

Abstract
Background
Immunotherapy is one promising therapeutic strategy against glioma, an aggressive form of brain cancer. Previous studies have demonstrated that multiple tumor antigens exist and can be used to induce tumor specific T cell responses. Furthermore, recently it was shown that TLR4-primed mesenchymal stem cells (MSCs), also known as MSC1, mostly elaborate pro-inflammatory mediators. Compared to MSCs, MSC-derived microvesicles (MVs) have advantageous properties that present them as stable, long lasting effectors with no risk of immune rejection. Therefore, peripheral blood monocyte derived dendritic cells (MoDCs) have been used to load tumor antigens and stimulate T cell mediated responses in the presence of MSC1-derived MVs in vitro.
Methods
The B92 tumor cell line was heated to 43°C for 90 min prior to preparation of tumor cell lysates. MVs were purified by differential ultracentrifugation after isolation, stimulation of proliferation and treatment of MSCs. Autologous T cells isolated from non-adherent cells were harvested during the procedure to generate MoDCs and then incubated with heat stressed tumor cell lysate pulsed DCs in the presence of MSC1-derived MVs. T cells were then co-cultured with tumor cells in 96-well plates at a final volume of 200 μl CM at an effector: target ratio of 100:1 to determine their specific cytotoxic activity.
Results
Flow cytometric analysis, T cell mediated cytotoxicity showed that heat stressed tumor antigen pulsed MoDCs and MSC1-derived MVs primed T cells elicited non-significantly enhanced cytotoxic activity toward B92 tumor cells (P≥0.05).
Conclusion
These findings may offer new insights into tumor antigen presenting technology involving dendritic cells and MSC1-derived MVs. Further exploration of the potential of such nanoscale particles in immunotherapy and in novel cancer vaccine settings appears warranted.
Keywords: Glial cells, tumor cell lysate, dendritic cells, MSC1-derived MVs, cancer immunotherapy
Introduction
Glial cells are present in the brain and spine, as they surround neurons and support them. Any uncontrollable and excessive growth in glial cells can lead to an aggressive form of brain cancer called glioma (Stupp et al., 2009; Stupp and Roila, 2009; Haar et al., 2012). Radiotherapy, chemotherapy and surgery are the currently used treatment option for people with glioma. However, cellular immunotherapy is a novel proven treatment which has raised hopes for therapy of several cancers (Yajima et al., 2005; Platten et al., 2016). In cancer immunotherapy, dendritic cells (DCs) and peptides are used for inducing anti-glioma responses which are capable of harnessing the power and specificity of the immune system to treat tumors (Liau et al., 2005). DCs are the most potent antigen-presenting cells of the body sensitizing T cells toward all acquired antigens and tumor derived peptides. DCs present tumor-derived peptides to native CD8+ T cells and then these T cells can initiate a cytotoxic T lymphocyte (CTL) differentiation programme after countering DCs (Li et al., 2016). To activate the immune system in cancer immunotherapy, DCs are loaded with tumor derived peptides ex vivo, which can subsequently activate the endogenous immune system upon injection (Radford et al., 2014). There are several mice models of cancer reports proving that DCs can capture tumor antigens of tumor cells and cross-present these antigens to T cells in tumor-draining lymph nodes that leads to the generation of tumor specific CTLs and contribute to tumor rejection (Richters et al., 2002; Pellegatta et al., 2006). Thus, DCs represent themselves as an important target for therapeutic interventions in cancer therapy and can be generated in vitro from monocytes by using GM-CSF and IL-4, and are therefore, called monocyte-derived DC (MoDC) (Tuyaerts et al., 2007; Guo et al., 2016).
Heat shock proteins-peptide complexes (HSP-PC) from tumors have proven to be extremely effective in inducing antitumor immunity. This is because lysates from heat-stressed tumor cells prepare an optimal source of tumor antigens to generate DC with mediated cross-presentation and thus can be used in clinical orders for DC cell-based vaccination against tumors (Schnurr et al., 2001; Nakai et al., 2006; Aguilera et al., 2011). Moreover, it has been reported that in large numbers of glial cells, heat stress up to 43°C for 90 min could induce HSP72 expression (Satoh and Kim, 1994).
Tumors are complicated tissues and contain multiple types of cells such as mesenchymal, immune, and vascular endothelial cells. Accordingly, extensive studies have been carried out to explain the interaction between cancer cells and their microenvironment. Multipotent mesenchymal stromal cells (formerly known as MSC) are increasingly used in cell-based therapies (Murphy et al., 2016). They are simply separated from other bone marrow-derived cells by their tendency to adhere to plastic (Nakamizo et al., 2005; Vu et al., 2016). Upon residing in the tumor microenvironment, MSCs targeted cancers are expected to release many bioactive factors, like mitogens, extracellular matrix proteins, angiogenic and inflammatory factors, as well as exosomes or MVs (Waterman et al., 2012; Senst et al., 2013). MSCs appear to affect the immune system by changing the proliferation and differentiation of dendritic cells, monocytes, macrophages, B and T cells, NK cells, including mast cells (Klopp et al., 2010; Klopp et al., 2011; Waterman et al., 2012). Evidence from several studies, have shown that MSCs can recruit and regulate T cells in both cell to cell contact and paracrine signaling (Beyth et al., 2005; Wang et al., 2014). However, there is a growing concern over the use of MSCs because they host tumors and can sustain tumor growth and metastasis. Nonetheless, recent studies defined a new approach for the polarization of MSCs in to a pro-inflammatory MSC1 and an immunosuppressive MSC2 phenotype (Waterman et al., 2010; Klopp et al., 2011). This is explained as the stimulation of TLR4 (Toll Like Receptor 4) in MSCs with lipopolysaccharides (LPS) resulting in MSC1 phenotype which has an anti-tumor effect and decrease tumor growth and metastasis (Waterman et al., 2012).
Experimental studies have detected that the useful effects of MSCs is relatively indirect and rely on the paracrine activity of MSCs and not on their engraftment. Therefore, at the core of the development of cell free strategies are novel therapeutic visions based on the application of MSCs-derived MVs as a safe and more beneficial alternative to cell therapy approaches (Collino et al., 2010; Bruno et al., 2012). These cell-derived MVs include survival signals such as growth factors and cytokines, therefore, providing promising results that indicate MSC-derived MVs have the same protective and supportive features as their original cells in tissue repair and anticancer therapy (Parekkadan et al., 2007). Unlike MSCs, MVs have some properties that that present them as stable, long lasting effectors with no risk of immune rejection (Lai et al., 2010), generate severe signals (Farsad, 2002), and their functions do not exhaust over time (Uccelli et al., 2008). However, the consequence of MSCs-derived MVs have not yet been studied in T cells mediated responses in the field of cancer therapy.
Therefore, in this study, we utilized MSC1-derived MVs as a pro-inflammatory cell free vesicle to form a viable basis for the development of novel cancer vaccines, via antigen-presenting cell technology. Furthermore, the study addresses the potential application of MSC1-derived MVs as a novel nanoscale immunotherapy treatment for cancer, by stimulating effective antitumor T-cell immunity.
Materials and Methods
Primary cell culture
B92 rat glial cell line was purchased from National Cell Bank of Iran (NCBI, Tehran, Iran) and was sub-cultured in RPMI 1640 supplemented with 10% FBS at an atmosphere of 5% CO2 and 37 °C to reach 80% confluency.
Heat stress
We used heat stressed tumor lysates as a source of tumor-associated antigens for pulsing DCs. Heat treatment was applied according to the method described before (Satoh and Kim, 1994). In brief, culture flasks were closed and immersed at 43 °C water bath for 90 min, subsequently incubated for 8 h at 37°C and 5% CO2. The viability of the heat stressed cells was more than 95%.
Preparation of tumor lysates
To obtain tumor lysates 3 × 106 B92 cells were trypsinized and washed in medium twice and then followed by four freezes (liquid nitrogen) and thaw (37 °C water bath) cycles. Large particles were removed by centrifugation (2,000 g for 10 min, followed by 13,000 g for 60 min at 4 °C) then lysate was filtered through a 0.22 μm mesh. The protein content was determined by the Coomassie blue protein assay (Biorad, London, UK) and aliquots were stored at – 80 °C. These steps were done according to previous methods (Aguilera et al., 2011).
Isolation, proliferation and identification of MSCs
MSCs were isolated as explained previously with some modifications (Mokarizadeh et al., 2012). Briefly, bone marrow was flush out of tibias and femurs from deeply anesthetized Wistar rats. The cells were suspended and centrifuge at 2,00 g, 4°C for 5 min then plated in flasks using low-glucose Dulbecco’s Modified Eagle’s Medium containing 15% FBS and incubated in humidified 5% CO2 at 37 °C. Three days after primary culture, the culture medium (CM) was collected and centrifuged, then the pellets were replated in a fresh flask. The cultures were changed with fresh ones twice a week and after reaching 70% confluence, the cells were removed using Trypsin-EDTA, counted, and passed at T75 flask. Cells of third passage were used to TLR priming. The immunophenotype of 3rd passage of MSCs were analyzed by flow cytometry using DAKO flow cytometry (Partec, Germany) as described before. The fallowing monoclonal Antibodies were used: FITC- labeled anti-CD45, PE-labeled anti-CD29 and PCY5-labeled anti-CD90 (Becton Dickinson). Isotype control were used in all cases. Moreover, the capabilities of the 3 passages of MSCs to differentiate in to osteocytes and adipocytes were examined as previously described (Abbasi et al., 2018).
TLR priming
The 3rd passage of MSCs was cultured and upon 60–70% confluence, LPS (10 ng/ml) (Sigma-Aldrich) was added to fresh CM as TLR4 agonist and incubated for 1 h as describe before (Waterman et al., 2010). The cells were then washed twice in CM without the TLR-agonists and used in the next steps of experiments.
Isolation of MVs
MVs were isolated by various centrifuge and ultracentrifuge as explained previously (Thery et al., 2006). Briefly, the collected supernatant of MSCs CM was centrifuged at 300 g for 10 min, 1,000 g for 20 min and 10,000 g for 30 min respectively. The final supernatant was ultra-centrifuged at 100,000 g for 2 h in the ultracentrifuge (Beckman coulter optimaTMXL-100K ultracentrifuge). The MV pellet was washed in phosphate buffer saline (PBS) and again centrifuged at 100,000 g for 2 h. The resultant pellet was re-suspended in PBS and stored at – 80 °C.
Generation of DCs
Dendritic cells were generated as previously described with minor modifications (Agger et al., 2000). Rats were anaesthetized and 15 ml heparinized blood was obtained by heart puncture. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation on ficoll/hypaque (Lymphoprep, Norway). PBMC were then plated into T25 flask and incubated for 2 h at 37 °C to remove the non-adherent cells. The adherent cells were cultured for 7 days in medium containing rat recombinant GM-CSF (Peprotech, USA) and IL-4 (Peprotech, USA) at a concentration of 50 ng/ml. Cytokine containing medium was renewed every 2 days. At day 4 of experiment heat stressed tumor lysate was added to MoDC at final protein concentration of 50 μg/mL and incubated overnight. To induce maturation of the MoDC, 20 μg/ml LPS (Sigma-Aldrich) was added as a maturation factor on day 5. Mature DCs were harvested on day 7, and viability of cells was determined 95% as using trypan blue exclusion assay.
Cell priming
The rats were anaesthetized again and the same amount of heparinized blood was obtained by heart puncture. PBMCs were plated into T25 flask and incubated for 2 h and non-adherent cells were harvested to isolate the autologous T cells. 1 × 105 tumor lysate pulse MoDCs were cultured with 1 × 106 autologous T cells (ratio = 1:10) in CM supplemented with 10% FBS in the presence of 20 µg/ml MSC1-derived MVs in 24 well microtiter plate for 7 days at 37 °C and 5% CO2. The culture was fed every 3 days with fresh medium and recombinant rat IL-2 (20 ng/ml) (Peprotech, USA). On day 7 tumor antigen primed T cells were harvested and applied to T cell cytotoxicity assay, respectively.
T cell cytotoxicity assay
Tumor antigen primed T cells were co-cultured with B92 tumor cells in 96-well V-bottom plates at an effector: target ratio of 100:1 (T cells: B92) in a final volume of 200 μl at 37°C and incubated for 3 h. The incubation was done in shaking incubator to prevent the adherent of B92 tumor cells. After co-incubation, cells were harvested and washed with PBS and re-suspended in 200 µl ABB (Annexin Binding Buffer). Then were stained in 5 μg/ml annexin-FITC for 20 min at room temperature in the dark and immediately before flow cytometric analysis, 5 μg/ml propidium iodide (PI) was added. Three color FACS analysis was carried out on a Coulter cytometer (Partec Germany). Unstained and annexin-FITC/PI stained B92 cells were used as respective controls. Duplicate wells were averaged and the percentage of specific lysis was calculated as fallow:
Specific Lysis= (sample-control)/ (total-control) ×100
In this formula control and total indicate target cells.
Ethics Statement
This study was conducted with permission from the Animal Ethics Committee of Ardabil University of Medical Sciences (Ethical code: IR.ARUMS.REC.1394.133). The animal procedures followed in this study were in accordance with the standards set forth in the Guide for the Care and Use of Laboratory Animals.
Statistical analysis
Means of at least 3 independent experiments were reported. The data were analyzed by SPSS software One Way ANOVA and LSD test. The level of significance was set at p<0.05 and results were presented as mean ±SD.
Results
Morphological appearance of monocyte derived mature DCs
PBMCs were incubated for 2 h at 37 °C and adherent population were cultured in the presence of GM-CSF and IL-4 for three days. On day 4, immature DCs were pulsed with tumor antigen to obtain two test groups: non-heated stressed tumor lysate group (unheated ones) and heat stressed tumor lysate group. On day 5, LPS as a maturation factor was added and by day 7, 60% of cells appeared to loosely adhere to each other in clumps or were isolated floating cells with typical dendritic morphology. On day 7, mature DCs were harvested and reviewed using invert microscopy. At this stage, there were no significant differences between the unheated stressed tumor lysate and heat stressed tumor lysate or both DCs of the test groups that exhibited similar typical cytological features, i.e., large irregular cells with numerous cell membrane processes under light microscopy (Figure 1). In our previous studies, immunophenotyping of monocyte-derived DCs and primed T cells was performed (Delirezh et al., 2009; GanjiBakhsh et al., 2011).
Figure 1.
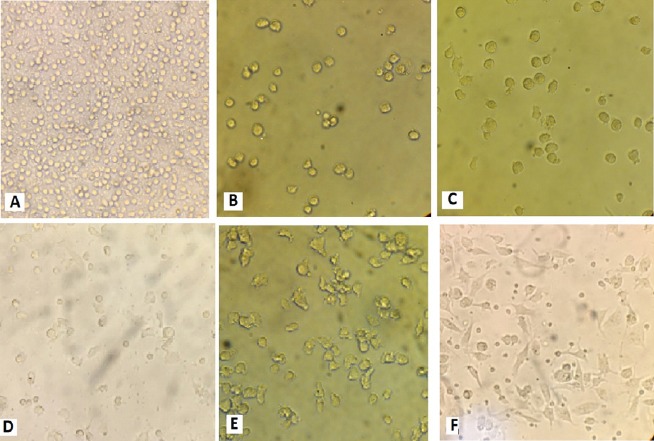
Monocyte- Derived Tumor Lysate Pulsed Dendrite Cells in 7 Days. Adherent population in the first day(A), immature DCs before pulsing with tumor lysate on day 4(B), immature DCs pulsed with tumor lysate on day 5(C), immature DCs pulsed with heat stressed tumor lysate on day 5(D), mature DCs pulsed with tumor lysate & LPS on day 7(E), mature DCs pulsed with heat stressed tumor lysate & LPS on day 7(F), all samples were reviewed by invert microscopy (400×).
Characterization and morphological appearance of MSCs
This study was carried out simultaneously with another study and therefore, the cells which were characterized and described were used (Abbasi et al., 2018). MSCs were distinguished by their fibroblast like appearance, colony forming tendency, and their tendency to adhere plastic (Figure 2).
Figure 2.
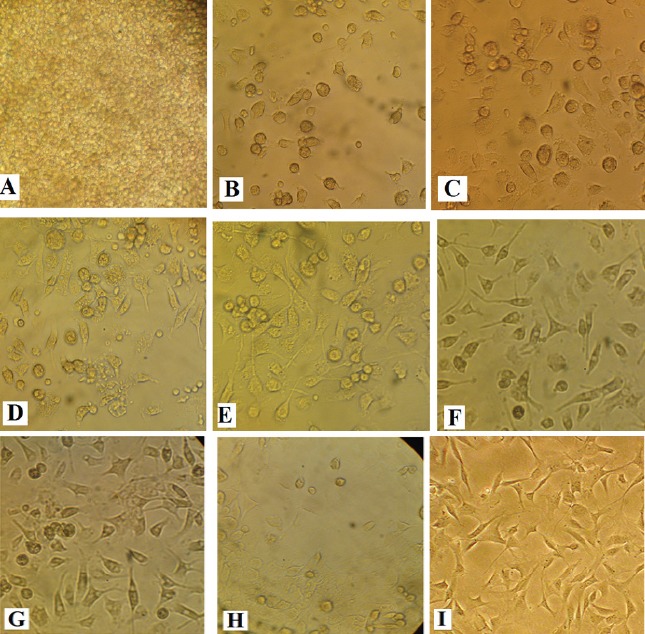
Culture of MSCs on Days 1-14 and 3rd Passages. Pooled bone marrow-derived cells (A), bone marrow-derived adherent cells on day 1(B), adherent cells on day 2(C), cells were becoming spindle-like on day 4 (D), a representative colony formed by MSCs on day 6 (E), high confluency at clonal density on day 8 (F), on day 10 (E), on day 12 (H) and on day 14 (I) after 3rd passages
Electron microscopy analysis of microvesicles
Analyses of MVs by electron microscopy showed the presence of nano-sized vesicles. Size of majorities were between 50 to 200 nm in several micrographs (Figure 3).
Figure 3.
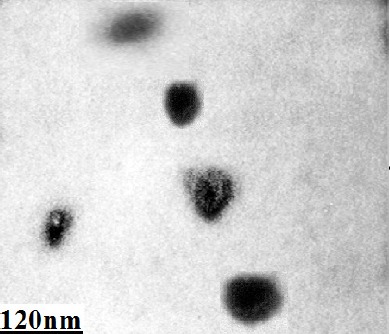
Structural Analysis of MVs Derived from Bone Marrow-Derived Mesenchymal Stem Cells. MVs were isolated from CM of MSCs by differential centrifugation and observed by a transmission electron microscope (Philips Bio Twin, CM100, Netherlands) at 80 kV. MVs were seen as nano-sized vesicles in electron micrograph.
Cell Cytotoxic Activity
We stimulated autologous T cells with antigen pulsed DCs to examine the capacity of heat stressed tumor lysate pulsed DC and MSC1-derived MVs to elicit cytotoxic T cell response in vitro. Subsequently the cytotoxic activity of primed T cells was analyzed using flow cytometry.
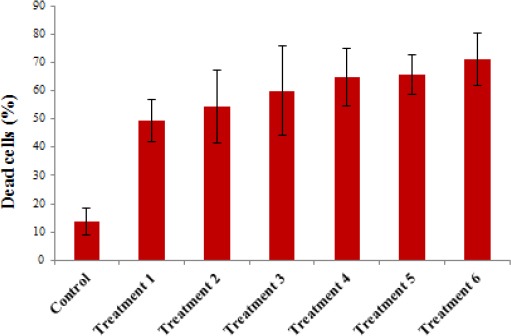
Cytotoxicity assays were conducted with primed T cells at 7 days after stimulation of T cell with tumor lysate pulsed DCs in the presence of polarized MVs or MSC1-derived MVs and non-polarized MSC-derived MVs. cytotoxicity against tumor cells is shown in figures 4 and 5 for 6 different groups at an effector: target (T cell: B92) ratio of 100:1 as follows: Treatment 1: glial tumor cells co-cultured with autologous T cells stimulated with tumor lysate pulsed DCs. Treatment 2: glial tumor cells co-cultured with autologous T cells stimulated with tumor lysate pulsed DCs in the presence of non-polarized MVs. Treatment 3: glial tumor cells co-cultured with autologous T cells stimulated with tumor lysate pulsed DCs in the presence of polarized MVs. Treatment 4: glial tumor cells co-cultured with autologous T cells stimulated with heat stressed tumor lysate pulsed DCs. Treatment 5: glial tumor cells co-cultured with autologous T cells stimulated with heat stressed tumor lysate pulsed DCs in the presence of non-polarized MVs. Treatment 6: glial tumor cells co-cultured with autologous T cells stimulated with heat stressed tumor lysate pulsed DCs in the presence of polarized MVs.
Figure 4.
Flow Cytometric Analysis of the Cytotoxic Activity of Autologous T Cells Stimulated with Heat Stressed Tumor Lysate pulsed DCs in the Presence of MSC-derived MVs. this causes more death in the targeted tumor cells in comparison with the unheated ones (P≥ 0.05).
Figure 5.
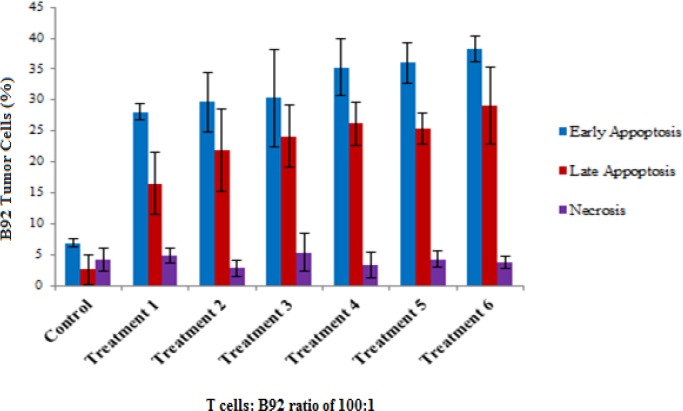
Early and Late Apoptosis as Well as Necrosis in Target Cells (B92 Tumor Cells) Induced by Tumor Lysate Pulsed DC Primed Cytotoxic T Lymphocytes in the Presence of MSC-Derived MVs (P≥ 0.05). The T cell mediated cytotoxicity assay was done using flow cytometry and data were analyzed by one-way-ANOVA.
The results showed that heat stressed tumor lysate pulsed DC primed T cells, produced more cytotoxic activity than normal tumor lysate pulsed DCs non-significantly (P≥ 0.05). Moreover, polarized MSC-derived MVs induced higher cytotoxic activity than non-polarized MSC-derived MVs non-significantly as well as (P≥ 0.05) (Figures 4 and 6). Apoptosis and necrosis analyses also showed the aforementioned pattern of responses (Figure 5).
Figure 6.
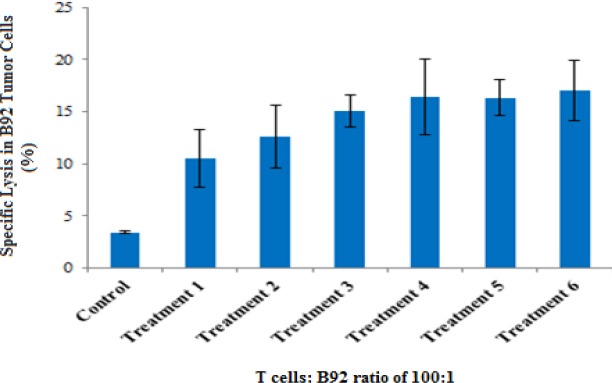
Specific Lysis of Tumor Cells Induced by Tumor Lysate Pulsed DC Primed Cytotoxic T Lymphocytes in the Presence of MSC-Derived MVs (P≥ 0.05). The flow cytometry by annexin-FITC/ PI for stained and unstained B92 tumor cells was used to determine specific lysis.
Discussion
Cellular immunotherapy is currently being considered as a potential new treatment modality that may manipulate the immune system and perhaps in combination with other therapies raise hope for the survival of patients with glioma. Cancer immunotherapy primes the patient’s own immune system to raise effective antitumor responses against glial cancer cells. Recently, cancer vaccine studies have been based on autologous dendritic cells loaded with glial peptides or whole tumor lysates which can subsequently trigger the immune system upon injection (Sakai et al., 2015). DCs are the most effective antigen presenting cells for initiating cellular immune responses through the stimulation of naive T cells (Finocchiaro and Pellegatta, 2016). These DCs play a central role in various immunotherapy protocols as they induce CTLs. CD8+ and CD4+ T lymphocytes identify antigens in the context of MHC class I and class II molecules, respectively. MoDCs are a crucial reservoir of specialized antigen presenting cells that are recruited in immune responses to certain tumors (Saito et al., 2006).
In the field of cancer therapy, the most notable properties of MSCs is their inherent ability to migrate towards tumor cells and provide many bioactive factors once resident (Sasportas et al., 2009). The migration of MSCs to tumors has been demonstrated in malignant glioma (Hai et al., 2012). It is remarkable that Waterman and his colleagues could separate MSCs from at least six different donors into two distinct phenotypes MSC1 and MSC2. MSC1 could reduce tumor growth and metastasis (Waterman et al., 2012). Various studies have shown that MSC-derived MVs possess equal features as their cellular counterparts in anti-cancer therapy (Sasportas et al., 2009).
Some methods have been developed to produce DC in vitro from the bone marrow (Inaba et al., 1992) or peripheral blood monocytes (Sallusto and Lanzavecchia, 1994). The method based on the use of MoDCs, seems to be the most practical strategy for clinical trials to induce antitumor responses. Hence, in the present study, MoDCs were loaded with tumor lysates in two forms; heated and unheated lysates and then matured in the presence of LPS. Previous studies (Adams et al., 2005; Bagheri et al., 2008) as well as ours have described the matured DCs as non-adherent, large irregular veiled cells with numerous cell membrane processes. Morphological examination of floated cells using inverted microscopy at 7 days of culture revealed no obvious differences among these heated and unheated cell lysates loaded DCs (Figure 1). Therefore, we precede the work with the comparison of functional properties and the role of MSC1s-derived MVs on DC maturation and cytotoxicity of stimulated T cells afterwards.
In the current study we used heat stressed tumor lysate as tumor antigens. Our results showed that this kind of tumor cell lysate as HSP-PC may increases the tumor specific cytotoxicity of T cells non-significantly (P≥ 0.05). Lysates from heat-stressed tumor cells provide an optimal source of tumor antigens and generate DCs with improved cross-presentation potential (Aguilera et al., 2011). Chaperone-enriched tumor lysate seems to be more effective than purified HSP alone in inducing tumor specific responses in various murine models (Zeng et al., 2006; Prins et al., 2013). Therefore, chaperone-rich cell lysate vaccine elicits more immunological effect than any single chaperone proteins and is thus used independently as vaccines. Obviously, tumor lysate can mediate antigen cross-presentation and activate CD8+ T lymphocytes. However, the presentation of tumor antigens by DC is not sufficient for CTL programming in the absence of co-stimulatory molecules and innate immunity, since the “helpless” CD8+ cells will cease to proliferate abundantly and will most likely undergo apoptosis (Schurich et al., 2009; Kurts et al., 2010). One mechanism for enhancing CTL programming involves the activation of TLR pathways that result in the synthesis of co-stimulatory molecules (Rudd et al., 2009; Yamamoto and Takeda, 2010). Furthermore, tumor cells treated with elevated temperatures release inflammatory chemokines in an Hsp70 and TLR4 dependent mechanisms and this effect may be significant in CTL programming and tumor cell death (Chen et al., 2009). Flow cytometric annexin-FITC/PI was used to the determination of apoptosis in B92 tumor cells. Previous studies of human MSCs have shown that co-culturing of MSC1 with cancer cells result in fewer tumor growth and much smaller angiogenesis in various cancer cell lines (Waterman et al., 2012). Also, MSC-derived MVs may reprogram tumor behavior by transferring their molecular contents and suppress angiogenesis by transferring anti-angiogenic molecules (Lee et al., 2013). Moreover, evidences suggest that MSCs are able to inhibit proliferation of K562 cells in a humoral microenvironment by secreting DKK-1 (dickkopf-1) (Zhu et al., 2009). In all, our studies on MSC/MSC1 in co-culture showed that MSC1 derived MVs with heat stressed tumor lysate pulsed DCs may change the tumor specific cytotoxicity of T cells and may enhance T lymphocyte cytotoxicity against respective tumor cells non-significantly.
Acknowledgments
This work was conducted by the financial support of Ardabil University of Medical Sciences, Ardabil, Iran. We are grateful to Ardabil University of Medical Sciences for financial support; Mr. Aliyari at Immunology Laboratory of faculty of Veterinary Medicine, Urmia University and Dr. Heydari at Urmia University of Medical sciences, department of pharmacology.
References
- 1.Abbasi A, Kukia NR, Froushani SMA, Hashemi SM. Nicotine and caffeine alter the effects of the LPS- primed mesenchymal stem cells on the co-cultured neutrophils. Life Sci. 2018;199:41–7. doi: 10.1016/j.lfs.2018.03.009. [DOI] [PubMed] [Google Scholar]
- 2.Adams S, O'Neill DW, Bhardwaj N. Recent advances in dendritic cell biology. J Clin Immunol. 2005;25:177–88. doi: 10.1007/s10875-005-4086-2. [DOI] [PubMed] [Google Scholar]
- 3.Agger R, Petersen MS, Toldbod HE, et al. Characterization of murine dendritic cells derived from adherent blood mononuclear cells in vitro. Scand J Immunol. 2000;52:138–47. doi: 10.1046/j.1365-3083.2000.00760.x. [DOI] [PubMed] [Google Scholar]
- 4.Aguilera R, Saffie C, Tittarelli A, et al. Heat-shock induction of tumor-derived danger signals mediates rapid monocyte differentiation into clinically effective dendritic cells. Clin Cancer Res. 2011;17:2474–83. doi: 10.1158/1078-0432.CCR-10-2384. [DOI] [PubMed] [Google Scholar]
- 5.Bagheri K, Delirezh N, Moazzeni SM. PPD extract induces the maturation of human monocyte-derived dendritic cells. Immunopharmacol Immunotoxicol. 2008;30:91–104. doi: 10.1080/08923970701812654. [DOI] [PubMed] [Google Scholar]
- 6.Beyth S, Borovsky Z, Mevorach D, et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005;105:2214–9. doi: 10.1182/blood-2004-07-2921. [DOI] [PubMed] [Google Scholar]
- 7.Bruno S, Grange C, Collino F, et al. Microvesicles derived from mesenchymal stem cells enhance survival in a lethal model of acute kidney injury. PLoS One. 2012;7:1–11. doi: 10.1371/journal.pone.0033115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen T1, Guo J, Han C, Yang M, Cao X. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J Immunol. 2009;182:1449–59. doi: 10.4049/jimmunol.182.3.1449. [DOI] [PubMed] [Google Scholar]
- 9.Collino F, Deregibus MC, Bruno S, et al. Microvesicles derived from adult human bone marrow and tissue specific mesenchymal stem cells shuttle selected pattern of miRNAs. PLoS One. 2010;5:1–15. doi: 10.1371/journal.pone.0011803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delirezh N1, Moazzeni SM, Shokri F, et al. Autologous dendritic cells loaded with apoptotic tumor cells induce T cell-mediated immune responses against breast cancer in vitro. Cell Immunol. 2009;257:23–31. doi: 10.1016/j.cellimm.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Farsad K. Exosomes: novel organelles implicated in immunomodulation and apoptosis. Yale J Biol Med. 2002;75:95–101. [PMC free article] [PubMed] [Google Scholar]
- 12.Finocchiaro G, Pellegatta S. Immunotherapy with dendritic cells loaded with glioblastoma stem cells: from preclinical to clinical studies. Cancer Immunol Immunother. 2016;65:101–9. doi: 10.1007/s00262-015-1754-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.GanjiBakhsh M, Nejati V, Delirezh N, Asadi M, Gholami K. Mixture of fibroblast, epithelial and endothelial cells conditioned media induce monocyte-derived dendritic cell maturation. Cell Immunol. 2011;272:18–24. doi: 10.1016/j.cellimm.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Guo X, Zhou Y, Wu T, et al. Generation of mouse and human dendritic cells in vitro. J Immunol Methods. 2016;432:24–9. doi: 10.1016/j.jim.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 15.Haar CP, Hebbar P, Wallace GC, et al. Drug resistance in glioblastoma: a mini review. Neurochem Res. 2012;37:1192–200. doi: 10.1007/s11064-011-0701-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hai C, Jin YM, Jin WB, et al. Application of mesenchymal stem cells as a vehicle to deliver replication-competent adenovirus for treating malignant glioma. Chin J Cancer. 2012;31:233–40. doi: 10.5732/cjc.011.10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F. Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2011;29:11–19. doi: 10.1002/stem.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klopp AH, Lacerda L, Gupta A, et al. Mesenchymal stem cells promote mammosphere formation and decrease E-cadherin in normal and malignant breast cells. PLoS One. 2010;5:1–9. doi: 10.1371/journal.pone.0012180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–14. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 21.Lai RC, Arslan F, Lee MM, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4:214–22. doi: 10.1016/j.scr.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Lee JK, Park SR, Jung BK, et al. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS One. 2013;8:1–11. doi: 10.1371/journal.pone.0084256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Zhang Z, Lin G, et al. Antigen-specific T cell response from dendritic cell vaccination using side population cell-associated antigens targets hepatocellular carcinoma. Tumour Biol. 2016;37:11267–78. doi: 10.1007/s13277-016-4935-z. [DOI] [PubMed] [Google Scholar]
- 24.Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11:5515–25. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 25.Mokarizadeh A, Delirezh N, Morshedi A, et al. Microvesicles derived from mesenchymal stem cells: Potent organelles for induction of tolerogenic signaling. Immunol Lett. 2012;147:47–54. doi: 10.1016/j.imlet.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Murphy N, Lynch K, Lohan P, Treacy O, Ritter T. Mesenchymal stem cell therapy to promote corneal allograft survival: current status and pathway to clinical translation. Curr Opin Organ Transplant. 2016;21:559–67. doi: 10.1097/MOT.0000000000000360. [DOI] [PubMed] [Google Scholar]
- 27.Nakai N, Asai J, Ueda E, et al. Vaccination of Japanese patients with advanced melanoma with peptide, tumor lysate or both peptide and tumor lysate-pulsed mature, monocyte-derived dendritic cells. J Dermatol. 2006;33:462–72. doi: 10.1111/j.1346-8138.2006.00110.x. [DOI] [PubMed] [Google Scholar]
- 28.Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–18. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 29.Parekkadan B, van Poll D, Suganuma K, et al. Mesenchymal stem cell-derived molecules reverse fulminant hepatic failure. PLoS One. 2007;2:1–6. doi: 10.1371/journal.pone.0000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pellegatta S, Poliani PL, Corno D, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006;66:10247–52. doi: 10.1158/0008-5472.CAN-06-2048. [DOI] [PubMed] [Google Scholar]
- 31.Platten M, Bunse L, Wick W, Bunse T. Concepts in glioma immunotherapy. Cancer Immunol Immunother. 2016;65:1269–75. doi: 10.1007/s00262-016-1874-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prins RM, Wang X, Soto H, et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J Immunother. 2013;36:152–7. doi: 10.1097/CJI.0b013e3182811ae4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radford KJ, Tullett KM, Lahoud MH. Dendritic cells and cancer immunotherapy. Curr Opin Immunol. 2014;27:26–32. doi: 10.1016/j.coi.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Richters CD, Mayen I, Havenith CE, Beelen RH, Kamperdijk EW. Rat monocyte-derived dendritic cells function and migrate in the same way as isolated tissue dendritic cells. J Leukoc Biol. 2002;71:582–7. [PubMed] [Google Scholar]
- 35.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saito H1, Frleta D, Dubsky P, Palucka AK. Dendritic cell-based vaccination against cancer. Hematol Oncol Clin North Am. 2006;20:689–710. doi: 10.1016/j.hoc.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 37.Sakai K, Shimodaira S, Maejima S, et al. Dendritic cell-based immunotherapy targeting Wilms'tumor 1 in patients with recurrent malignant glioma. J Neurosurg. 2015;123:989–97. doi: 10.3171/2015.1.JNS141554. [DOI] [PubMed] [Google Scholar]
- 38.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–18. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sasportas LS, Kasmieh R, Wakimoto H, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci U S A. 2009;106:4822–7. doi: 10.1073/pnas.0806647106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satoh J, Kim SU. HSP72 induction by heat stress in human neurons and glial cells in culture. Brain Res. 1994;653:243–50. doi: 10.1016/0006-8993(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 41.Schnurr M, Galambos P, Scholz C, et al. Tumor cell lysate-pulsed human dendritic cells induce a T-cell response against pancreatic carcinoma cells: an in vitro model for the assessment of tumor vaccines. Cancer Res. 2001;61:6445–50. [PubMed] [Google Scholar]
- 42.Schurich A, Böttcher JP, Burgdorf S, et al. Distinct kinetics and dynamics of cross-presentation in liver sinusoidal endothelial cells compared to dendritic cells. Hepatology. 2009;50:909–19. doi: 10.1002/hep.23075. [DOI] [PubMed] [Google Scholar]
- 43.Senst C, Nazari-Shafti T, Kruger S, et al. Prospective dual role of mesenchymal stem cells in breast tumor microenvironment. Breast Cancer Res Treat. 2013;137:69–79. doi: 10.1007/s10549-012-2321-0. [DOI] [PubMed] [Google Scholar]
- 44.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study:5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 45.Stupp R, Roila F. Malignant glioma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20:126–8. doi: 10.1093/annonc/mdp151. [DOI] [PubMed] [Google Scholar]
- 46.Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;3:1–29. doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- 47.Tuyaerts S, Aerts JL, Corthals J. Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol Immunother. 2007;56:1513–37. doi: 10.1007/s00262-007-0334-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–36. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 49.Vu MQ, Der Sarkissian S, Borie M, Bessette PO, Noiseux N. Optimization of mesenchymal stem cells to increase their therapeutic potential. Methods Mol Biol. 2016;1416:275–88. doi: 10.1007/978-1-4939-3584-0_16. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, Chen X, Cao W, Shi Y. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat Immunol. 2014;15:1009–16. doi: 10.1038/ni.3002. [DOI] [PubMed] [Google Scholar]
- 51.Waterman RS, Henkle SL, Betancourt AM. Mesenchymal stem cell 1 (MSC1)-based therapy attenuates tumor growth whereas MSC2-treatment promotes tumor growth and metastasis. PLoS One. 2012;7:1–11. doi: 10.1371/journal.pone.0045590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waterman RS, Morgenweck J, Nossaman BD, et al. Anti-inflammatory mesenchymal stem cells (MSC2) attenuate symptoms of painful diabetic peripheral neuropathy. Stem Cells Transl Med. 2012;1:557–65. doi: 10.5966/sctm.2012-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS One. 2010;5:1–14. doi: 10.1371/journal.pone.0010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yajima N, Yamanaka R, Mine T, et al. Immunologic evaluation of personalized peptide vaccination for patients with advanced malignant glioma. Clin Cancer Res. 2005;11:5900–11. doi: 10.1158/1078-0432.CCR-05-0559. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto M, Takeda K. Current views of toll-like receptor signaling pathways. Gastroenterol Res Pract. 2010;2010:1–8. doi: 10.1155/2010/240365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zeng Y, Graner MW, Katsanis E. Chaperone-rich cell lysates, immune activation and tumor vaccination. Cancer Immunol Immunother. 2006;55:329–38. doi: 10.1007/s00262-005-0694-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu Y, Sun Z, Han Q, et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia. 2009;23:925–33. doi: 10.1038/leu.2008.384. [DOI] [PubMed] [Google Scholar]