H]FIEx/+; Gt(ROSA26)SorCreERT2/+/, were serum-starved for 2 hours and then treated with BMP6 or activin A for 1 hour. (D) Comparison of BMP6- or activin A-stimulated signaling in Acvr1+/+ (+/+), Acvr1[R206H]FlEx/+ ([R206H]FIEx/+), Acvr1[R206H]/+ ([R206H]/+), Acvr1[R206H]FlEx/nuII ([R206H]FIEx/null), and Acvr1[R206H1/nuII ([R206H]/null) ES cells. All lines also carry Gt(ROSA26)SorCreERT2/+. Response to ligands was visualized by Western blotting for phospho-Smad1/5. Total Smad5 and β-tubulin (loading control) are shown for comparison. RLU, relative light unit. RLUs are background-subtracted; error bars represent SD from samples run in triplicate.
Table 1.
Activity of TGF-β and BMP family ligands in HEK293/BRE-Luc reporter lines. —, median effective concentration (EC50) > 1 × 10−7 or no Cmax; Cmax (fold change), fold change at Cmax. GDF, growth and differentiation factor; DPPIV, dipeptidyl peptidase IV.
| Acvr1.WT |
Acvr1.R206H |
|||
|---|---|---|---|---|
| EC550 | Cmax (fold change) |
EC50 | Cmax (fold change) |
|
| Signal observed only in ACVR1[R206H]-expressing cells | ||||
| Activin A | — | — | 1.28 × 10−9 | 2.11 |
| Activin AB | — | — | 2.70 × 10−10 | 2.07 |
| Activin AC | — | — | 1.23 × 10−9 | 2.02 |
| Activin B | — | — | 1.63 × 10−10 | 2.41 |
| BMP15 | — | — | 1.78 × 10−8 | 2.29 |
| Identical signal observed in both lines | ||||
| BMP5 | 4.35 × 10−9 | 2.49 | 5.99 × 10−9 | 2.94 |
| BMP6 | 3.99 × 10−10 | 3.4 | 6.26 × 10−10 | 3.8 |
| BMP2/BMP7 | 1.59 × 10−9 | 2.81 | 1.77 × 10−9 | 2.7 |
| BMP4/BMP7 | 2.20 × 10−9 | 2.69 | 2.03 × 10−9 | 2.67 |
| ACVR1[R206H]-expressing cells are more responsive | ||||
| BMP2 | 9.95 × 10−9 | 1.5 | 1.32 × 10−9 | 2.9 |
| BMP4 | 4.79 × 10−9 | 2.35 | 1.76 × 10−9 | 4.2 |
| BMP7 | 2.99 × 10−9 | 2.65 | 5.72 × 10−9 | 3.64 |
| BMP9 | 1.05 ×10−11 | 1.62 | 1.61 × 10−11 | 2.61 |
| BMP10 | 1.00 × 10−9 | 1.52 | 6.97 × 10−10 | 3.11 |
| No signal observed in either cell line | ||||
| BMP3 | — | — | — | — |
| BMP3b | — | — | — | — |
| BMP8a | — | — | — | — |
| TGF-ß1 | — | — | — | — |
| TGF-ß2 | — | — | — | — |
| TGF-ß3 | — | — | — | — |
| GDF-3 | — | — | — | — |
| GDF-8 | — | — | — | — |
| GDF-11 | — | — | — | — |
| GDF-15 | — | — | — | — |
| DPPIV | — | — | — | — |
To exclude the possibility that the observed results were an artifact of overexpression, we examined whether the altered signaling properties of ACVR1[R206H] would reproduce in knock-in Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ ES cells (Fig. 2; see below). During the course of these experiments, we discovered that before activation by Cre, Acvr1[R206H]FLEx is a hypomorphic allele with about half of its transcripts missing exon 5 (fig. S3 and Discussion). Therefore, from a functional standpoint, Acvr1[R206H]FlEx/+ cells are nearly equivalent to Acvr1null/+. Nonetheless, in agreement with the data obtained in human embryonic kidney (HEK) 293 cells, comparison between Acvr1[R206H]FlEx+; Gt(ROSA26)SorCreERT2/+ ES cells and their Cre-activated counterparts [Acvr1[R206H]/+; Gt(ROSA26)SorCreERT2/+ ES cells] showed that the latter had become responsive to activin A (Fig. 1C). This altered responsiveness was also observed in Acvr1[R206H]nul1; Gt(ROSA26)SorCreERT2/+ ES cells, indicating that one allele of Acvr1[R206H] is sufficient to confer altered signaling and that the presence of a wild-type allele does not markedly modify this aberrant response, although it may have contributed to an increase in the signal (Fig. 1D). Signaling via activin A-Acvr1[R206H] activated Smad1/5 and did not switch the signaling to Smad2/3. Furthermore, the presence of the Acvr1[R206H] variant in these cells did not appear to markedly alter activin A signaling via its canonical receptor and Smad2/3 (fig. S4).
Fig. 2. A conditional-on knock-in allele of ACVR1R206H.
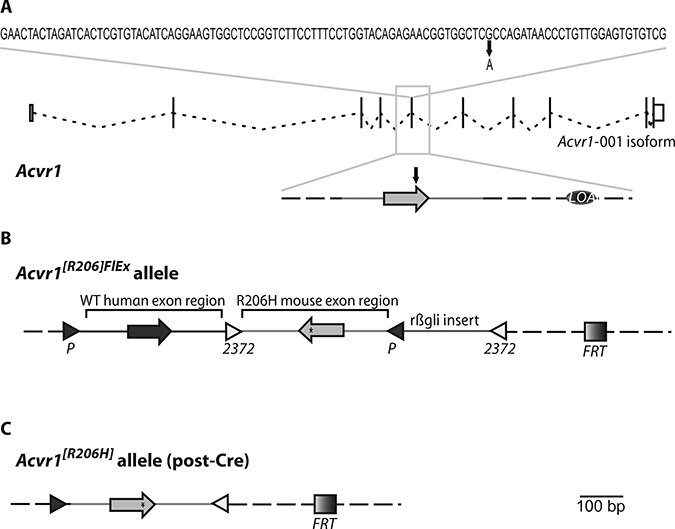
(A) Sequence of exon 5 along with the exon-intron structure of Acvr1 isoform 001 (adapted from www.Ensembl.org). The G-to-A missense mutation that results in the R206H amino acid change is marked. LOAp marks the small intronic region deleted for genotyping purposes (see Materials and Methods). (B) Structure of the Acvr1[R206H]FlEx allele. Mouse Acvr1 exon 5 and associated intronic sequence (R206H mouse exon region) have been placed in the antisense strand of Acvr1 within introns 5 to 6 and have been altered to encode R206H (*). The corresponding human exon and associated intronic sequence have been inserted in the sense strand to replace its corresponding mouse counterpart [wild-type (WT) human exon region]. These two regions have been flanked by FlEx arrays, arranged in a manner such that, upon action of Cre, the introduced human sequence will be deleted and the R206H mouse exon region will be brought to the sense strand, thereby giving rise to an Acvr1[R206H] allele. To ensure efficient action by Cre, the loxP and lox2372 sites of the 3′FlEx array have been separated by a small insert derived from rabbit hemoglobin β intron 2 (rβgli insert). An FRT site also remains after removal of the drug selection cassette (not shown). (C) Structure of the Acvr1[R206H] allele. Mouse Acvr1 exon 5 and associated intronic sequence have been placed in the sense strand of Acvr1.Note that the only extraneous elements that remain after action by Cre are two lox sites, an FRT site, and the small deletion of the region initially marked as LOAp in (A). Light gray, mouse sequences; dark gray, human sequences; dotted line, unaltered mouse genomic sequence in the Acvr1 locus.
Abrogating FKBP12 binding does not convert ACVR1 into an activin-responsive receptor
The above data indicate that an amino acid change in the intracellular domain of a type I BMP receptor is capable of altering the responsiveness of that receptor in a qualitative way, rendering it permissive to activation by normally nonstimulatory ligands. One possible explanation of how this happens might be the reduced affinity that ACVR1[R206H] displays for FKBP12 (4,5). Therefore, we tested whether wild-type ACVR1 could be converted into an activin-responsive receptor if FKBP12 binding was reduced by FK506. This was not the case. The only effect of FK506 was to enhance signaling from canonical ligands. It did not enable wild-type ACVR1 to respond to activin A (fig. S5).
Activin A normally binds to but acts as an antagonist of ACVR1
We further examined whether the responsiveness to nonstimulatory ligands that we observed could be explained by ACVR1[R206H] gaining the ability to bind to them. We tested this in assays that used artificial fusion receptors that report, by lacZ complementation, association between type I and type II BMP receptors, which is triggered by ligand binding. We demonstrated that ACVR1, but not the closely related ACVRL1, formed heterodimers with ACVR2A or ACVR2B in the presence of nonstimulatory activing A, AB, AC, and BB (fig. S6), indicating that the wild-type ACVR1 receptor can bind nonstimulatory activins, even though these ligands cannot trigger signaling through the receptor. Therefore, the altered responsiveness of ACVR1[R206H] could not be attributed to newly acquired binding properties, because wild-type ACVR1 is already able to bind these nonstimulatory ligands. The ability of activins to form nonsignaling complexes with ACVR1 in conjunction with the type II receptors ACVR2A and ACVR2B suggests that activins could act as competitive inhibitors of BMP signaling through ACVR1. Indeed, activin A inhibited BMP6-induced signaling both in a competition assay using HEK293/BRE-Luc reporter cells overexpressing wild-type ACVR1 (Fig. 3A) and in tests of BMP6-induced Smad1/5 phosphorylation in mouse ES cells in which Acvr1 is expressed from its native locus (Fig. 3B). Furthermore, this competition could not be attributed to inhibition of Smad1/5 phosphorylation by phospho-Smad2/3 because the levels of the latter were highest at the lowest concentration of activin A (Fig. 3C), which was also the least effective dose for inhibiting Smad1/5 phosphorylation (Fig. 3, B and C). Therefore, activin A may function as a competitive inhibitor of BMP-initiated signaling via ACVR1 by competing with BMPs for binding and generating an ACVR1-activin-ACVR2 complex that does not signal. Together, these results indicate that the most prominent property of ACVR1[R206H] differentiating it from wild-type ACVR1 is its ability to transduce signal from ligands that normally act as inhibitors ofBMP-initiated signaling through ACVR1. During the review of our manuscript, an independent group also reported that activin A can act as an inhibitor of ACVR1-mediated signaling (13).
Fig. 3. Activin A inhibits BMP6 signaling via ACVR1.
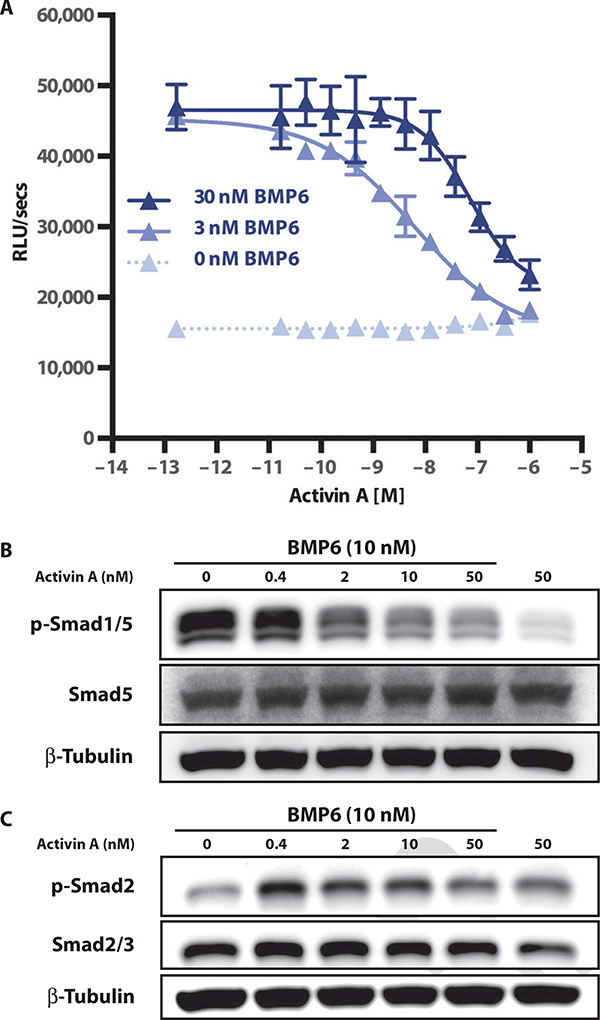
(A) The HEK293/BRE-Luc reporter line expressing WT ACVR1 was stimulated with 0,3, or 30 nM BMP6 and varying amounts (dose curve) of activin A. Error bars represent SD from samples run in triplicate. Lowest concentration on dose response represents no addition of activin A. (B and C) Competition assays were performed in WT ES cells to examine whether the activin competition for BMP6 signaling could result from an increase in Smad2 phosphorylation. Samples were analyzed for phospho-Smad1/5 (B) and phospho-Smad 2 (C) by Western blotting.
A conditional-on knock-in model of ACVR1R206H develops HO
To determine whether there is physiologic significance to the altered properties of ACVR1[R206H], we engineered a Cre-dependent conditional-on knock-in allele of this variant, Acvr1[R206H]FlEx (Fig. 2). This decision was driven in part by the need to bypass the perinatal lethality observed with the previous knock-in mouse (11) and in part to enable definition of the cell types involved in ACVR1[R206H]-driven HO. A detailed description of Acvr1[R206H]FlEx appears in Materials and Methods. Briefly, Acvr1[R206H]FIEx, by virtue of having the R206H-coding exon in the antisense strand (Fig. 2B), cannot express Acvr1[R206H] until it has been moved into the sense strand by Cre. This rearrangement is enabled by the use of FlEx arrays (14), which guide Cre to invert the R206H-encoding exon into the sense strand while simultaneously deleting the corresponding wild-type exon (Fig. 2C). In this manner, with Acvr1[R206H]FlEx, the FOP-causing Acvr1[R206H] variant could be turned on in specific cells and at specific times by using appropriate Cre drivers (15). We used a globally expressed, tamoxifen-inducible CreERT2 line, Gt(ROSA26)SorCreERT2/+ (16), which allowed the FOP variant to be turned on in adult animals at all sites of endogenous Acvr1 expression.
We enabled the expression of Acvr1[R206H] by treating Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ with tamoxifen, alongside Acvr1+/+; Gt(ROSA26)SorCreERT2/+ controls. The health of the mice was monitored daily, whereas development of HO was monitored every 2 weeks with in vivo μCT (computed tomography). Imaging revealed that tamoxifentreated Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice developed HO in the sternum (Fig. 4A), caudal vertebrae (Fig. 4B), hip joint (Fig. 4C), and hindlimb (Fig. 4D), mirroring sites that commonly ossify in FOP patients. HO was detectable as early as 2 weeks (fig. S7) and did not require induction by injury. HO was also progressive (fig. S8), and as observed in FOP, the heterotopic bone fused with native skeletal elements, thereby locking these elements (for example, a tibia with a femur and pelvis) into one position (Fig. 4, E and F, and fig. S8). Also mirroring FOP, the mature heterotopic bone contained bone marrow and histologically resembled normal bone (Fig. 4G). Developing HO lesions showed evidence of muscle destruction, inflammatory infiltration, fibroblast proliferation, and cartilage and bone formation, similar to the processes reported in FOP lesions (fig. S9) (17).
Fig. 4. Heterotopic boneformation in Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice after tamoxifen treatment.
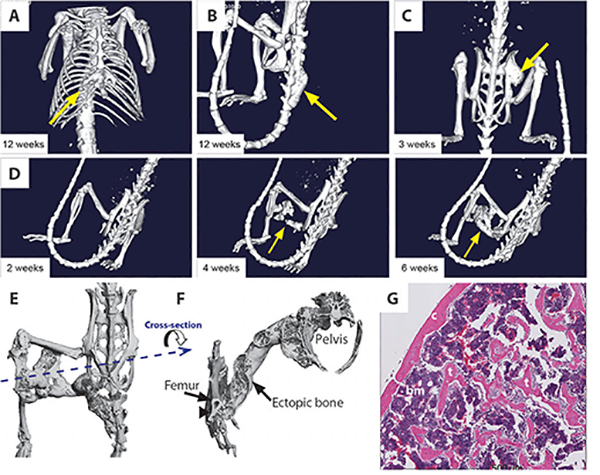
(A to D) Representative examples of in vivo μCT images from Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice 2 to 12 weeks after tamoxifen administration. Ectopic bone growth was found at a number of different sites including adjacent to the existing skeleton in the (A) sternum, (B) caudal vertebrae, and (C) hip joint. (D) Ectopic bone growth formed between 2 and 4 weeks after tamoxifen injection and could occur distal to the existing skeleton. (E) Ex vivo μCT image of an ectopic bone lesion from the dorsal side showing bridging from the femur to the pelvis. (F) A transverse view through the ectopic bone shows that the newly formed bone has both cortical and trabecular like structures. At the region of intersection of the ectopic bone and the endogenous skeleton (arrow), there is evidence of remodeling of the cortical bone, but no evidence of bone marrow sharing. (G) Hematoxylin and eosin (H&E)-stained histological sections of ectopic bone lesion demonstrating cortical (c) and trabecular (t) bone-like structures and bone marrow (bm).
ACVR1[R206H]-driven HO is a ligand-dependent process
One of the open questions regarding the HO process driven by ACVR1 [R206H] was whether it requires activation by ligand. Given that ACVR1[R206H] displays two altered signaling properties—(i) enhanced responsiveness to certain (but not all) canonical ligands and (ii) gain of responsiveness to normally inhibitory ligands—we used the broad-acting BMP and activin blockers, ACVR2A-Fc and ACVR2B-Fc (18–20), as a first test. ACVR2A-Fc and ACVR2B-Fc block the action of BMPs and activins by binding BMPs and activins, thereby blocking association of these ligands with their natural receptors. As in our previous experiments, all but one of the tamoxifen-treated Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice that were given an Fc control developed HO within 4 weeks. However, 30 of 33 mice treated with ACVR2A-Fc and ACVR2B-Fc, either in combination or alone, had no detectable HO; the 3 that did show HO displayed a greatly reduced HO severity (table S1 and fig. S10). These results indicated that ACVR1[R206H]-initiated HO is ligand-dependent because ACVR2A-Fc and ACVR2B-Fc interfere with binding of these ligands to their natural receptors.
Activin A is a ligand in vivo of ACVR1[R206H]-driven HO
We sought to identify the ligand responsible for inducing HO through ACVR1[R206H]. Given that HO in FOP patients is often initiated by an inflammatory flare up (21) and that activin A is secreted by a number of different cell types in response to injury and inflammation (22,23), together with our finding that responsiveness to activin A was one of the most notable changes in function of the mutated ACVR1 receptor, we hypothesized that activin A could initiate the HO process. We then tested whether activin A induced HO in an ACVR1[R206H]-dependent manner in tamoxifen-treated Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice. These experiments were carried out with collagen sponge implants adsorbed either with BMP2 (an osteogenic BMP) or with activin A (which has not been reported to cause bone formation in normal animals). Whereas BMP2-containing collagen sponges ossified in all implant-recipient mice, the activin A-containing implants drove ossification only in the tamoxifen- activated Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice (fig. S11), indicating that the mutated ACVR1 receptor confers in vivo HO-initiating responsiveness to activin A.
Given that activin A induced HO in tamoxifentreated Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ but not wild-type mice, we first excluded the possibility that tamoxifen treatment itself could induce expression of activin A (fig. S12) and then tested whether activin A is required for HO. We generated a fully human antibody against activin A using our VelocImmune mouse platform (24, 25). Tamoxifen- treated Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice were divided into three groups (seven to eight mice per group) and treated with either an isotype control antibody or the anti-activin A (and activin AB and AC)-blocking monoclonal antibody (mAb) (fig. S13) or ACVR2A-Fc (as a positive control). Whereas all the mice in the isotype control group developed HO as early as 3 weeks after initiation of treatment (Fig. 5, A, D, E, and G), the group treated with anti-activin F5 A mAb did not develop HO even at 6 weeks after treatment (Fig. 5, C and G). Nearly identical results were obtained with ACVR2A-Fc in which only two ofeight mice treated displayed a very small nodule of HO each (Fig. 5, B, F, and G). Collectively, our results indicate that activin A is an obligate ligand in the HO process observed in this model of FOP and that it likely acts by directly activating Acvr1[R206H].
Fig. 5. ACVR2A-Fc and activin A antibodies prevent HO in Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice.
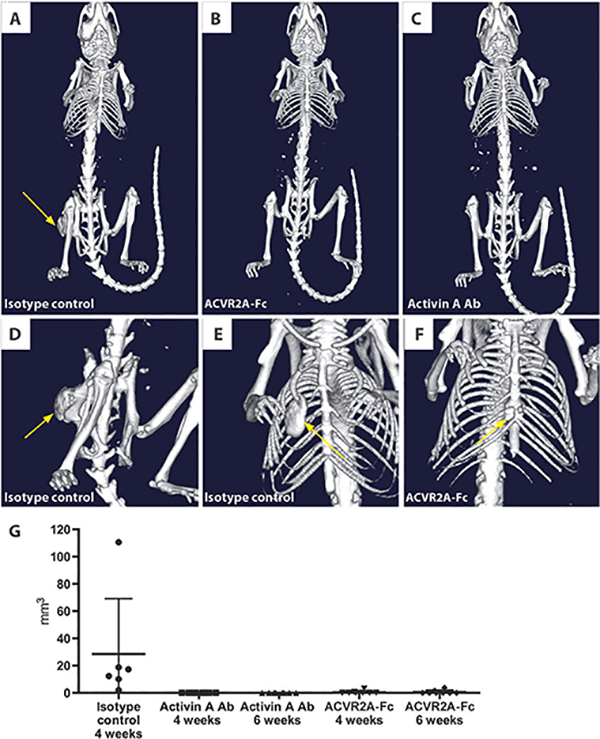
Representative in vivo μCT images of mice at 3 weeks after initiation of the FOP model. (A) HO formation in a mouse treated with isotype control antibody (25 mg/kg, twice weekly). (B) No evidence of HO formation in a mouse treated with ACVR2A-Fc (10 mg/kg, twice weekly). (C) No evidence of HO in mice treated with activin A antibody (Ab) (25 mg/kg). (D and E) Large HO lesions at various locations including the hip joint and rib cage in mice treated with an isotype control antibody. (F) A small HO lesion found in two of the mice treated with ACVR2A-Fc. (G) Quantification of the total volume of HO lesions in each mouse at 4 weeks (isotype control, n = 6; activin A antibody, n = 8; ACVR2B-Fc, n = 8) and 6 weeks (activin A antibody; ACVR2B-Fc) after initiation of the model.
DISCUSSION
A conditional-on knock-in Acvr1[R206H] model of FOP
We describe here a physiologic, versatile, genetically humanized mouse model of FOP, Acvr1[R206H]FlEx. These mice develop HO at anatomical sites that mirror those in FOP patients, as well as those observed in the unregulated knock-in line, Acvr1tm1Emsh (11,26). Being a conditional-on model, Acvr1[R206H]FlEx enables time- and cell type-controlled querying of the function of Acvr1[R206H] and bypasses the early perinatal lethality of F1 generation Acvr1tm1Emsh/+ mice.
Another conditional-on transgenic mouse—Tg(CAG-LacZ,-ACVR1*,-EGFP)35–1Mis (27, 28)—has been used to model FOP (29) by using a constitutively active (and largely ligand-independent) artificial variant, ACVR1[Q207D] (30), to induce Smad1/5/8 signaling after Cre treatment. This mouse likely models only one aspect of FOP, that is, the HO process resulting from Smad1/5/8 signaling. In contrast, Acvr1[R206H]FlEx is a knock-in of the most common FOP allele and involves minimal alterations of the native locus, which are further reduced after conversion to Acvr1[R206H] by Cre. After Cre activation, other than the desired R206H-coding mutation, the only other alterations are two lox sites, one FRT site, and a small deletion of nonconserved intronic sequence. We expect that the mouse model presented here should closely mirror ACVR1[R206H] and its role in FOP.
Note that although Acvr1[R206H]FlEx was intended to function as a wild-type allele before activation of the R206H-coding variant by Cre, Acvr1[R206H]FlEx is hypomorphic. Acvr1[R206H]FlEx/+ mice cannot be bred to homozygosity (table S2), and furthermore, ES cells that are Acvr1[R206H]FlEx/nul1 show a disproportional reduction in correctly spliced Acvr1 message (fig. S3). About half of the Acvr1 mRNA expressed from the Acvr1[R206H]FlEx allele lacks exon 5 and instead generates an aberrant mRNA in which exon 4 splices into exon 6, thereby generating a null, frameshift variant. However, reduction of Acvr1 expression from Acvr1[R206H]FlEx does not appear to have any obvious physiologic consequences for Acvr1[R206H]FlEx/+ mice because they are healthy and do not display any features that differentiate them from their Acvr1+/+ littermates.
ACVR1[R206H] is a gain-of-function variant that responds to normally inhibitory ligands
ACVR1[R206H] unexpectedly displayed a heretofore unrecognized gain-of-function property: the ability to respond to activin A and other ligands that normally inhibit signaling through wild-type ACVR1 while also retaining its ability to respond to canonical ligands (Fig. 6). The F6 responsiveness of ACVR1[R206H] to normally inhibitory ligands was demonstrated in vivo by showing that activin A-containing collagen implants ossify only in Acvr1[R206H]/+ mice. The implication of this finding is that the mutation-containing intracellular domain of ACVR1, particularly the GS subdomain, regulates the responsiveness of this receptor with respect to both the magnitude of the response and the identity of the activating ligands.
Fig. 6. An intracellular domain amino acid change (R206H) transforms ACVR1 into an activin-responsive receptor.
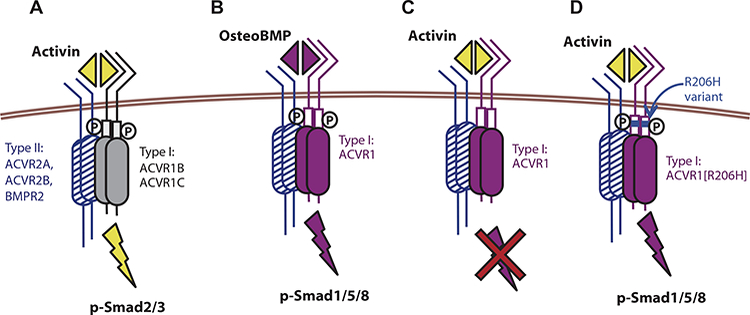
(A) Activin signals via the type I receptors ACVR1B/1C and Smad2/3 phosphorylation but shares type II receptors (ACVR2A, ACVR2B, and BMPR2) with BMPs. (B) ACVR1, together with the type II receptors, recognizes BMPs and stimulates phosphorylation of Smad1/5/8. (C) ACVR1, together with the type II receptors, binds activin, but the resulting complex does not stimulate phosphorylation of Smad1/5/8; instead, activin acts as an inhibitor of canonical BMP-mediated signaling through ACVR1. (D) The R206H variant of the ACVR1 receptor responds to activin, inducing Smad1/5/8 phosphorylation, just like a BMP, effectively converting the ACVR2-ACVR1-activin complex from a dead end complex into a signaling complex. The R206H variant does not lose its ability to respond to canonical BMPs. The ACVR2-ACVR1-activin complex is shown here to contain a heterodimer of ACVR1-ACVR1[R206H]; however, this is not an obligate arrangement because a homodimer of ACVR1[R206H] is also capable of transducing the signal.
The molecular mechanism whereby ACVR1[R206H] gains this property is currently not known; however, we have excluded reduced affinity to FKBP12 or a newly gained ability to bind normally inhibitory ligands as explanations. The ability of ACVR1 to form complexes with activin A in the presence of ACVR2A and ACVR2B has been reported previously (31), but these results were later considered to be an experimental artifact and were not examined further (32). Here, we attach functional significance to these findings as we show that activin A can act as a competitive inhibitor of canonical signaling from wild-type ACVR1 by locking ACVR1 into a nonactive complex with ACVR2, a property that is independent of the activin A-mediated activation of Smad2/3 phosphorylation from its canonical receptors. It is currently unclear whether loss of the inhibitory effects of activin A on this receptor system has physiologic significance. Generally, the finding that activin can act as an inhibitor of canonical BMP signaling via ACVR1 calls for a reassessment of existing data obtained using activins. We note that the reciprocal situation, where BMPs compete with activins for shared type II receptors, has already been reported (33).
Activin A is a potential therapeutic target in FOP
We further explored the physiologic significance of the ability of ACVR1[R206H] to respond to activin A in our mouse model of FOP. Inhibition of activin A with a blocking antibody completely inhibited development of HO. Although our results do not exclude the possibility that other ligands may participate, they indicate that activin A (and perhaps the activin A-containing heterodimers, activin AB and AC) must play a major, indeed obligate, role. Moreover, our data were consistent with the idea that activin A, normally produced by cells of the immune system during inflammation (22, 23), is co-opted and reinterpreted by ACVR1[R206H]-expressing cells with osteogenic potential. Hence, activin A may provide the missing link between inflammation and HO in FOP.
We would like to caution, however, that there is a paucity of data implicating activin A as the driver of HO in FOP patients per se; this is largely due to the inability to safely biopsy patients in between, or during, attacks. We cannot therefore formally exclude the possibility that other ligands play a role in the development of HO in FOP patients. Nonetheless, given the high degree of evolutionary conservation in this signaling system as well as in the process of bone formation overall, together with the fidelity of our genetically humanized mouse model in mimicking the human disease, we are cautiously optimistic about exploring activin A blockade in FOP patients.
Several additional aspects of HO in FOP should be taken into consideration for clinical development of potential therapies. A significant component of HO in FOP is its episodic nature, with long periods of quiescence of HO activity and marked patient-to-patient variability in the sites and degree of HO. These facts may complicate development of therapeutics, as the periods of activity and quiescence are currently not predictable in part because of a paucity of suitable biomarkers. Therefore, continuous treatment has been contemplated. Given the chronic and mostly early-onset (pediatric) nature of FOP, continuous and chronic treatment would require the drug to have a very good safety profile and a minimal number of adverse events. Broad-acting agents —such as ACVR2A-Fc or ACVR2B-Fc, which block multiple BMP family members—could be considered, but their broad actions may lead to nonspecific toxicities; indeed, published studies point to a number of untoward effects (34, 35). On the other hand, our identification of an apparent single causative ligand, activin A, provides opportunity for targeted blockade that may limit side effects and prove therapeutically useful. Furthermore, acute treatment of FOP with a targeted therapeutic could provide important benefit; for example, patients undergoing elective surgeries, which would normally be avoided due to the risk of inducing further HO, could be protected during the perioperative period with the activin A blocking antibody. Last, in addition to FOP, ACVR1 [R206H] has been discovered as a second hit somatic mutation in diffuse intrinsic pontine glioma, suggesting that exploration of activin A or other normally inhibitory ligands in this rare malignancy could be informative.
MATERIALS AND METHODS
Study design
The goals of this study were to elucidate the mechanism by which the R206H mutation in ACVR1 causes FOP. To determine whether ACVR1 [R206H] signaling is ligand dependent, we modeled this variant in an overexpressing HEK293 system, and we confirmed our results in an ES cell system where the mutant allele was expressed from its endogenous locus. We then tested whether our in vitro findings were physiologically relevant in a mouse model of FOP. All in vitro experiments were performed at least three times and with triplicate samples.
To study the efficacy of decoy receptor-Fc fusion proteins or antibodies in preventing HO, Acvrl[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice were injected intraperitoneally with tamoxifen to induce inversion of the mutant allele and concurrently treated subcutaneously with the potential therapeutic or appropriate control. Mice were grouped to ensure age and gender matching between treatments. The experimenter was not blinded to group identity, but analysis of HO formation was independently assessed, blinded to mouse and group identity. The experimental endpoint for in vivo treatment studies was determined to be 4 weeks, based on the fact that most of the mice in a control treated group had developed HO by this time.
Engineering Acvr1[R206H]FlEx
A conditional-on knock-in allele, Acvr1[R206H]FlEx, of ACVR1 [R206H] was engineered using FlEx (14). The R206H-causing mutation was introduced into mouse Acvr1 exon ENSMUSE00001134574—exon 5 (e5) in isoform 001. The resulting mutant e5 plus flanking intronic sequence (Chr2:58472046–58476368; Ensembl release 71) was placed in the antisense strand of Acvr1; simultaneously, the corresponding region from human ACVR1 (Chr2:158628593–158632803; Ensembl release 71) was introduced into the sense strand, upstream of the inverted mouse exon (Fig. 1B). Because the amino acid sequence encoded by mouse e5 differs from that of human by one amino acid—D182 in human, E182 in mouse—D182 was recoded to E182 in the introduced human e5.
To enable Cre-dependent replacement of the introduced human sequence with its R206H counterpart, the human sequence was placed inside a FlEx array (composed of lox2372 and loxP sites in a parallel orientation), and an additional FlEx array (3′ FlEx array) was placed downstream of the R206H-bearing exon in antiparallel orientation with respect to the 5′ FlEx array. To ensure efficient recombination by Cre, the lox sites of the 3′ FlEx array were separated from each other by a 182-base pair (bp) fragment from rabbit hemoglobin β (HBB1; Chr1:146,237,271–146,237,469), which has been shown to be transcriptionally inert (36). This arrangement of FlEx arrays enables Cre-mediated inversion of the R206H-coding exon into the sense strand and simultaneous deletion of the introduced human sequence (Fig. 1).
Knock-in Acvr1[R206H]FlEx/+ ES cells were generated with VelociGene technology (37). The targeting vector was built with bacterial homologous recombination (38) to modify the bacterial artificial chromosome bMQ_214f04. To facilitate selection of recombinants in Escherichia coli and in ES cells, an FRT-flanked neomycin phoshotransferase antibiotic/ drug selection cassette (DSC) was placed within introns 5 to 6, in the sense strand, simultaneously replacing a small nonconserved region encompassed by coordinates Chr2:58473769–58473884 to accommodate probes for genotyping by loss of allele assay (LOA). The sequence of the targeting vector is available upon request. Targeting was performed in VGB6 ES cells to generate Acvr1[R206H]FlEx(neo)/+ (VG1648) ES cells. One targeted clone, VG1648D-E2, was treated with FLP(o) (39) to remove the DSC, to generate Acvr1[R206H]FlEx/+ (VG1649) ES cells. In a parallel, to generate ES cells for in vitro experiments, Acvr1[R206H]FlEx(neo)/+ was also targeted into ES line 2192L-F4, which is Gt(ROSA26)SorCreERT2-hyg/+, as described (36). The resulting Acvr1[R206H]FlEx(neo)/+; Gt(ROSA26) SorCreERT2-hyg/+ line was treated with FLP(o) (39) to remove the DSCs and generate Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ ES cells. These ES cells were also rendered Acvr1[R206H]FlEx/null by targeting the remaining wild-type Acvr1 locus with the same targeting vector as that used to generate KOMP allele VG13048 (40).
Genotyping of targeted ES cells and mice by LOA
Targeting was determined by LOA (37), using T aqMan probe 1649TUP (5′-TCTGGATAGTAAGGTCAGTTGCTGCG-3′) and primers 1649TUF (5′-GGCTGACTGATCTGAAGGAAATGG-3′) and 1649TUR (5′-AGAGGAAGGAGACGCTAAGAATC-3′), as well as probe 1649TDP (5′-AAGGTCAGTTGCTGCGTCTTCCC-3′) and primers 1649TDF (5′-TGAAGGAAATGGGCTTCTGGATAG-3′) and 1649TDR (5′-CATACTCACTCTTCCTGTTAGAGGA-3′). The same probes were used to determine the genotype of the resulting mice and their progeny.
Inducing FOP in Acvr1[R206H]FlEx mice
Acvr1[R206H]FlEx/+ mice were generated from clone VG1649C-A2 using VelociMouse (41). To enable time-controlled yet whole-body inversion of the Acvr1[R206H]FlEx allele, Acvr1[R206H]FlEx/+ mice were mated with Gt(ROSA26)SorCreERT2/+ mice to generate Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+. These were maintained in heterozygosity on a mixed C57BL/6NTac-129S6/SvEvTac background. All experiments were performed in accordance with the Institutional Animal Care and Use Committee of Regeneron. Both male and female mice were used between 9 and 15 weeks of age, although mice were age- and sex-matched between groups. No age- or sex-related phenotypes were noted. The model was initiated by inversion of the R206H-encoding exon into the sense strand by treating Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice with tamoxifen (40 mg/kg, intraperitoneally) (Sigma) in oil daily for 8 days (to activate CreERT2). To assess heterotopic bone formation, mice were anesthetized by isoflurane and whole body-scanned, with a field of view at 60 mm × 120 mm, using in vivo μCT (Quantum FX, PerkinElmer). The x-ray source was set to a current of 160 μA, voltage of 90 kVp, with a voxel size at 120 or 240 μm. The CT imaging was visualized and quantified using Analyze software (AnalyzeDirect). Whole-body bone tissue was autosegmented by threshold, the heterotopic bone was manually segmented from the original bone, and then bone volume and density were calculated by Analyze.
Quantification of inversion of the Acvr1[R206H]FlEX allele in vivo
The degree of inversion of the R206H exon into the sense strand and conversion of Acvr1[R206H]FIEx to Acvr1[R206H] were determined with digital polymerase chain reaction (PCR) and using the following probes and primers: Acvr1_R206H_Pre-Inversion: probe, CTGAGCG- GATTGCTGCCCTTCATGTGAG; primers, 5′-GACCGGTATAACT- TCGTATAATGTATGC-3′ (forward) and 5′-TTTAAGACA- AGGGGGAAAAGAAACG-3′ (reverse); Acvr1_R206H_Post-Inversion: probe, 5′-TACCTGGAAATCCTGTGGGTTCCCAC-3′; primers, 5′- CCAGAGTTGACCGGTATAACTTCG-3′ (forward) and 5′-CAA- CAGTGCCCGCTTCAGC-3′ (reverse); Acvr1_WT: probe, 5′-CCA- CAGCATGTATTGCAGGA-3′; primers, 5′-GCTGGAAA- TCCTGTGGGTTC-3′ (forward) and 5′-AGCAACAGTGC- CCGCTTC-3′ (reverse). DNA concentration was normalized to 8 ng/μl in nuclease-free water. Assay master mixes were made using Biosearch assays (11.25 nmol primer/2.5 nmol probe mix in 625 μl of TE), QuantStudio 3D Digital PCR Master Mix (Life Technologies), and water. Master mix (10.4 μl) was added to 7 μl of normalized sample. Sample (14.5 μl) was loaded onto QuantStudio 3D digital PCR 20K Chips using the QuantStudio 3D digital chip loader (Life Technologies). Cycling was performed on the ProFlex 2×Flat PCR System (Life Technologies) under the following conditions: 96°C for 10 min, 39 cycles of 60°C (2 min) and 98°C (30 s), followed by holding at 60°C for 2 min, and then at 10°C until analysis. Chips were read on the QuantStudio 3D Digital PCR Instrument (Life Technologies). Inversion rates—about 80% in whole blood and about 50% in sampled muscle tissue—were comparable across treatment groups.
Statistical analysis of mouse crosses
χ2 and P values were calculated with an online calculator (www.graphpad.com/quickcalcs/chisquared1.cfm).
Recombinant proteins and treatment of mice
ACVR2A-Fc and ACVR2B-Fc were generated in Chinese hamster ovary cells and purified. ACVR2A-Fc consists of the extracellular domain of ACVR2A (Swiss-Prot P27037 S25-P134) linked to the human immunoglobulin G1 (IgG1) Fc domain (D104-K330). ACVR2B-Fc consists of the extracellular domain of ACVR2B (Swiss-Prot Q13705 E23-P133; R64) linked to the human IgG1 Fc domain (D104-K330). A neutralizing antibody generated at Regeneron against human activin A was produced using VelocImmune (fig. S13) (24, 25). For treatment studies, mice were separated to ensure that age and sex matching across groups and treatments were initiated on the same day as tamoxifen administration. Mice were injected subcutaneously with either ACVR2A-Fc, ACVR2B-Fc, or Fc (10 mg/kg) or with an activin A blocking antibody or an isotype control antibody (25 mg/kg) twice a week for 4 weeks. Heterotopic bone formation was monitored weekly by in vivo μCT imaging.
In vivo implantation of activin A- or BMP2-containing collagen sponges
Collagen sponges (4 mm diameter) (SpongeCol, Advanced BioMatrix) were adsorbed with 10-μl suspension of either 20 μg of recombinant human activin A (rhActivin A) or 5 μg of recombinant human BMP2 (rhBMP2) (R&D Systems) or saline for 30 min at room temperature before implantation. Mice were anesthetized with xylazine and ketamine. Their right gluteus region was shaved and wiped with alcohol and betadine. Muscle tissue was exposed by making a small incision on the skin and fascia. Muscle fibers were separated using blunt forceps to create a pouch. Collagen sponges were implanted into the muscle pouch, and separated muscle fibers were sutured with 5.0 absorbable suture (Visorb). Skin incisions were closed with instant tissue adhesive (Surgi-Lock 2oc, Meridian Animal Health). Mice were given two doses ofthe analgesic Buprenex at a dose of0.1 mg/kg. Two days after implantation, Acvr1[R206H]FlEx/+; Gt(ROSA26)Sor CreERT2/+ mice were injected with tamoxifen as described above to initiate the model. Heterotopic bone formation was assessed at 2 weeks after implantation by in vivo mCT imaging.
Histological analyses
HO lesions and surrounding tissues were dissected, fixed in formalin, decalcified with ImmunoCal (Decal), embedded in paraffin, and sectioned at 7 μm. Sections were stained with hematoxylin (Richard-Allan Scientific Signature Series Hematoxylin 2 7231, Thermo Scientific) and eosin Y (Thermo Scientific) and imaged on a whole-slide imager (SCN400, Leica) at ×20 magnification.
Generation of ACVR1 and ACVR1[R206H]-expressing HEK293/BRE-Luc cells
HEK293 cells were transfected with pFA (Stratagene) harboring 10 tandem repeats of a GC-rich BMP responsive element (BRE; 5′- GCCGCCGCAGC-3′) and a 168-bp minimal promoter from mouse osteocalcin (Bglap) driving firefly luciferase (Luc). The corresponding stable cell line was generated by transfection of pFA.BRE-Luc using Lipofectamine 2000 (Invitrogen) followed by selection with G418 at 500 μg/ml. Unless otherwise noted, cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% (v/v) fetal bovine serum (FBS), penicillin/streptomycin (50 U/ml), and 2 mM L-glutamine. Complementary DNA for human ACVR1 was cloned into pCMV (pCMV.ACVRl). The ACVR1[R206H] variant was introduced into pCMV.ACVRl and confirmed by sequencing. To generate ACVR1 wild-type and R206H overexpressing HEK293/BRE-Luc stable cell lines, HEK293/BRE-Luc cells were transfected with pCMV.ACVR1 or pCMV.ACVR1[R206H] using TransIT-LT1 transfection reagent (Mirus). Stable lines were generated by selection with hygromycin B (100 μg/ml). Pools of stably transfected cells expressing nearly identical levels of ACVR1 and ACVR1[R206H] were generated by fluorescence activated cell sorting (FACS) after surface staining with an anti-ACVR1 antibody (see below). The resulting pools were used for signaling assays.
FACS to select ACVR1-expressing cells
For live cell staining, 15 × 106 cells were treated with TrypLE Express Enzyme (Life Technologies). Harvested cells were washed with
phosphate-buffered saline (PBS) containing 2% FBS and stained with mouse monoclonal anti-ACVR1 antibody MAB637 (5 μg/ml) (R&D Systems) for 1 hour at 4°C. Cells were then washed once with PBS containing 2% FBS, and goat anti-mouse IgG Alexa Fluor 647 (Invitrogen) was used for detection. After two washes in PBS containing 2% FBS, cells were filtered with 70-μm cell strainer. DAPI (4′,6-diamidino- 2-phenylindole)-positive, live cells were sorted for surface expression of ACVR1. Pools were collected with MoFlo XDP (Beckman Coulter). Expression of ACVR1 was confirmed after FACS-based cell sorting by an additional round of surface staining (fig. S1).
Signaling assays in HEK293/BRE-Luc reporter lines
HEK293/BRE-Luc reporter cell lines were plated in 96-well plates (Thermo Scientific) at 10,000 cells/0.33 cm2, incubated at 37°C for 3 hours, and then treated with recombinant proteins as described. For competition assays, rhActivin A and rhBMP6 (R&D Systems) were premixed at indicated concentrations before addition to cells. For experiments that included anti-activin A antibody, reporter cells were plated as above and anti-activin A antibody was preincubated for 30 min with recombinant activins before addition to cells. For all reporter assays, luciferase expression was measured 16 hours after treatments with Bright-Glo Luciferase Assay System (Promega).
Inversion of the R206H-containing exon in mouse ES cells
Mouse ES cells were cultured on irradiated MEF feeders and passaged twice on gelatin before being used for experiments. Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ and Acvr1iR206H]FlEx/null; Gt(ROSA26)SorCreERT2/+ mouse ES cells were treated with 100 nM (Z)-4-hydroxytamoxifen (Sigma-Aldrich) for 24 hours to induce inversion of the R206H-containing exon. To assess inversion, genomic DNA was isolated from the wild-type ES cells as a control and both Acvr1[R206H]FlEx/+ and Acvr1[R206H]FlEx/null cells. PCR was performed using primers 5′-ACACAGAACAGA- GACCCCCA-3′ and 5′-GCCTTTCTAATGTGCGCGTT-3′ designed to yield a PCR product of 1277 bp when the Acvr1 allele is uninverted and a PCR product of 675 bp when inverted (fig. S14).
Reverse transcription polymerase chain reaction analysis of Acvr1[R206H]FlEx/+
RNA was isolated using QIAzol Lysis Buffer (Qiagen), and reverse transcription polymerase chain reaction was performed using SuperScript III One-Step (Invitrogen) to amplify exons 4 to 6 of Acvr1 isoform 1 (ENSMUST00000112599), using 5′-TGGAGTTGCTCTCAG- GAAGT-3′ and 5′-GAGAAGATCTTCACAGCGACA-3′ as primers.
Cell treatments and immunoblotting
Mouse ES cells were serum-starved for 2 hours before a 1-hour treatment with either rhBMP6 or rhActivin A (R&D Systems) at indicated concentrations. For competition assays, in ES cells, rhActivin A and rhBMP6 (R&D Systems) were premixed at indicated concentrations before addition. Cells were harvested in an isotonic buffer containing 1% NP-40 substitute (Fluka) and a 2× concentration of protease and phosphatase inhibitor cocktail (Thermo Scientific). HEK293/BRE-Luc cell lines expressing wild-type ACVR1 or ACVR1[R206H] were plated on six-well dishes (Corning) at 1 × 105 cells per well and grown overnight in complete medium. rhBMP6 or rhActivin A was added at indicated concentrations for 20 min. Cells were lysed in hypotonic buffer containing 1% NP-40 substitute and 2× protease and phosphatase inhibitor cocktail.
Whole-cell lysates were prepared in SDS-containing buffer, and proteins were resolved on 10% or 4 to 20% gradient SDS-polyacrylamide gel electrophoresis gels (Invitrogen) and transferred to polyvinylidene difluoride membranes (Millipore). Immunoblotting was performed using standard procedures. Primary antibodies toward Smad5 (Cell Signaling, 9517), phospho-Smad1/5 (Cell Signaling, 9516S), and β- tubulin (Sigma T5201) were used at a dilution of 1:1000. Binding was visualized with horseradish peroxidase-conjugated secondary antibodies (Promega) and ECL substrate (EMD Millipore). Blots were imaged with ImageQuant LAS 4000 (GE Healthcare) instrument or x-ray film (Kodak).
Supplementary Material
www.sciencetranslationalmedicine.org/cgi/content/full/7/303/303ra137/DC1
Materials and Methods
Fig. S1. Expression of ACVR1 in HEK293/BRE-Luc cells is detected by surface staining.
Fig. S2. BMP family ligands parse into two classes with respect to the response they elicit in ACVR1-overexpressing compared to ACVR1[R206H]-overexpressing lines.
Fig. S3. Acvr1 exon 5 can be skipped during splicing of the Acvr1[R206H]FlEx pre-mRNA.
Fig. S4. Activin A–induced Smad2/3 signaling in ACVR1[R206H]-expressing cells is similar to wild-type ES cells.
Fig. S5. FKBP12 inhibition by FK506 does not render wild-type ACVR1 into an activin-responsive receptor.
Fig. S6. Activins and BMPs bind to wild-type ACVR1 and form complexes with ACVR2A.
Fig. S7. Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+mice develop HO as early as 2 weeks after tamoxifen administration.
Fig. S8. Quantification of HO formation Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice from 2 to 6 weeks after tamoxifen administration.
Fig. S9. H&E sections from HO lesions shows a variety of different cell types, mirroring those found in FOP lesions.
Fig. S10. ACVR2A-Fc and ACVR2B-Fc prevent ectopic bone formation in Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice.
Fig. S11. Activin A causes HO in tamoxifen-treated Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ but not wild-type mice.
Fig. S12. Tamoxifen does not alter serum activin A levels.
Fig. S13. Anti–activin A blocking mAb blocks the activity of inhibin A–containing activins but not activin B.
Fig. S14. In mouse ES cells, the Acvr1[R206H]FlEx allele is inverted by 24 hours after 100 nM tamoxifen treatment.
Table S1. Number of mice with HO lesions in treatment experiments.
Table S2. Acvr1[R206H]FlEx/+ mice cannot be bred to homozygosity
Acknowledgments:
We thank the VelociGene team for generating the mouse ES cells and all the mouse cohorts used in this study, E. Latres for advice on the use of the anti-activin A blocking antibody, A. Rafique and D. Buckler for characterization of the anti-activin A blocking antibody, and K. Daniels for assistance with FACS.
Footnotes
Author contributions: A.N.E. conceived the project and, together with AJ.M., C.S., SJ.H., and V.I., developed the research plan and designed and supervised the experiments. A.N.E., S.J.H., V.I., P.B.Y., G.D.Y., and A.J.M. co-authored the text. K.F. built the knock-in allele. D.M.A.W., L.H., X.W., L.W., N.D., HJ.K., R.A.C., R.C., and D.D. performed the experiments, analyzed the data, and prepared the corresponding figures and Materials and Methods subsections.
Competing interests: All authors except P.B.Y. are employees of Regeneron Pharmaceuticals Inc. and hold stock in the company. Regeneron has filed patent applications—US2014/0283158, which pertains to the mouse model, and US2015/0037339, which pertains to the use of anti-activin A neutralizing antibodies as therapy for FOP.
Data and materials availability: All materials (including the conditional-on knock-in mouse line Acvr1) [R206H]FlEx)generated for the research described herein can be obtained by the completion of a material transfer agreement.
REFERENCES AND NOTES
- 1.Hüning I, Gillessen-Kaesbach G, Fibrodysplasia ossificans progressiva: Clinical course, genetic mutations and genotype-phenotype correlation. Mol. Syndromol 5, 201–211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS,A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet 38, 525–527 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Groppe JC, Shore EM, Kaplan FS, Functional modeling of the ACVR1 (R206H) mutation in FOP. Clin. Orthop. Relat. Res 462, 87–92 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Groppe JC, Wu J, Shore EM, Kaplan FS, In vitro analyses of the dysregulated R206H ALK2 kinase-FKBP12 interaction associated with heterotopic ossification in FOP. Cells Tissues Organs 194, 291–295 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaikuad A, Alfano I, Kerr G, Sanvitale CE, Boergermann JH, Triffitt JT, von Delft F, Knapp S, Knaus P, Bullock AN, Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem 287, 36990–36998 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bagarova J, Vonner AJ, Armstrong KA, Börgermann J, Lai CS, Deng DY, Beppu H, Alfano I, Filippakopoulos P, Morrell NW, Bullock AN, Knaus P, Mishina Y, Yu PB, Constitutively active ALK2 receptor mutants require type II receptor cooperation. Mol. Cell Biol 33, 2413–2424 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujimoto M, Ohte S, Shin M, Yoneyama K, Osawa K, Miyamoto A, Tsukamoto S, Mizuta T, Kokabu S, Machiya A, Okuda A, Suda N, Katagiri T, Establishment of a novel model of chondrogenesis using murine embryonic stem cells carrying fibrodysplasia ossificans progressiva- associated mutant ALK2. Biochem. Biophys. Res. Commun 455, 347–352 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Fujimoto M, Ohte S, Osawa K, Miyamoto A, Tsukamoto S, Mizuta T, Kokabu S, Suda N, Katagiri T, Mutant activin-like kinase 2 in fibrodysplasia ossificans progressiva are activated via T203 by BMP type II receptors. Mol. Endocrinol 29, 140–152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yano M, Kawao N, Okumoto K, Tamura Y, Okada K, Kaji H, Fibrodysplasia ossificans progressiva-related activated activin-like kinase signaling enhances osteoclast formation during heterotopic ossification in muscle tissues. J. Biol. Chem 289, 16966–16977 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Culbert AL, Chakkalakal SA, Theosmy EG, Brennan TA, Kaplan FS, Shore EM, Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification. Stem Cells 32, 1289–1300 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, Maidment AD, Kaplan FS, Shore EM, An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner Res 27,1746–1756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaplan FS, Chakkalakal SA, Shore EM, Fibrodysplasia ossificans progressiva: Mechanisms and models of skeletal metamorphosis. Dis. Model. Mech 5, 756–762 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olsen OE, Wader KF, Hella H, Mylin AK, Turesson I, Nesthus I, Waage A, Sundan A, Holien T, Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun. Signal 13, 27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnütgen F, Doerflinger N, Calléja C, Wendling O, Chambon P, Ghyselinck NB, A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat. Biotechnol 21, 562–565 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Nagy A, Mar L, Watts G, Creation and use of a cre recombinase transgenic database. Methods Mol. Biol 530, 365–578 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Seibler J, Zevnik B, Küter-Luks B, Andreas S, Kern H, Hennek T, Rode A, Heimann C, Faust N, Kauselmann F, Schoor M, Jaenisch R, Rajewsky K, Kühn R, Schwenk F, Rapid generation of inducible mouse mutants. Nucleic Acids Res. 31, e12 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glaser DL, Economides AN, Wang L, Liu X, Kimble RD, Fandl JP, Wilson JM, Stahl N, Kaplan FS, Shore EM, In vivo somatic cell gene transfer of an engineered Noggin mutein prevents BMP4-induced heterotopic ossification. J. Bone Joint Surg. Am 85-A, 2332–2342 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Souza TA, Chen X, Guo Y, Sava P, Zhang J, Hill JJ, Yaworsky PJ, Qiu Y, Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol. Endocrinol 22, 2689–2702 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sako D, Grinberg AV, Liu J, Davies MV, Castonguay R, Maniatis S, Andreucci AJ, Pobre EG, Tomkinson KN, Monnell TE, Ucran JA, Martinez-Hackert E, Pearsall RS, Underwood KW, Seehra J, Kumar R, Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J. Biol. Chem 285, 21037–21048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.del Re E, Sidis Y, Fabrizio DA, Lin HY, Schneyer A, Reconstitution and analysis of soluble inhibin and activin receptor complexes in a cell-free system. J. Biol. Chem 279, 53126–53135 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Pignolo RJ, Shore EM, Kaplan FS, Fibrodysplasia ossificans progressiva: Clinical and genetic aspects. Orphanet J. Rare Dis 6, 80 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aleman-Muench GR, Soldevila G, When versatility matters: Activins/inhibins as key regulators of immunity. Immunol. Cell Biol 90, 137–148 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Hedger MP, Winnall WR, Phillips DJ, de Kretser DM, The regulation and functions of activin and follistatin in inflammation and immunity. Vitam. Horm 85, 255–297 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Macdonald LE, Karow M, Stevens S, Auerbach W, Poueymirou WT, Yasenchak J, Frendewey D, Valenzuela DM, Giallourakis CC, Alt FW, Yancopoulos GD, Murphy AJ, Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc. Natl. Acad. Sci. U.S.A 111, 5147–5152 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy AJ, Macdonald LE, Stevens S, Karow M, Dore AT, Pobursky K, Huang TT, Poueymirou WT, Esau L, Meola M, Mikulka W, Krueger P, Fairhurst J, Valenzuela DM, Papadopoulos N, Yancopoulos GD, Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc. Natl. Acad. Sci. U.S.A 111, 5153–5158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. www.informatics.jax.org/allele/MGI:5471642.
- 27. www.informatics.jax.org/allele/MGI:3821884.
- 28.Fukuda T, Scott G, Komatsu Y, Araya R, Kawano M, Ray MK, Yamada M, Mishina Y, Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis 44, 159–167 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD, BMP type I receptor inhibition reduces heterotopic ossification. Nat. Med 14, 1363–1369 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wieser R, Wrana JL, Massagué J, GS domain mutations that constitutively activate TßR-I, the downstream signaling component in the TGF-ß receptor complex. EMBOJ. 14,2199–2208 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Attisano L, Carcamo J, Ventura F, Weis FM, Massagué J, Wrana JL, Identification of human activin and TGFß type I receptors that form heteromeric kinase complexes with type II receptors. Cell 75, 671–680 (1993). [DOI] [PubMed] [Google Scholar]
- 32.Macias-Silva M, Hoodless PA, Tang SJ, Buchwald M, Wrana JL, Specific activation ofSmadl signaling pathways bythe BMP7 type I receptor, ALK2. J. Biol. Chem 273,25628–25636 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Lowery JW, Intini G, Gamer L, Lotinun S, Salazar VS, Ote S, Cox K, Baron R, Rosen V, Loss of BMPR2 leads to high bone mass due to increased osteoblast activity. J. Cell Sci 128, 1308–1315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruckle J, Jacobs M, Kramer W, Pearsall AE, Kumar R, Underwood KW, Seehra J, Yang Y, Condon CH, Sherman ML, Single-dose, randomized, double-blind, placebo-controlled studyofACE-011 (ActRIIA-IgG1) in postmenopausal women. J. BoneMiner. Res 24,744–752 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Relizani K, Mouisel E, Giannesini B, Hourdé C, Patel K, Gonzalez SM, Jülich K, Vig- naud A, Piétri-Rouxel F, Fortin D, Garcia L, Blot S, Ritvos O, Bendahan D, Ferry A, Ventura- Clapier R, Schuelke M, Amthor H, Blockade of ActRIIB signaling triggers muscle fatigabilityand metabolic myopathy. Mol. Ther 22, 1423–1433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Economides AN, Frendewey D, Yang P, Dominguez MG, Dore AT, Lobov IB, Persaud T, Rojas J, McClain J, Lengyel P, Droguett G, Chernomorsky R, Stevens S, Auerbach W, Dechiara TM, Pouyemirou W, Cruz JM Jr., Feeley K, Mellis IA, Yasenchack J, Hatsell SJ, Xie L, Latres E, Huang L, Zhang Y, Pefanis E, Skokos D, Deckelbaum RA, Croll SD, Davis S, Valenzuela DM, Gale NW, Murphy AJ, Yancopoulos GD, Conditionals by inversion provide a universal method for the generation of conditional alleles. Proc. Natl. Acad. Sci. U.SA 110, E3179–E3188 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, Auerbach W, Poueymirou WT, Adams NC, Rojas J, Yasenchak J, Chernomorsky R, Boucher M, Elsasser AL, Esau L, Zheng J, Griffiths JA, Wang X, Su H, Xue Y, Dominguez MG, Noguera I, Torres R, Macdonald LE, Stewart AF, DeChiara TM, Yancopoulos GD, High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol 21, 652–659 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Buchholz F, Muyrers JP, Stewart AF, A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet 20, 123–128 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Raymond CS, Soriano P, High-efficiency FLP and FC31 site-specific recombination in mammalian cells. PLOS One 2, e162 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. https://www.komp.org/pdf.php?projectID=VG13048.
- 41.Poueymirou WT, Auerbach W, Frendewey D, Hickey JF, Escaravage JM, Esau L, Doré AT, Stevens S, Adams NC, Dominguez MG, Gale NW, Yancopoulos GD, DeChiara TM, Valenzuela DM, F0 generation mice fully derived from gene-targeted embryonic stem cells allowing immediate phenotypic analyses. Nat. Biotechnol 25, 91–99 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
www.sciencetranslationalmedicine.org/cgi/content/full/7/303/303ra137/DC1
Materials and Methods
Fig. S1. Expression of ACVR1 in HEK293/BRE-Luc cells is detected by surface staining.
Fig. S2. BMP family ligands parse into two classes with respect to the response they elicit in ACVR1-overexpressing compared to ACVR1[R206H]-overexpressing lines.
Fig. S3. Acvr1 exon 5 can be skipped during splicing of the Acvr1[R206H]FlEx pre-mRNA.
Fig. S4. Activin A–induced Smad2/3 signaling in ACVR1[R206H]-expressing cells is similar to wild-type ES cells.
Fig. S5. FKBP12 inhibition by FK506 does not render wild-type ACVR1 into an activin-responsive receptor.
Fig. S6. Activins and BMPs bind to wild-type ACVR1 and form complexes with ACVR2A.
Fig. S7. Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+mice develop HO as early as 2 weeks after tamoxifen administration.
Fig. S8. Quantification of HO formation Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice from 2 to 6 weeks after tamoxifen administration.
Fig. S9. H&E sections from HO lesions shows a variety of different cell types, mirroring those found in FOP lesions.
Fig. S10. ACVR2A-Fc and ACVR2B-Fc prevent ectopic bone formation in Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ mice.
Fig. S11. Activin A causes HO in tamoxifen-treated Acvr1[R206H]FlEx/+; Gt(ROSA26)SorCreERT2/+ but not wild-type mice.
Fig. S12. Tamoxifen does not alter serum activin A levels.
Fig. S13. Anti–activin A blocking mAb blocks the activity of inhibin A–containing activins but not activin B.
Fig. S14. In mouse ES cells, the Acvr1[R206H]FlEx allele is inverted by 24 hours after 100 nM tamoxifen treatment.
Table S1. Number of mice with HO lesions in treatment experiments.
Table S2. Acvr1[R206H]FlEx/+ mice cannot be bred to homozygosity