

Abstract
The striatum, the input structure of the basal ganglia, is a major site of learning and memory for goal-directed actions and habit formation. Spiny projection neurons of the striatum integrate cortical, thalamic, and nigral inputs to learn associations, with cortico-striatal synaptic plasticity as a learning mechanism. Signaling molecules implicated in synaptic plasticity are altered in alcohol withdrawal, which may contribute to overly strong learning and increased alcohol seeking and consumption. To understand how interactions among signaling molecules produce synaptic plasticity, we implemented a mechanistic model of signaling pathways activated by dopamine D1 receptors, acetylcholine receptors, and glutamate. We use our novel, computationally efficient simulator, NeuroRD, to simulate stochastic interactions both within and between dendritic spines. Dopamine release during theta-burst and 20Hz stimulation was extrapolated from fast-scan cyclic voltammetry data collected in mouse striatal slices. Our results show that the combined activity of several key plasticity molecules correctly predicts the occurrence of either LTP, LTD or no plasticity for numerous experimental protocols. To investigate spatial interactions, we stimulate two spines, either adjacent or separated on a 20 μm dendritic segment. Our results show that molecules underlying LTP exhibit spatial specificity, whereas 2-arachidonylglycerol exhibits a spatially diffuse elevation. We also implement changes in NMDA receptors, adenylyl cyclase and G protein signaling that have been measured following chronic alcohol treatment. Simulations under these conditions suggest that the molecular changes can predict changes in synaptic plasticity; thereby accounting for some aspects of alcohol use disorder.
Keywords: computational model, LTP, LTD, signaling pathways, basal ganglia, striatum
Graphical Abstract
We created a comprehensive model of signaling pathways underlying synaptic plasticity. The model shows that a combination of molecules in the spine and dendrite predicts the development of LTP or LTD.
Introduction
Alcohol use disorder is a costly, societal burden with several behavioral characteristics that impair treatment success. First, chronic alcohol use exhibits characteristics of compulsive use (Everitt and Robbins 2005), hindering the ability to avoid drinking in the face of negative consequences. In addition, the tendency to relapse when exposed to environmental or sensory inputs that were previously associated with alcohol use impairs long term success of withdrawal treatments (Seo and Sinha 2014). In both humans and rodent models of withdrawal, relapse in response to environmental cues, despite extinction of instrumental responding (Ciccocioppo et al 2001; Nic Dhonnchadha and Kantak 2011), suggests overly strong goal-directed learning (Heinz et al 2009). On the other hand, persistent behavior (e.g. alcohol use) despite reward devalulation (i.e. negative consequences) is characteristic of habitual behavior (Barker et al 2015; Yin et al 2004).
Numerous studies have highlighted the involvement of the basal ganglia in both habitual behavior and reward learning. In particular, association of environmental or sensory cues with rewards (goal-directed learning) occurs not only in ventral striatum (nucleus accumbens) but also in dorsal medial striatum (Devan et al 1999; Gremel and Costa 2013; Her et al 2016). In addition, behavior that becomes habitual is learned by or transferred to the dorsal striatum (Goodman et al 2016; Gremel and Costa 2013; O’Hare et al 2016; Smith and Graybiel 2016; Yin et al 2004). Thus, understanding how alcohol produces overly strong associations, habits and relapse requires a deeper understanding of how goal-directed and habitual memories are stored in the striatum.
Striatal synaptic plasticity is a mechanism underlying striatal dependent learning and adaptive changes in behavior (Hawes et al 2015; Shan et al 2014; Yin et al 2009). Depending on the temporal pattern of synaptic inputs, either strengthening (LTP: long term potentiation) or weakening (LTD: long term depression) occurs (Pawlak et al 2010; Reynolds and Wickens 2002). The molecular mechanisms underlying synaptic plasticity are diverse and depend on the stimulation pattern (Kreitzer and Malenka 2008). For example, some types of LTD are N-methyl D-aspartic acid (NMDA) receptor dependent (Pawlak and Kerr 2008), whereas other types of LTD, e.g. those induced with high frequency stimulation, are NMDA receptor independent (Calabresi et al 1992; Paille et al 2013; Shindou et al 2011). The diversity of stimulation patterns and molecular mechanisms hinders a comprehensive understanding of striatal synaptic plasticity and memory storage.
Converging evidence from alcohol studies (DePoy et al 2013; Ma et al 2017; Shen et al 2011) reveals aberrant synaptic plasticity following chronic alcohol treatment, both in dorsomedial and dorsolateral striatum. Altered synaptic plasticity during alcohol intoxication is thought to contribute to overly strong habit learning, whereas alterations after withdrawal are thought to contribute to relapse as well as other cognitive and behavioral changes (Gremel and Lovinger 2016). Several of the molecular mechanisms underlying striatal synaptic plasticity are altered after withdrawal from chronic alcohol (Abrahao et al 2017). Calcium influx through NMDA type glutamate receptors is enhanced (Chen et al 2011; Ma et al 2017; Wang et al 2010), several molecules in the G protein coupled pathways are altered (Kashem et al 2012; Lucchi et al 1983; May et al 1995; Nestby et al 1999), and cannbinoid receptor type 1 (CB1R) signaling is down-regulated after withdrawal from alcohol (DePoy et al 2013). A key question is whether these identified molecular changes can contribute to relapse or other behavioral and cognitive changes including enhanced alcohol seeking and consumption.
To better understand molecular mechanisms underlying striatal-dependent learning, we developed a comprehensive model of signaling pathways to determine whether the activation of the set of critical molecules can predict and explain the set of synaptic plasticity publications. Furthermore, we created a chronic alcohol treated model to identify which of the altered molecular mechanisms contribute most significantly to changes in synaptic plasticity that may be associated with pathological behavior such as enhanced alcohol seeking and consumption.
Methods
Ethical Approval
All animal handling and procedures were in accordance with the National Institutes of Health animal welfare guidelines. The voltammetry experiments were performed at the National Institute on Alcohol Abuse and Alcoholism and were approved by the National Institute on Alcohol Abuse and Alcoholism Animal Care and Use Committee (protocol LIN-DL-1).
Model
To investigate how temporal patterns of synaptic activation control the development of potentiation versus depression, we created a multi-compartmental, stochastic reaction-diffusion model of signaling pathways activated by calcium influx and metabotropic receptor activation (Fig. 1A). The model incorporated the signaling pathways from several prior models (Kim et al 2013; Oliveira et al 2012) and added in muscarinic type m1 and m4 receptors (Nair et al 2015), as well as exchange factor activated by cAMP (Epac1). The list of reactions and rate constants of the model is available on github (https://github.com/neurord/D1pathways/blob/master/Rxn_SPNspineAChm4R_Gshydr5_AC1_GiGsfast-GapD.xml).
Figure 1.

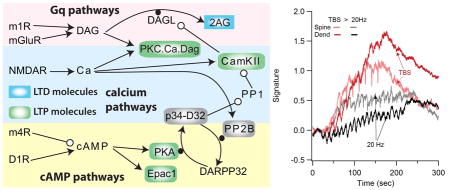
Signaling pathways underlying synaptic plasticity in striatal direct pathway spiny projection neuron. (A) Dopamine D1 receptors (D1R) are coupled to the stimulatory subtype of GTP binding protein (GolfGTP), and lead to production of cAMP via adenylyl cyclase (AC). cAMP activates Exchange Protein Activated by cAMP (Epac) and protein kinase A (PKA). Both metabotropic glutamate (mGluR1/5) receptors and acetylcholine muscarinic type 1 (m1) receptors are coupled to Gq subtype of GTP binding protein (GqGTP) which activates phospholipase C (PLC), which produes of diacylgycerol (DAG), which is a substrate for diacylclyercol lipase (Dgl) and also activates calcium bound protein Kinase C (PKC). Acetylcholine also activates muscarinic type 4 (m4) receptors, which are coupled to inhibitory subtype of GTP binding protein (GiGTP) and inhibit the production of cAMP by adenylyl cyclase. Calcium (Ca) binds to various buffers including calmodulin which can activate protein phosphatase 2B (calcineurin; PP2B), phosphodisterase 1B (PDE1B) to degrade cAMP, and calcium calmodulin dependent protein kinase type 2 (CamKII). There are several interactions between these pathways: CamKII phosphorylates and inactivates diacylclyercol lipase, which produces the endocannbinoid 2 arachidonylgycerol (2AG). PKA phosphorylation of DARPP32 inhibits the protein phosphatase type 1 (PP1) that dephosphorylates CamKII. PLC: phospholipase C, PIP2: phosphoinositol bis-phosphate, PP2A: protein phosphatase 2A. (B) Morphology used for simulations, visualized using NeuroRDViz.py (https://www.gihub.com/neuroRD/NeuroRDviz). Most results used the 2 μm dendrite with single spine; simulations on spatial specifity used the 20 μm long dendrite with 10 spines (some attached to the same voxel). In both morphologies, the submembrane voxels are 0.1 μm high, the next layer of voxels are 0.2 μm high, and the central core voxels are 0.3 μm high. Diffusion is axial in the spines and two dimensional in the dendrite.
The signaling pathways were simulated within one of two morphologies: either a 2 μm long piece of dendrite with one attached spine or a 20 μm long dendrite with ten randomly distributed spines. Both dendrites had a diameter of 0.6 μm (Fig. 1B). In both cases, the dendrite and spines were subdivided into voxels to accurately simulate spatial aspects of signaling molecules. The layer of voxels adjacent to the membrane was labeled the submembrane domain.
As this is a spatial model, some molecules were localized to the spine or submembrane domain whereas others were freely diffusible. Metabotropic receptors, G proteins, adenylyl cyclase, phospholipase C, diacylglycerol lipase, phosphoinositol bis-phosphate, and protein kinase A (PKA) holoenzyme were localized and/or anchored both in this submembrane domain and the spine head. The diffusible molecules included cyclic adenosine monophosphate (cAMP), adenosine triphosphate (ATP), calcium, all forms of calmodulin (Cam), calcium-calmodulin dependent protein kinase type 2 (CamKII), dopamine- and cAMP-regulated neuronal phosphoprotein (DARPP32) and Epac. The diffusion constants of diffusible molecules are listed in the reaction rates file.
Molecule quantities were either taken from prior striatal models (Kim et al 2013; Oliveira et al 2012), experimental publications or adjusted to reproduce experimentally measured concentrations of downstream molecules, e.g. the balance of adenylyl cyclase and phosphodiesterase was adjusted to produce a 30 nM basal cAMP concentration (Bacskai et al 1993; Mironov et al 2009). Changes in molecule quantity due to chronic alcohol treatment was based on published experiments showing increase of Gbg (Bowers et al 2008), increase of metabotropic glutamate receptor types 1/5 (mGluR1/5) (Meinhardt et al 2013; Obara et al 2009), lower evoked dopamine (Hirth et al 2016; Siciliano et al 2017), changes in Gs coupled signaling (Kashem et al 2012; Lucchi et al 1983; May et al 1995; Nestby et al 1999) or increased calcium influx due to increases in NMDA receptor 2B (NR2B) subunits relative to NR2A subunits (Chen et al 2011; Ma et al 2017; Wang et al 2010). Initial conditions specifying molecule quantities are available on github (https://github.com/neurord/D1pathways/blob/master/IC_SPNspineAChm4R_AC1_coupleGqhigh.xml).
The model was activated using either theta burst stimulation (TBS) to produce LTP (Hawes et al 2013) or 20 Hz stimulation to produce LTD (Hawes et al 2013; Lerner and Kreitzer 2012). In addition, two spike timing dependent plasticity (STDP) protocols were used to provide an additional protocol that produces LTP. In brain slices, theta burst stimulation consists of a burst of 4 stimulation pulses at 50 Hz, with the burst repeated 10 times at a theta frequency of 10.5 Hz. Four of these trains of 10 bursts are delivered 15 sec apart. 20 Hz stimulation consists of 1 sec of 20 Hz stimulation repeated 20 times with a 10 sec interval. Experimentally, the STDP protocol consisted of 60 pairings of a pre-synaptic stimulation with a post-synaptic action potential, the latter elicited with a 30 ms depolarizing current injection. Glutamate release by electrical stimulation was modeled as 1 mM glutamate for each stimulation pulse (for binding to mGluRs), calcium influx into the spine produced by activation of post-synaptic NMDA receptors and calcium influx into the dendrite caused by activation of voltage-dependent calcium channels. The amount of calcium influx was adjusted to reproduce the calcium concentration simulated using a biophysically based model of a spiny projection neuron (Jedrzejewska-Szmek et al 2016), which showed that calcium concentration was ~5x higher for TBS than 20Hz, and that STDP with a +10 ms interval produced 15 μM whereas a -30 ms interval produced <2 μM peak calcium concentration. Electrical stimulation also evokes release of dopamine (Rice and Cragg 2004; Salinas et al 2016), and firing of acetylcholinergic neurons. The acetylcholine concentration profile followed the burst and pause pattern observed in vivo and in vitro (Bennett et al 2000; Goldberg and Reynolds 2011; Morris et al 2004). The dopamine concentration for the first train was 0.8 μM for 20Hz and 1.2 μM for TBS, based on voltammetric measurements (described below). For subsequent trains, the dopamine concentration was lower, consistent with the time interval dependent decrease in evoked dopamine during repeated stimulation (Mamaligas et al 2016). Thus, subsequent trains of dopamine for TBS (with a 15s inter-train interval) were ~0.8 μM (a 30% decrease), and subsequent trains of dopamine for 20 Hz (with a 10s inter-train interval) were either ~0.4 μM (for moderate dopamine) or decreased to ~0.25 μM (for low dopamine). For STDP, both +10ms and −30ms intervals used the same dopamine, acetylcholine and glutamate. Fig 2 shows the dopamine, acetylcholine, calcium and glutamate profiles for 20Hz and theta burst stimulation.
Figure 2.

Synaptic inputs to the model. The amount of calcium influx (A) was adjusted to reproduce the calcium concentration simuated using a biophysical model of a spiny projection neuron (Jedrzejewska-Szmek et al 2016). The concentration is considerably higher in the spine than in the dendrite, as observed experimentally. The concentration is higher for TBS than for 20Hz because the lower frequency stimulation depolarizes the neuron less than TBS. The acetylcholine profile (B) exhibited the burst and pause pattern observed in vivo. (C) Dopamine concentration used as input to the model. The reduction in dopamine for subsequent trains was based on measures of dopamine recovery kinetics (Mamaligas et al 2016), which shows that recovery of dopamine release is less after 10s intervals (used for 20Hz) than after 15s intervals (used for TBS). (D). Voltammetric measurements of dopamine used to constrain model dopamine input. A single burst of 4 pulses produces ~0.8 μM of dopamine. Giving 40 pulses of either TBS or 20Hz increases the dopamine transient by ~30% compared to the 4 pulse burst. Based on this data, dopamine input (shown in C) for TBS had a concentration of 1.2 μM. Since a train of 20Hz uses only 20 pulses, the dopamine transient for 20Hz was lowered compared to TBS.
We used a stochastic simulation technique, as many molecular populations are small resulting in large fluctuations about the mean within small compartments such as spines (Anderson et al 2010; Grima 2009). Similarly, diffusion of second messenger molecules within and between spines and dendrites occurs stochastically. The model was implemented using an efficient mesoscopic stochastic reaction-diffusion simulator NeuroRD (Jedrzejewski-Szmek and Blackwell 2016) because the large numbers of molecules in the morphology described (Fig. 1) made tracking individual molecules in microscopic stochastic simulators computationally expensive. NeuroRD version 3.2.3 (https://github.com/neurord/stochdiff/releases) adaptively switches between exact and leap without partitioning the system based on propensity of each reaction or diffusion event. The accuracy control parameter was set to 0.002 for all simulations. This simulator uses reflective boundary conditions (molecules that would have diffused beyond the edge of the morphology remained within the morphology). A single simulation of 300 sec (of the dendrite with 1 spine) takes ~18 hours on a Intel®Xeon®CPU E5-2650 processor. Simulations were repeated 3 times each, and analyzed in python 2.7. All model files and analysis code are freely available (https://github.com/neurord/D1pathways).
To screen for sensitive model parameters, the model was implemented in SimBiology toolbox of MATLAB (MathsWork) and solved with ode15s solver with a maximum time step of 0.01s in a one compartment morphology using a deterministic approach as in previous work (Gutierrez-Arenas et al 2014; Nair et al 2016; Nair et al 2015). We calculated local sensitivities for every model output variable with respect to every model parameter. To obtain the numeric approximations of the change in output variables, each parameter was perturbed one at a time by ±10% of its initial value. We performed local sensitivity analysis as it has been successfully used previously to identify analytical mechanisms in various biological models (Gutierrez-Arenas et al 2014; Nair et al 2016; Nair et al 2015). All the rate constants were investigated to determine how sensitive the output variables were to deviation of parameters. This simplified model will be deposited in BioModels database (Chelliah et al 2015) in SBML format following acceptance of this study.
Fast-scan Cyclic Voltammetry
Brain slices for fast-scan cyclic voltammetry were prepared as described in (Crowley et al 2014; Hawes et al 2013). Briefly, four to eight week old C57BL6/J male mice (n=5, Jackson Laboratory, Bar Harbor, ME; stock #000664) were anesthetized with isoflurane and sacrificed by rapid decapitation. Brains were extracted quickly and placed in oxygenated ice-cold sucrose slicing solution (in mM: 194 Sucrose, 30 NaCl, 4.5 KCl, 26 NaHCO3, 1.2 NaH2PO4, 10 dextrose, and 1 MgCl2). Coronal slices were cut 300μm thick on a Leica vibratome (VT1200S), bisected between hemispheres, and placed in an incubation chamber containing aCSF (in mM: NaCl 126, NaH2PO4 1.25, KCl 2.8, CaCl2 2, MgCl2 1, NaHCO3 26, Dextrose 11) heated to 33°C for 30 minutes and then removed to room temperature (21–23°C) for one hour before recording. Following the incubation period, slices were transferred to the slice chamber and a twisted, bipolar, stainless steel stimulating electrode (Plastics One) was placed in the white matter overlying the dorsal medial striatum. The carbon fiber electrode was then positioned in the dorsal medial striatum approximately 300 μm from the stimulating electrode.
Carbon fiber electrodes were made as previously described (Crowley et al 2014) and cut to ~150 μm in length. The carbon fiber electrode potential was linearly scanned as a triangle waveform from −0.4V to 1.2V and back to −0.4V at 400V/s. Cyclic voltammograms were collected at 10 Hz using a Chem-Clamp (Dagan Corporation) and DEMON voltammetry software (Yorgason et al 2011). Dopamine release was evoked using 40 200 μs monophasic electrical stimuli delivered with a Constant Current Stimulator (Digitimer) either in a theta burst or 20Hz pattern. In all slices, dopamine release was measured first in response to 4 pulses at 50 Hz. Then, the response to 40 pulses using the protocol was measured, and reported as a ratio of the train response to the 4 pulse response. For theta burst protocol, a burst of four pulses was delivered at 50Hz, and this burst was repeated 10 times with an interburst interval of 95 msec. For the 20Hz protocol, 40 pulses were delivered at 20 Hz for 2 sec. The stimulation intensity for each slice was determined from a preliminary input-output curve and set to yield a peak dopamine transient approximately 50% of the maximal response (between 500–800 μA).
Statistical Analysis
To develop a predictive model of LTP versus LTD, we used a linear discriminant analysis applied to the key molecules for two sets of simulations: those whose experimental outcomes were known and those whose experimental outcomes were unknown. We sampled the concentration of five different molecules (2AG, Epac, PKA, CamKII, PKC) each at three time points (the average concentration using a 60s window centered at 60s, 120s and 180s) for both the spine and the dendrite. To introduce a non-linearity, two separate two-outcome discriminant analyses were performed and the results combined. The first outcome was LTP or not, the second outcome was LTD or not. The overall result was LTP if the two outcomes were (LTP, not LTD); LTD if the two outcomes were (not LTP, LTD) and no plasticity otherwise. Because many of these 30 molecule-time-space samples are correlated, we used a two-part procedure, in which the first part selects a subset of the 30 samples to use in the second part. In part 1, the two stepwise discriminant analyses utilized the SAS procedure STEPDISC applied to 3 repeats of each of 10 conditions (Table 1). The stepwise discriminant using LTP versus no LTP as the outcome provided one set of samples, and the stepwise discriminant using LTD versus no LTD as the outcome provided a second set of samples. In part 2, two discriminant analyses (SAS procedure DISCRIM) are performed (again, one for LTP versus no LTP and one for LTD versus no LTD) using the two sets of samples identified in part 1. The results of the two discriminant analyses in part 2 are combined to classify both the training set of 3 repeats of 10 conditions (listed in Table 1), and the test set of conditions (3 repeats each) with unknown outcomes (listed in Tables 2 and 3). For both part 1 and part 2, all three repeats of all 10 conditions (Table 1) are used as the “training” set; however, the results reported from part 2 for these 10 conditions are from the leave-one-out cross-validation method used by the SAS procedure DISCRIM.
Table 1.
Cross-validation outcome of 3 trials for each condition predicted by linear discriminant analysis. The outcome of two discriminant analyses was used for the predictions. The LTD (or no LTD) discriminant analysis used spine PKC and dendritic pCamKII at 1 min, and dendritic 2AG at 3 min. The LTP (or no LTP) discriminant analysis used dendritic 2AG, spine Epac1, dendritic pCamKII, and spine pCamKII at 1 min; spine PKC and dendritic pCamKII at 2 min, and phosphorylated PKA targets in the spine at 3 min. Experimental Outcome gives the reported EPSP or population spike (PopSpike) amplitude from the publication in Source. NP indicates no plasticity determined as non-significant difference to the experimental control.
| Condition | LTD | LTP | No Plasticity | Experimental Outcome | Source |
|---|---|---|---|---|---|
| 20Hz, block PKA | 3 | 0 | 0 | LTD | (Lerner and Kreitzer 2012)* |
| 20Hz, Control moderate Da | 3 | 0 | 0 | LTD (80%,86%) | (Hawes et al 2013) |
| 20Hz, Control low Da | 3 | 0 | 0 | LTD (80%,86%) | (Hawes et al 2013) |
| TBS, Block m1 | 0 | 0 | 3 | NP (85%) | (Hawes et al 2013) |
| TBS, block mGlu | 0 | 0 | 3 | NP (85%) | (Hawes et al 2013) |
| TBS, block PKA | 0 | 0 | 3 | NP (105%) | (Hawes et al 2013) |
| TBS, block DaD1R | 0 | 0 | 3 | NP (97%) | (Hawes et al 2013) |
| TBS, Control | 0 | 3 | 0 | LTP (135%) | (Hawes et al 2013) |
| STDP, ISI=−30ms | 3 | 0 | 0 | LTD (84%) | (Fino et al 2010) |
| STDP, ISI=10ms | 0 | 3 | 0 | LTD (176%) | (Fino et al 2010) |
This condition is extrapolated from 100 Hz and the effect of A2A agonist at 20 Hz, both experiments performed in D2R neurons.
Table 2.
Outcome of 3 trials predicted by discriminant analysis for conditions not yet tested experimentally.
| Condition | LTD | LTP | NC |
|---|---|---|---|
| 20Hz, block m1 | 3 | 0 | 0 |
| 20Hz, block m4 | 1 | 0 | 2 |
| 20Hz, block DaD1R | 3 | 0 | 0 |
| 20Hz, block mGlu* | 3 | 0 | 0 |
| 20Hz, block CamKII | 3 | 0 | 0 |
| TBS, block m4 | 0 | 3 | 0 |
| TBS, block CamKII | 1 | 0 | 2 |
considered unknown because LTD was observed using 10hz (not 20Hz) stimulation in unidentified neurons (Ronesi and Lovinger 2005) and no LTD was observed using 20hz stimulation in D2R (not D1R) neurons (Lerner and Kreitzer 2012).
Table 3.
Predictions of alcohol withdrawn models. In all cases, the stimulation protocol delivered only 40% of the trains used in previous simulations.
| Condition | LTD | LTP | No Plasticity |
|---|---|---|---|
| 20Hz, control | 3 | 0 | 0 |
| 20Hz, withdrawn | 0 | 3 | 0 |
| 20Hz, NMDA alone | 0 | 1 | 2 |
| 20Hz, G protein signaling | 2 | 0 | 1 |
| TBS, control | 2 | 0 | 1 |
| TBS, withdrawn | 0 | 3 | 0 |
| TBS, NMDA alone | 0 | 3 | 0 |
| TBS, G protein signaling | 0 | 1 | 2 |
Results
Model agrees with numerous data sets
We created a spatial, stochastic reaction-diffusion model of signaling pathways of direct pathway (Dopamine D1 receptor-containing) spiny projection neurons to investigate the control of synaptic plasticity by spatio-temporal patterns. The model included pathways activated by calcium influx, as well as several metabotropic receptors: Dopamine D1 receptors coupled to GolfGTP, muscarinic type 4 acetylcholine receptors coupled to GiGTP, and both metabotropic glutamatergic types 1/5 receptors and muscarinic type 1 acetylcholine receptors that are coupled to GqGTP (Fig. 1A). These reactions, together with diffusion of second messengers, were implemented in a spatial model of a dendrite with one or more spines (Fig 1B). Model parameters were adjusted to reproduce diverse sets of data that measured DARPP32 phosphorylation in response to various agonists and antagonists, and depolarization induced suppression of inhibition (DSI). The model was simulated stochastically using NeuroRDv3, open source software for computational efficient simulation of reaction-diffusion systems (https://github.com/neurord/stochdiff/releases).
Figure 3 illustrates that the model indeed reproduces many of the experiments reported in the literature. Stimulation of dopamine receptors, calcium influx, inhibition of phosphodiesterases, and inhibition of phosphatases each produces the appropriate increase in DARPP32 phosphorylation on Threonine (Thr) 34 and Thr75 (Fig 3A). In addition, the model exhibits the correct basal quantities of several molecules, including cAMP (Mironov et al 2009), calcium and phosphorylation of DARPP-32 on Thr34 and Thr75, which have been reported in the literature (Nishi et al 2002; Nishi et al 1999). Activation of mGluR1/5 receptors enhances 2AG production for brief durations of calcium influx but not for long durations of calcium influx (Uchigashima et al 2007). The increase in 2AG by mGluR1/5 activation in the model (Fig 3B) is comparable to the 2AG dependent increase in short term depression of inhibitory inputs produced mGluR5/1 receptor stimulation (Uchigashima et al 2007).
Figure 3.

Model reproduces the experimentally observed change in DARPP32 phosphorylation, and the experimentally observed effect of mGluR agonists on 2AG production. A. Bath application of dopamine agonists (DA) produces an increase in phosphorylation of DARPP32 on Thr34, which is reduced by NMDA agonists (Ca). In contrast, bath application of both dopamine and calcium agonists together reduce phosphorylation of DARPP32 on Thr75. These changes are consistent with experimental measurements (Nishi et al 2002; Nishi et al 1999). Bath application of the type 10 phosphodiesterase inhibitor papaverine (Pap) but not the type 4 phosphodiesterase inhibition, rolipram (Roli), increases phosphorylation of DARPP32 on Thr34. These changes are consistent with experimental measurements (Nishi et al 2008). Both inhibitors of protein phosphatase 2B by Cyclosporin A (CyA) and protein phosphatase 2A by Okadaic Acid (OA) elevate phosphorylation of DARPP32 on Thr34 whereas only the inhibitor of protein phosphatase 2A elevates phosphorylation of DARPP32 on Thr75. These changes are consistent with experimental measurements (Nishi et al 2002; Nishi et al 1999). B. DHPG facilitates the production of 2AG by calcium, but such facilitation is smaller for the 5s duration calcium influx, as reported in (Uchigashima et al 2007).
A single set of molecular signatures predicts the correct direction of synaptic plasticity
To evaluate the ability of the model to predict synaptic plasticity, we simulated a theta burst paradigm (Hawes et al 2013) that produces LTP and a 20Hz paradigm (Hawes et al 2013; Lerner and Kreitzer 2012) that produces LTD. These stimulation paradigms were selected because both LTP and LTD can be produced in the same brain region and magnesium concentration, with only a change in stimulation pattern. We evaluated both the control conditions, and the effect of various receptor and kinase antagonists, both to demonstrate the ability of the model to predict synaptic plasticity direction and to make experimentally testable predictions.
The synaptic inputs used to activate the model (calcium influx, glutamate, dopamine and acetylcholine) were adjusted based on several data sources. The amount of calcium influx (Fig 2A) was adjusted to reproduce the calcium concentration simulated using a biophysical model of a spiny projection neuron (Jedrzejewska-Szmek et al 2016), which itself was constrained by calcium imaging experiments (Carter and Sabatini 2004; Shindou et al 2011). Note that the concentration is considerably higher in the spine than in the dendrite, as observed experimentally. The acetylcholine profile (Fig 2B) followed the burst and pause pattern observed in vivo (Goldberg and Reynolds 2011; Morris et al 2004). Dopamine concentration (Fig 2C) for the first train of stimulation was based on published experiments measuring the dopamine concentration in response to several stimulation pulses (Threlfell et al 2012; Zhang and Sulzer 2004), and further constrained by voltammetry experiments measuring dopamine concentration in response to a train of 40 pulses (Fig 2D). The latter experiments demonstrate that a train of 40 pulses delivered at theta burst produces a dopamine elevation that is slightly higher than the 40 pulses delivered at 20Hz. Because both patterns used the same number of pulses, this shows that the theta burst pattern is slightly more efficient than the 20Hz train. Since the 20Hz train in the model delivered only 20 pulses, the dopamine model input in response to the first train of 20Hz was correspondingly reduced below the value measured for 40 pulses. In addition to these constraints for dopamine in response to the first train of stimulation, the dopamine for subsequent trains was reduced (Fig 2C), especially for the 20Hz stimulation delivered at 10s intervals because dopamine recovery is lower for 10s than 15 s interval (Mamaligas et al 2016).
Figure 4 shows that key molecules implicated in synaptic plasticity differ between TBS and 20Hz. Both phosphorylated CamKII (Fig 4A) and activated PKC (Fig 4B) are greater in response to TBS than 20Hz. In addition, both PKC and active CamKII (both phosphorylated or calmodulin bound CamKII) are greater in the spine than in the dendrite due to a higher spine calcium concentration. Both PKA and Epac1 depend on dopamine D1 receptor activation producing an elevation in cAMP concentration, and both molecules are greater for TBS than 20Hz (Fig 4C, D). The differential response of all four of these molecules to TBS versus 20Hz is consistent with experiments showing their role in various forms of striatal LTP. The elevation of 2AG (Fig 4E) does not differ between TBS and 20Hz early during the stimulation protocol, however, the 2AG elevation is more persistent for 20Hz than TBS, consistent with its role in LTD.
Figure 4.

Response of key plasticity molecules differs between TBS and 20Hz stimulation. Traces of (A) phosphorylated CamKII, (B) active PKC, (C) PKA phosphorylated targets (phosphodiesterases, protein phosphatase 2A, and DARPP32 phosphorylated on Thr34): in the spine (Left column) and the dendrite (right column). Traces of (D) 2AG and (E) Epac1 in the dendrite were qualitatively similar to the spine traces, which were too noisy to distinguish individually. PKA, CamKII, PKC and Epac1 were greater for TBS than for 20Hz, whereas 2AG exhibited a more prolonged elevation for 20Hz than for TBS. PKC was much greater in the spine than in the dendrite because it binds to the membrane associated diacylgylcerol for activation.
To develop a predictive model of LTP versus LTD, we simulated the effect of various antagonists on molecule dynamics during TBS or 20 Hz stimulation, and then used a discriminant analysis applied to the key molecules. We sampled the mean (within a 60s window) molecule concentration at three time points: 60s, 120s and 180s after stimulation onset, in both the spine and the dendrite. To eliminate correlated samples and reduce the dimensionality of the space, a stepwise linear discriminant analysis was performed first to identify the important samples, and then a second linear discriminant analysis used the identified molecule-time-space samples and known outcomes to associate regions in multi-dimensional space with a particular outcome.
Table 1 lists the simulation experiments, the experimentally observed outcome, the prediction by the discriminant analysis, as well as the source of the experimental data. These results show that using a small set of molecule-time-space samples can correctly predict all outcomes. The LTD (or no LTD) discriminant analysis used spine PKC and dendritic pCamKII at 1 min, and dendritic 2AG at 3 min, which samples are consistent with the differences seen in Fig 4. The LTP (or no LTP) discriminant analysis used dendritic 2AG, spine Epac1, both dendritic and spine pCamKII at 1 min; spine PKC and dendritic pCamKII at 2 min, and phosphorylated PKA targets in the spine at 3 min. The number of pCamKII samples is consistent with the prolonged difference between TBS and 20Hz seen in Fig 4A, and also the phosphorylated PKA targets differ more at 3 min after stimulation than at other times (Fig 4C). Fig 5A shows two 2-D slices through the multi-dimensional discriminant space to illustrate how the combination of key molecules can predict the occurrence of LTP, LTD or no plasticity. For example, the plot of PKC versus pCamKII suggests that moderate PKC and low pCamKII concentrations are associated with LTD, whereas the plot of PKA versus 2AG confirms that a high 2AG concentration is needed for LTD. Fig 5A, B also shows that LTP requires elevated PKC, CamKII and PKA, and that the elevated PKC cannot be accompanied by either an elevated 2AG or a low PKA.
Figure 5.

Mean molecule concentration measured 60s after stimulation onset illustrates plasticity outcome predicted by discriminant analysis. (A1, B1) PKC in the spine versus phosphoCamKII (pCamKII) in the dendrite. (A2, B2) PKA phosphorylated proteins in the spine versus 2AG in the dendrite. (A) Prediction of the discriminant analysis for conditions for which experimental outcomes are known. (B) Predictions of the discriminant analysis for which experimental outcomes are unknown. For all panels, red symbols indicate outcome of LTP, blue symbols indicate no plasticity and black symbols indicate LTD.
Predictions for conditions whose experimental outcomes are unknown were made by using time samples from the corresponding simulations as test data for the discriminant analyses. Table 2 gives the outcomes for these conditions, and Figures 5B and 6 graphically illustrate the change in molecule concentrations for these simulations. The discriminant analysis predicts that blocking m4R will still produce LTP for theta burst, but will eliminate 20Hz LTD, which is logical because the Gi coupled muscarinic m4 receptors inhibit adenylyl cyclase; thus, blocking m4R would enhance cAMP (Nair et al 2015). Blocking CamKII will prevent LTP induced by TBS, but will not hinder LTD induced by 20Hz, a prediction consistent with the inhibition of diacylglycerol lipase by CamKII phosphorylation (Shonesy et al 2013) and the reports of CamKII inhibitors preventing other types of striatal LTP (Cui et al 2016). Blocking dopamine D1 receptors will not disrupt 20Hz LTD, as the Golf coupled D1R activates adenylyl cyclase and increases cAMP, a molecule not known to be needed for LTD. Blocking mGluR is predicted to have no effect on LTD, which is similar to the lack of effect on 10 Hz LTD (Ronesi and Lovinger 2005), but differs from that shown for 20Hz LTD in D2R neurons (Lerner and Kreitzer 2012). In summary, by collectively considering the concentration of five key molecules implicated in synaptic plasticity, the simulations can account for known synaptic plasticity experiments and makes experimentally testable predictions regarding the effect of antagonists.
Figure 6.

Plasticity signatures illustrate how blocking receptors or kinases blocks plasticity. Plasticity signatures were created based on the discriminant analysis result. Thus, the spine signature (right column) is the normalized sum of PKC activity and PKA phosphorylated proteins in the spine, and the dendrite signature (left column) is the normalized sum of Epac1 and phosphorylated CamKII minus 2AG. (A) response to 20 Hz stimulation, (B) response to theta burst stimulation. The effect of antagonists is to reduce either the dendrite or spine signature compared to control, both for theta burst and 20 Hz stimulation protocols.
The stochastic reaction-diffusion model used here contains several 100 parameters, some of which are not directly measured but inferred to fit experimental data. To gain additional insight we carried out local sensitivity analysis which facilitated identifying the sensitive parameters. To achieve this, we first re-implemented the model reactions in a deterministic, single compartment model. We assumed the same total amounts of the molecules, similar reaction kinetics, and typically we used the averaged input signals from the NeuroRD model. Fig 7A shows an example using activated PKC, which is comparable to Fig 4B. The output was compared with the average behavior of the NeuroRD model. The single compartment SimBiology model responses correlated with the NeuroRD model for both TBS and 20 Hz, but of course did not capture differences due to location of the molecules and also the different input signals, for instance calcium experienced by these molecules in the spine versus the dendrite. As comparisons of the changes in the signature molecules for 20Hz and theta burst are the most important, we normalized the output by first estimating the area under the curve (AUC) and then compared the ratio of the response for TBS to 20 Hz for the signature molecules (Fig 7B). Then we perturbed the parameters one at a time by ±10%, repeated the simulations, and again looked at the same target molecules for 20 Hz and TBS. A large majority of the 10% perturbations produced a much smaller effect in the target molecules. However, there are a few parameters that if varied changed the output over 10%. Fig 7C and D illustrates this for 20Hz and TBS respectively.
Figure 7.

One compartment simulations in SimBiology demonstrates robustness to parameter changes. (A) The dynamical changes in the active PKC concentration during the course of simulation performed in NeuroRD (here the averaged values in spine and dendrite shown) and SimBiology show good agreement. (B) The area under the curve (AUC) for molecule activations are estimated for TBS and 20 Hz, and then the ratios of molecule activation for TBS relative to 20Hz are calculated. These ratios are quite similar for NeuroRD and SimBiology simulations. (C, D): Principal sensitive parameters affecting the signature molecules for 20Hz (C) and TBS (D). Y-axis represents the fractional response change for the indicated molecules due to each respective parameter. The following parameter naming convention is used for the x-axis: Kcat_S1xS2 is catalytic rate of the enzyme S1 on substrate S2; kf_S1xS2 is association rate constant (kf) between two molecules S1 and S2; kb_S1xS2 is dissociation rate constant (kb) between two molecules S1 and S2.
Spatial patterns of stimulation produce spatially specific LTP and heterosynaptic LTD
To evaluate the spatial specificity of synaptic plasticity, the morphology was extended into a 20 μm long dendrite with 10 randomly placed spines (Fig 1C). We stimulate two spines, either 1 and 2 (adjacent) or 1 and 7 (separated). To evaluate spine specificity, we compare the molecule concentrations (PKA plus PKC – spine molecules identified in the stepwise discriminant analysis) of spines 2 and 7 for both the adjacent and separated stimulation condition. Comparing the response of spine 2 with spine 7, tests for cooperative effect of having a stimulated spine adjacent or further away.
Fig 8A shows that spine signature exhibits spatial specificity: the spine signature is higher when the spine is stimulated for both spines 2 and 7, though only for theta burst stimulation. In addition, the spines exhibit a small degree of cooperativity: the spine signature is slightly higher when an adjacent spine is simulated than when a distant spine is stimulated, again only for the theta burst stimulation. In all cases, the spine signature was greater for TBS (left) than for 20Hz (right). The spine signature does not differ between stimulated and non-stimulated spines for 20 Hz stimulation, suggesting a lack of spatial specificity at this spatial scale for LTD.
Figure 8.

Simulations in a long dendrite with multiple spines reveals spine spatial specificity for TBS but not for 20 Hz. In all simulations, spine 0 and either spine 1 or spine 6 are stimulated. (A) Spine signature (normalized sum of PKC activity and PKA phosphorylated proteins) reveal spatial specificity for theta burst stimulation, but not 20 Hz stimulation. Spine 1 is stimulated for the adjacent (adj) case and spine 6 is stimulated for the separated (sep) case. Only plots of spine 1 (black and gray traces) and spine 6 (red and pink traces) are shown, with the dark traces indicating the stimulated condition, and the lighter traces indicating the non-stimulated condition. (B) Dendritic signature differs between TBS and 20 Hz, but does not exhibit spatial specificity. Dendritic signature is calculated as normalized phosphoCamKII plus Epac1 minus 2AG. B1: two adjacent spines stimulated, B2: two separated spines stimulated.
To evaluate spatial and temporal specificity in the dendrite, we plotted dendritic molecules used: Epac1+CamKII – 2AG, since CamKII contribute to LTP whereas 2AG is needed for LTD. Figure 8B, C the dendritic signature is greater for TBS than for 20Hz, similar to the effect of temporal pattern in the 1 spine case. In addition, comparison of Fig 8B with 8C shows a lack of spatial specificity at this scale, as the dendritic signature for both cases (spines separated or spines adjacent) do not differ. Overall, these results suggest that LTP changes in the spine will exhibit spatial specificity; whereas LTD will not exhibit spatial specificity on this spatial scale. In addition, the signature for non-stimulated spines during theta burst stimulation resembles that off 20 Hz stimulation suggesting that heterosynaptic LTD may occur during theta burst stimulation.
Effect of alcohol is to enhance LTP and reduce LTD
A troubling aspect of alcohol use disorder is the tendency to relapse in response to cues previously associated with alcohol ingestion. The convergence of cue-related cortical inputs with drug-related dopamine inputs in the striatum during alcohol taking functions as a conditioned reinforcer and results in aberrant synaptic plasticity. After chronic alcohol exposure, cue-induced reinstatement of alcohol seeking or other cognitive deficits contributing to relapse may be caused by this aberrant synaptic plasticity (as well as other mechanisms).
To evaluate whether the molecular changes of chronic alcohol enhance development of plasticity of cortical-striatal synapses, we simulated partial TBS and 20Hz stimulations protocols (4 instead of 10 theta burst trains and 8 instead of 20 trains of 20 Hz stimulation), while continuing to keep number of pulses equivalent for protocols. The glutamatergic inputs of these stimulation paradigms are a surrogate for the drug-paired cues. The reduced number of trains represents the very few (i.e. only one) cues needed for reinstatement, and also prevent a ceiling effect in detecting an increase in signaling molecules. Predictions for these simulations were made by using time samples from the corresponding simulations as test data for the original discriminant analysis.
Figure 9 shows that molecules predictive of plasticity are enhanced in the chronic alcohol model compared to the control model. The discriminant analysis predicted both TBS and 20 Hz would produce LTD in the control model, and LTP in the chronic alcohol model. Additional simulations evaluated which molecular changes contributed most to the change in plasticity. Table 3 lists the predictions from the control and chronic alcohol simulations together with the predictions from changes in NMDA alone or changes in G protein signaling (mGluR, Gprot and AC) alone. Changes in NMDA alone are not sufficient to account for LTP in response to the reduced stimulation, whereas changes in G protein signaling give the same predictions as the full chronic alcohol model.
Figure 9.

Effect of alcohol withdrawal on synaptic plasticity. Arrows show a change in molecule concentration from that predicting LTD to that predicting LTP.
Discussion
We created a unified model of signaling pathways of striatal, direct pathway (dopamine D1 receptor-containing) spiny projection neurons to investigate the control of synaptic plasticity by spatio-temporal patterns of stimulation. Model parameters were constrained by a large and diverse set of data to provide credibility and robustness to the results. We simulated a set of ten LTP and LTD stimulation protocols with known outcomes, including stimulation in the presence of antagonists of various receptors or kinases, and were able to account for the plasticity results of all protocols. We evaluated molecule concentrations during the first few minutes after stimulation because experiments in other brain regions have revealed a short temporal window for induction of synaptic plasticity (Murakoshi et al 2017; Ronesi et al 2004). Most prior models have investigated only a small subset of different protocols; thus the ability of our model to account for both STDP and frequency dependent protocols, and many pharmacological agents, strengthens the model predictions. The unique, spatial aspects of the model enabled evaluation of spatial specificity and heterosynaptic plasticity.
The use of a spatial model permitted inspection of molecule concentrations separately in the spine and in the dendrite. Molecules in these different spatial compartments may serve different roles during induction of synaptic plasticity, analogous to synaptic tagging and capture that has been proposed in the hippocampus (Frey and Morris 1997; Redondo and Morris 2011). Kinases in the spine may “tag” specific spines as the ones to undergo long term plasticity, via changes in spine volume or receptor insertion (Okamoto et al 2004); whereas kinases in the dendrite initiate synthesis of proteins, either locally or by sending a signal to the soma (Redondo and Morris 2011). The newly synthesized proteins (which lack spatial specificity) are then “captured” by the spatially specific, tagged spines to convert the short term changes into long term changes.
Many of the molecules used by the model to predict plasticity have been implicated in the processes needed for synaptic tagging or capture. Tagging is thought to involve re-organization of the actin cytoskeleton (Ramachandran and Frey 2009; Redondo and Morris 2011), which requires activation of cofilin for depolymerization, followed by inactivation of cofilin to facilitate re-polymerization (Gu et al 2010). Both cofilin and several other actin interacting proteins are regulated by PKA (Lamprecht and Ledoux 2004; Nadella et al 2009), CamKII (Araki et al 2015; Hoffman et al 2013) and Epac1 (Penzes and Cahill 2012; Penzes et al 2011). The capture part of synaptic tagging implies that new proteins are synthesized as part of LTP induction. Translation (protein synthesis) and transcription typically require extracellular signal regulated kinase (ERK) activation, which is critical for most forms of synaptic plasticity (Hawes et al 2013; Kelleher, et al 2004; Shalin et al 2006; Shiflett and Balleine 2011; Valjent et al 2006). Both PKA and Epac1 are key signaling molecules activated by cAMP that converge on ERK (Gelinas et al 2007; McAvoy et al 2009; Vossler et al 1997). PKC also contributes to ERK activation via phosphorylation of kinases that activate ERK (Bouschet et al 2003; Shalin et al 2006). CamKII has been implicated in the regulation of protein synthesis via phosphorylation of cytoplasmic polyadenylation element binding protein (Atkins et al 2005). Together, control of actin and protein translation provide downstream mechanisms that are consistent with the results.
Whereas most of the molecules act post-synaptically to produce LTP, both 2AG and anandamide act pre-synaptically to produce LTD (Gerdeman et al 2002; Ronesi et al 2004) (Lerner and Kreitzer 2012). Both of these endocannabinoids are produced post-synaptically in response to elevations in calcium and metabotropic glutamate receptor activation (though the Gq coupled muscarinic m1 receptors may also contribute), with anandamide implicated in 100Hz LTD (Ade and Lovinger 2007) and 2AG implicated in 20 Hz LTD (Lerner and Kreitzer 2012). Calcium is required not only to produce diacylglycerol but also either for diacylglycerol lipase activity or for exporting 2AG from the post-synaptic terminal (Shonesy et al 2015). Then, 2AG diffuses to the pre-synaptically located CB1 receptors to induce LTD through a decrease in neurotransmitter release (Atwood et al 2014; Shonesy et al 2015). A previous model (Kim et al 2013) demonstrated that 2AG is produced even during LTP induction protocols. That model did not include the more recently discovered inhibition of 2AG production by CamKII (Shonesy et al 2013). Including this pathway interaction resulted in suppression of 2AG production during the theta burst stimulation; however, the brief 2AG elevation may still be sufficient to induce hetero-synaptic LTD (Ronesi et al 2004).
Two types of spatial specificity were exhibited by the model. The concentration of molecules in the spine differed from the dendrite; and in the simulations of the long dendrite with multiple spines, molecule concentration differed between stimulated and non-stimulated spines. PKC exhibits strong spatial specificity because it is activated by binding to the membrane associated diacylglycerol (Oancea and Meyer 1998; Olds et al 1989). The spatial specificity of PKC resulted in spatial specificity of LTP; however, two other molecules implicated in LTP: PKA and CamKII, exhibited smaller gradients despite the low rate of diffusion measured experimentally (Lu et al 2014; Tillo et al 2017). The lack of gradients could be due to the stimulation protocols used for synaptic plasticity induction: spatially non-specific dopamine (Dreyer et al 2010) or calcium influx through dendritic voltage gated calcium channels (Higley and Sabatini 2010) can elevate PKA and CamKII in the dendrites. Alternatively, the lack of gradients could indicate the need to add additional mechanisms for spatially delimiting molecule activity, such as CamKII anchoring to NMDA receptors (Bayer et al 2001), or inactivation of diffusing PKA catalytic subunits by excess PKA regulatory subunit (Walker-Gray et al 2017). Not all molecules exhibited spatial specificity: the molecules that diffused freely, such as 2AG and Epac1, did not exhibit a spatial gradient. The lack of gradient for 2AG is consistent with experiments in other brain regions showing that endocannabindoids can affects synapses ~20 microns away from the site of generation (Wilson and Nicoll 2001), and suggests that LTD will not exhibit spatial specificity at the spatial resolution of our simulations. If the brief elevation of 2AG observed with TBS is sufficient to produce LTD, this would suggest that TBS LTP is accompanied by heterosynaptic LTD, and suggests a mechanism underlying homeostatic scaling (Turrigiano 2012).
There are several caveats to the present study. Ideally an experimental test of some of the predictions are required to better demonstrate the validity of the model, for example, testing the CaMKII dependence of theta burst LTP. The recent demonstration of CamKII dependence for other plasticity protocols (Cui et al 2016) suggest that this prediction is likely to be confirmed. In the absence of experimental validation, the plasticity outcomes predicted by the discriminant analysis are quite logical. For example, the prediction that enhanced (but not reduced) cAMP signaling would block 20 Hz LTD is consistent with results from 100Hz LTD experiments (Augustin et al 2014). Similarly, PKA is required for most forms of striatal LTP (Hawes et al 2013; Spencer and Murphy 2002), and thus blocking m4R should facilitate LTP, as predicted previously (Nair et al 2015). The one limitation of the model is that it did not show an LTD deficit when mGluRs were blocked. This is partly due to strong contribution of m1R in the production as well as diffusion of 2AG. The observation of a significant decrease in spine diacylglycerol after block of mGluRs suggest that restricted 2AG diffusion may solve this model prediction. Another possibility is that m1Rs are restricted to the spine (Olson et al 2005) instead of distributed in the dendrites. The restricted spatial distribution would decrease the dominance of m1Rs contribution to 2AG production, and improve the model veracity. Yet another possibility, is that m1R activated signaling molecules contribute to LTP by targeting KCNQ channels to enhance cell excitability (Shen et al. 2005). In otherwords, if m1R in the model does not contribute to the “plasticity pool” of Gq – PLC signaling, then the mGluR activated pool of Gq would increase in importance. Testing the latter hypothesis requires an integrated electrical – biochemical model.
In general, the model is robust to variation in parameters, providing confidence in the model predictions; however, model results are somewhat sensitive to parameters related to receptors, such as receptor quantity. On the other hand, the sensitivity analysis only changed single parameters at a time, despite the fact that some processes are coupled. For example, changes in cAMP caused by increases in adenylyl cyclase can be compensated by increases in phosphodiesterase. Similarly, it is possible that changes in receptor quantity could be partly compensated by changes in neurotransmitter concentration. Thus, better measurements of both neurotransmitter quantity and receptor quantity are needed. FSCV has good accuracy and temporal sensitivity, but it is a measure of overflow (and not synaptic) dopamine. In addition, there is currently no good method for measuring ACh with good temporal specificty in an intact brain or brain slices.
The linear discriminant analysis is an automatic method for determining how each molecule contributes to the plasticity outcome. The initial, stepwise discriminant analysis selected a subset of molecule-time-space samples to determine the occurrence of LTP or LTD. Consistent with the role of dopamine D1R in LTP, the molecules activated by cAMP signaling pathways were selected only for the LTP determination. The linear aspect of this statistical method is a disadvantage, since it is unlikely that plasticity depends on a linear combination of molecule activity. We introduced a single non-linearity by combining the outputs of two linear discriminants; however, sophisticated and modern methods, such as random forest (Diaz-Uriarte and Alvarez de Andrés 2006; Maroco et al 2011), may give better results. On the other hand, all statistical analyses would suffer from the problem of how to sample the wave forms. The initial stepwise discriminant analysis to select a subset of time-space-molecule samples is one solution to this difficulty, though the utility of additional time samples cannot be ruled out. Perhaps the ideal approach is to evaluate the effect of model kinases on key downstream “hub” signaling molecules, such as ERK activation (Shiflett and Balleine 2011), which is influenced by Epac1, PKA and PKC.
A comprehensive understanding of the molecular mechanisms of striatal synaptic plasticity could be of use for understanding several clinically relevant conditions. Numerous studies (Goodman et al 2016; Gremel and Costa 2013; Hawes et al 2015; Shan et al 2014; Yin et al 2004) have demonstrated that synaptic plasticity of the striatum underlies habit learning, and that aberrant synaptic plasticity occurs following chronic drug or alcohol treatment in rodents and primates (DePoy et al 2013; Ma et al 2017; Shen et al 2011). In particular, the reinstatement of drug taking behavior on exposure to a single drug-paired cue may be a form of fast re-learning, caused by molecular changes observed in withdrawn animals (Chen et al 2011; Kashem et al 2012; Lucchi et al 1983; Ma et al 2017; May et al 1995; Nestby et al 1999; Wang et al 2010). Alternatively aberrant synaptic plasticity may partly contribute to alcohol seeking behavior. To evaluate whether a set of identified molecular changes could account for the aberrant synaptic plasticity observed with chronic alcohol, we evaluated the plasticity outcome while using a brief simulation protocol, which produces LTD in the control model. Simulations using the chronic alcohol treatment model show that both a brief train of 20 Hz stimulation and a brief train of TBS will produce LTP, and that both increased calcium through NMDA receptors and changes in G protein signaling contributed to this result. The inability to produce LTD is consistent with experimental studies (Adermark et al 2011; DePoy et al 2013; Xia et al 2006), whereas the enhancement of LTP is a model prediction, though partly supported by a recent demonstration of enhanced 100Hz LTP in an alcohol treated rodent (Ma et al 2018). Because pre-synaptic CB1 receptor signaling is downregulated by ethanol abuse (DePoy et al 2013), a more sophisticated analysis of the model output would need to take into account the decreased effectiveness of 2AG for making predictions. Nonetheless, experimental confirmation of these model predictions would suggest that the withdrawn model could be a test bed to determine the key molecular mechanisms to target for preventing relapse.
Acknowledgments
This work was supported by ONR grant DURIP N00014-13-1-0745 and through the joint NIH-NSF CRCNS program through NIAAA grant R01AA16020. PT and JHK were additionally funded by the European Horizon 2020 Framework Programme under grant agreement no 720270 (Human Brain Project SGA1), the Swedish Research Council, and the Swedish e-Science Research Center. The authors declare that they have no conflict of interest.
Abbreviations
- 2AG
2 arachidonylgycerol
- ATP
adenosine triphosphate
- CamKII
calmodulin dependent protein kinase type 2
- cAMP
cyclic adenosine monophosphate,
- CB1R
endocannbinoid receptor type 1
- DAG
diacylgyclerol
- DARPP32
dopamine- and cAMP-regulated neuronal phosphoprotein
- D1R
Dopamine D1 receptors
- Epac
Exchange Protein Activated by cAMP
- ERK
extracellular signal regulated kinase
- GiGTP
alpha subunit of Gi (inhibitory) subtype of GTP binding protein
- GqGTP
alpha subunit of Gq (phospholipase C coupled) subtype of GTP binding protein
- GolfGTP
alpha subunit of Golf (stimulatory) subtype of GTP binding protein
- GTP
guanosine triphosphate
- LTP
long term potentiation
- LTD
long term depression
- mGluR
metabotropic glutamate receptors
- NMDA
N-methyl D-aspartic acid
- PKA
protein kinase A
- PKC
protein Kinase C
- PP1
protein phosphatase type 1
- PP2A
protein phosphatase type 2A
- PP2B
calcineurin
- TBS
theta burst stimulation
- Thr
Threonine
- VDCC
voltage dependent calcium channel
Footnotes
Conflict of Interest Statement: The authors declare no conflict of interest
Author Contributions:
NeuroRDViz software: BE
Voltammetry experiments AGS
Sensitivity analysis: PT and JHK
Model creation: KTB, PT, JHK
Model simulation and analysis: KTB
Prepared manuscript, AGS, PT, JHK, KTB, DML
Data Accessibility:All model files and analysis code are freely available (https://github.com/neurord/D1pathways). The simplified model will be deposited in BioModels database in SBML format following acceptance of this study.
Reference List
- Abrahao KP, Salinas AG, Lovinger DM. Alcohol and the Brain: Neuronal Molecular Targets, Synapses, and Circuits. Neuron. 2017;96:1223–1238. doi: 10.1016/j.neuron.2017.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ade KK, Lovinger DM. Anandamide regulates postnatal development of long-term synaptic plasticity in the rat dorsolateral striatum. J Neurosci. 2007;27:2403–2409. doi: 10.1523/JNEUROSCI.2916-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adermark L, Jonsson S, Ericson M, Soderpalm B. Intermittent ethanol consumption depresses endocannabinoid-signaling in the dorsolateral striatum of rat. Neuropharmacology. 2011;61:1160–1165. doi: 10.1016/j.neuropharm.2011.01.014. [DOI] [PubMed] [Google Scholar]
- Anderson JB, Anderson LE, Kussmann J. Monte Carlo simulations of single- and multistep enzyme-catalyzed reaction sequences: effects of diffusion, cell size, enzyme fluctuations, colocalization, and segregation. J Chem Phys. 2010;133:034104. doi: 10.1063/1.3459111. [DOI] [PubMed] [Google Scholar]
- Araki Y, Zeng M, Zhang M, Huganir RL. Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron. 2015;85:173–189. doi: 10.1016/j.neuron.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Davare MA, Oh MC, Derkach V, Soderling TR. Bidirectional regulation of cytoplasmic polyadenylation element-binding protein phosphorylation by Ca2+/calmodulin-dependent protein kinase II and protein phosphatase 1 during hippocampal long-term potentiation. J Neurosci. 2005;25:5604–5610. doi: 10.1523/JNEUROSCI.5051-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Lovinger DM, Mathur BN. Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends Neurosci. 2014;37:663–673. doi: 10.1016/j.tins.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin SM, Beeler JA, McGehee DS, Zhuang X. Cyclic AMP and afferent activity govern bidirectional synaptic plasticity in striatopallidal neurons. J Neurosci. 2014;34:6692–6699. doi: 10.1523/JNEUROSCI.3906-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Hochner B, Mahaut-Smith M, Adams SR, Kaang BK, Kandel ER, Tsien RY. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 1993;260:222–226. doi: 10.1126/science.7682336. [DOI] [PubMed] [Google Scholar]
- Barker JM, Corbit LH, Robinson DL, Gremel CM, Gonzales RA, Chandler LJ. Corticostriatal circuitry and habitual ethanol seeking. Alcohol. 2015;49:817–824. doi: 10.1016/j.alcohol.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Callaway JC, Wilson CJ. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci. 2000;20:8493–8503. doi: 10.1523/JNEUROSCI.20-22-08493.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouschet T, Perez V, Fernandez C, Bockaert J, Eychene A, Journot L. Stimulation of the ERK pathway by GTP-loaded Rap1 requires the concomitant activation of Ras, protein kinase C, and protein kinase A in neuronal cells. J Biol Chem. 2003;278:4778–4785. doi: 10.1074/jbc.M204652200. [DOI] [PubMed] [Google Scholar]
- Bowers MS, Hopf FW, Chou JK, Guillory AM, Chang SJ, Janak PH, Bonci A, Diamond I. Nucleus accumbens AGS3 expression drives ethanol seeking through G betagamma. Proc Natl Acad Sci USA. 2008;105:12533–12538. doi: 10.1073/pnas.0706999105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Chelliah V, Juty N, Ajmera I, Ali R, Dumousseau M, Glont M, Hucka M, Jalowicki G, Keating S, Knight-Schrijver V, Lloret-Villas A, Natarajan KN, Pettit JB, Rodriguez N, Schubert M, Wimalaratne SM, Zhao Y, Hermjakob H, Le NN, Laibe C. BioModels: ten-year anniversary. Nucleic Acids Res. 2015;43:D542–D548. doi: 10.1093/nar/gku1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Cuzon Carlson VC, Wang J, Beck A, Heinz A, Ron D, Lovinger DM, Buck KJ. Striatal involvement in human alcoholism and alcohol consumption, and withdrawal in animal models. Alcohol Clin Exp Res. 2011;35:1739–1748. doi: 10.1111/j.1530-0277.2011.01520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccocioppo R, Angeletti S, Weiss F. Long-lasting resistance to extinction of response reinstatement induced by ethanol-related stimuli: role of genetic ethanol preference. Alcohol Clin Exp Res. 2001;25:1414–1419. doi: 10.1097/00000374-200110000-00002. [DOI] [PubMed] [Google Scholar]
- Crowley NA, Cody PA, Davis MI, Lovinger DM, Mateo Y. Chronic methylphenidate exposure during adolescence reduces striatal synaptic responses to ethanol. Eur J Neurosci. 2014;39:548–556. doi: 10.1111/ejn.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Prokin I, Xu H, Delord B, Genet S, Venance L, Berry H. Endocannabinoid dynamics gate spike-timing dependent depression and potentiation. Elife. 2016;5:e13185. doi: 10.7554/eLife.13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePoy L, Daut R, Brigman JL, MacPherson K, Crowley N, Gunduz-Cinar O, Pickens CL, Cinar R, Saksida LM, Kunos G, Lovinger DM, Bussey TJ, Camp MC, Holmes A. Chronic alcohol produces neuroadaptations to prime dorsal striatal learning. Proc Natl Acad Sci USA. 2013;110:14783–14788. doi: 10.1073/pnas.1308198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devan BD, McDonald RJ, White NM. Effects of medial and lateral caudate-putamen lesions on place- and cue-guided behaviors in the water maze: relation to the thigmotaxis. Behavioral Brain Research. 1999;100:5–14. doi: 10.1016/s0166-4328(98)00107-7. [DOI] [PubMed] [Google Scholar]
- Diaz-Uriarte R, Alvarez de Andrés AS. Gene selection and classification of microarray data using random forest. BMC Bioinformatics. 2006;7:3. doi: 10.1186/1471-2105-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer JK, Herrik KF, Berg RW, Hounsgaard JD. Influence of phasic and tonic dopamine release on receptor activation. J Neurosci. 2010;30:14273–14283. doi: 10.1523/JNEUROSCI.1894-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM, Venance L. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol. 2010;588:3045–3062. doi: 10.1113/jphysiol.2010.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Morris RGM. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Gelinas JN, Banko JL, Hou L, Sonenberg N, Weeber EJ, Klann E, Nguyen PV. ERK and mTOR signaling couple beta-adrenergic receptors to translation initiation machinery to gate induction of protein synthesis-dependent long-term potentiation. J Biol Chem. 2007;282:27527–27535. doi: 10.1074/jbc.M701077200. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Reynolds JN. Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience. 2011;198:27–43. doi: 10.1016/j.neuroscience.2011.08.067. [DOI] [PubMed] [Google Scholar]
- Goodman J, Ressler RL, Packard MG. The dorsolateral striatum selectively mediates extinction of habit memory. Neurobiol Learn Mem. 2016;136:54–62. doi: 10.1016/j.nlm.2016.09.012. [DOI] [PubMed] [Google Scholar]
- Gremel CM, Costa RM. Orbitofrontal and striatal circuits dynamically encode the shift between goal-directed and habitual actions. Nat Commun. 2013;4:2264. doi: 10.1038/ncomms3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremel CM, Lovinger DM. Associative and sensorimotor cortico-basal ganglia circuit roles in effects of abused drugs. Genes Brain Behav. 2016 doi: 10.1111/gbb.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grima R. Investigating the robustness of the classical enzyme kinetic equations in small intracellular compartments. BMC Syst Biol. 2009;3:101. doi: 10.1186/1752-0509-3-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, Zheng JQ. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci. 2010;13:1208–1215. doi: 10.1038/nn.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Arenas O, Eriksson O, Kotaleski JH. Segregation and crosstalk of D1 receptor-mediated activation of ERK in striatal medium spiny neurons upon acute administration of psychostimulants. PLoS Comput Biol. 2014;10:e1003445. doi: 10.1371/journal.pcbi.1003445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes SL, Evans RC, Unruh BA, Benkert EE, Gillani F, Dumas TC, Blackwell KT. Multimodal Plasticity in Dorsal Striatum While Learning a Lateralized Navigation Task. J Neurosci. 2015;35:10535–10549. doi: 10.1523/JNEUROSCI.4415-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes SL, Gillani F, Evans RC, Benkert EA, Blackwell KT. Sensitivity to theta-burst timing permits LTP in dorsal striatal adult brain slice. J Neurophysiol. 2013;110:2027–2036. doi: 10.1152/jn.00115.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz A, Beck A, Grusser SM, Grace AA, Wrase J. Identifying the neural circuitry of alcohol craving and relapse vulnerability. Addict Biol. 2009;14:108–118. doi: 10.1111/j.1369-1600.2008.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Her ES, Huh N, Kim J, Jung MW. Neuronal activity in dorsomedial and dorsolateral striatum under the requirement for temporal credit assignment. Sci Rep. 2016;6:27056. doi: 10.1038/srep27056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–966. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirth N, Meinhardt MW, Noori HR, Salgado H, Torres-Ramirez O, Uhrig S, Broccoli L, Vengeliene V, Rossmanith M, Perreau-Lenz S, Kohr G, Sommer WH, Spanagel R, Hansson AC. Convergent evidence from alcohol-dependent humans and rats for a hyperdopaminergic state in protracted abstinence. Proc Natl Acad Sci USA. 2016;113:3024–3029. doi: 10.1073/pnas.1506012113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman L, Farley MM, Waxham MN. Calcium-calmodulin-dependent protein kinase II isoforms differentially impact the dynamics and structure of the actin cytoskeleton. Biochemistry. 2013;52:1198–1207. doi: 10.1021/bi3016586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrzejewska-Szmek J, Damodaran S, Dorman DB, Blackwell KT. Calcium dynamics predict direction of synaptic plasticity in striatal spiny projection neurons. Eur J Neurosci. 2017;45:1044–1056. doi: 10.1111/ejn.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrzejewski-Szmek Z, Blackwell KT. Asynchronous tau-leaping. J Chem Phys. 2016;144:125104. doi: 10.1063/1.4944575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashem MA, Ahmed S, Sarker R, Ahmed EU, Hargreaves GA, McGregor IS. Long-term daily access to alcohol alters dopamine-related synthesis and signaling proteins in the rat striatum. Neurochem Int. 2012;61:1280–1288. doi: 10.1016/j.neuint.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Kim B, Hawes SL, Gillani F, Wallace LJ, Blackwell KT. Signaling Pathways Involved in Striatal Synaptic Plasticity are Sensitive to Temporal Pattern and Exhibit Spatial Specificity. PLoS Comput Biol. 2013;9:e1002953. doi: 10.1371/journal.pcbi.1002953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R, Ledoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. RGS4 Is Required for Dopaminergic Control of Striatal LTD and Susceptibility to Parkinsonian Motor Deficits. Neuron. 2012;73:347–359. doi: 10.1016/j.neuron.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HE, MacGillavry HD, Frost NA, Blanpied TA. Multiple spatial and kinetic subpopulations of CaMKII in spines and dendrites as resolved by single-molecule tracking PALM. J Neurosci. 2014;34:7600–7610. doi: 10.1523/JNEUROSCI.4364-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchi L, Covelli V, Anthopoulou H, Spano PF, Trabucchi M. Effect of chronic ethanol treatment on adenylate cyclase activity in rat striatum. Neurosci Lett. 1983;40:187–192. doi: 10.1016/0304-3940(83)90300-2. [DOI] [PubMed] [Google Scholar]
- Ma T, Barbee B, Wang X, Wang J. Alcohol induces input-specific aberrant synaptic plasticity in the rat dorsomedial striatum. Neuropharmacology. 2017;123:46–54. doi: 10.1016/j.neuropharm.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Cheng Y, Roltsch HE, Wang X, Lu J, Gao X, Huang CCY, Wei XY, Ji JY, Wang J. Bidirectional and long-lasting control of alcohol-seeking behavior by corticostriatal LTP and LTD. Nat Neurosci. 2018 doi: 10.1038/s41593-018-0081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamaligas AA, Cai Y, Ford CP. Nicotinic and opioid receptor regulation of striatal dopamine D2-receptor mediated transmission. Sci Rep. 2016;6:37834. doi: 10.1038/srep37834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroco J, Silva D, Rodrigues A, Guerreiro M, Santana I, de MA. Data mining methods in the prediction of Dementia: A real-data comparison of the accuracy, sensitivity and specificity of linear discriminant analysis, logistic regression, neural networks, support vector machines, classification trees and random forests. BMC Res Notes. 2011;4:299. doi: 10.1186/1756-0500-4-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May T, Wolf U, Wolffgramm J. Striatal dopamine receptors and adenylyl cyclase activity in a rat model of alcohol addiction: effects of ethanol and lisuride treatment. J Pharmacol Exp Ther. 1995;275:1195–1203. [PubMed] [Google Scholar]
- McAvoy T, Zhou MM, Greengard P, Nairn AC. Phosphorylation of Rap1GAP, a striatally enriched protein, by protein kinase A controls Rap1 activity and dendritic spine morphology. Proc Natl Acad Sci USA. 2009;106:3531–3536. doi: 10.1073/pnas.0813263106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhardt MW, Hansson AC, Perreau-Lenz S, Bauder-Wenz C, Stahlin O, Heilig M, Harper C, Drescher KU, Spanagel R, Sommer WH. Rescue of infralimbic mGluR2 deficit restores control over drug-seeking behavior in alcohol dependence. J Neurosci. 2013;33:2794–2806. doi: 10.1523/JNEUROSCI.4062-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Skorova E, Taschenberger G, Hartelt N, Nikolaev VO, Lohse MJ, Kugler S. Imaging cytoplasmic cAMP in mouse brainstem neurons. BMC Neurosci. 2009;10:29. doi: 10.1186/1471-2202-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H. Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron. 2004;43:133–143. doi: 10.1016/j.neuron.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Murakoshi H, Shin ME, Parra-Bueno P, Szatmari EM, Shibata AC, Yasuda R. Kinetics of Endogenous CaMKII Required for Synaptic Plasticity Revealed by Optogenetic Kinase Inhibitor. Neuron. 2017;94:37–47. doi: 10.1016/j.neuron.2017.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadella KS, Saji M, Jacob NK, Pavel E, Ringel MD, Kirschner LS. Regulation of actin function by protein kinase A-mediated phosphorylation of Limk1. EMBO Rep. 2009;10:599–605. doi: 10.1038/embor.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair AG, Bhalla US, Hellgren KJ. Role of DARPP-32 and ARPP-21 in the Emergence of Temporal Constraints on Striatal Calcium and Dopamine Integration. PLoS Comput Biol. 2016;12:e1005080. doi: 10.1371/journal.pcbi.1005080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair AG, Gutierrez-Arenas O, Eriksson O, Vincent P, Hellgren KJ. Sensing Positive versus Negative Reward Signals through Adenylyl Cyclase-Coupled GPCRs in Direct and Indirect Pathway Striatal Medium Spiny Neurons. J Neurosci. 2015;35:14017–14030. doi: 10.1523/JNEUROSCI.0730-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestby P, Vanderschuren LJ, De Vries TJ, Mulder AH, Wardeh G, Hogenboom F, Schoffelmeer AN. Unrestricted free-choice ethanol self-administration in rats causes long-term neuroadaptations in the nucleus accumbens and caudate putamen. Psychopharmacology (Berl) 1999;141:307–314. doi: 10.1007/s002130050838. [DOI] [PubMed] [Google Scholar]
- Nic Dhonnchadha BA, Kantak KM. Cognitive enhancers for facilitating drug cue extinction: insights from animal models. Pharmacol Biochem Behav. 2011;99:229–244. doi: 10.1016/j.pbb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Bibb JA, Matsuyama S, Hamada M, Higashi H, Nairn AC, Greengard P. Regulation of DARPP-32 dephosphorylation at PKA- and Cdk5-sites by NMDA and AMPA receptors: distinct roles of calcineurin and protein phosphatase-2A. J Neurochem. 2002;81:832–841. doi: 10.1046/j.1471-4159.2002.00876.x. [DOI] [PubMed] [Google Scholar]
- Nishi A, Kuroiwa M, Miller DB, O’Callaghan JP, Bateup HS, Shuto T, Sotogaku N, Fukuda T, Heintz N, Greengard P, Snyder GL. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J Neurosci. 2008;28:10460–10471. doi: 10.1523/JNEUROSCI.2518-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Nairn AC, Greengard P. Role of calcineurin and protein phosphatase-2A in the regulation of DARPP-32 dephosphorylation in neostriatal neurons. J Neurochem. 1999;72:2015–2021. doi: 10.1046/j.1471-4159.1999.0722015.x. [DOI] [PubMed] [Google Scholar]
- O’Hare JK, Ade KK, Sukharnikova T, Van Hooser SD, Palmeri ML, Yin HH, Calakos N. Pathway-Specific Striatal Substrates for Habitual Behavior. Neuron. 2016;89:472–479. doi: 10.1016/j.neuron.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oancea E, Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- Obara I, Bell RL, Goulding SP, Reyes CM, Larson LA, Ary AW, Truitt WA, Szumlinski KK. Differential effects of chronic ethanol consumption and withdrawal on homer/glutamate receptor expression in subregions of the accumbens and amygdala of P rats. Alcohol Clin Exp Res. 2009;33:1924–1934. doi: 10.1111/j.1530-0277.2009.01030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Olds JL, Anderson ML, McPhie DL, Staten LD, Alkon DL. Imaging of memory-specific changes in the distribution of protein kinase C in the hippocampus. Science. 1989;245:866–8699. doi: 10.1126/science.2772638. [DOI] [PubMed] [Google Scholar]
- Oliveira RF, Kim M, Blackwell KT. Subcellular Location of PKA Controls Striatal Plasticity: Stochastic Simulations in Spiny Dendrites. PLoS Comput Biol. 2012;8:e1002383. doi: 10.1371/journal.pcbi.1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci. 2005;25:1050–1062. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paille V, Fino E, Du K, Morera-Herreras T, Perez S, Kotaleski JH, Venance L. GABAergic circuits control spike-timing-dependent plasticity. J Neurosci. 2013;33:9353–9363. doi: 10.1523/JNEUROSCI.5796-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak V, Wickens JR, Kirkwood A, Kerr JN. Timing is not Everything: Neuromodulation Opens the STDP Gate. Front Synaptic Neurosci. 2010;2:146. doi: 10.3389/fnsyn.2010.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME. Deconstructing signal transduction pathways that regulate the actin cytoskeleton in dendritic spines. Cytoskeleton (Hoboken) 2012;69:426–441. doi: 10.1002/cm.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Woolfrey KM, Srivastava DP. Epac2-mediated dendritic spine remodeling: implications for disease. Mol Cell Neurosci. 2011;46:368–380. doi: 10.1016/j.mcn.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran B, Frey JU. Interfering with the actin network and its effect on long-term potentiation and synaptic tagging in hippocampal CA1 neurons in slices in vitro. J Neurosci. 2009;29:12167–12173. doi: 10.1523/JNEUROSCI.2045-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo RL, Morris RG. Making memories last: the synaptic tagging and capture hypothesis. Nat Rev Neurosci. 2011;12:17–30. doi: 10.1038/nrn2963. [DOI] [PubMed] [Google Scholar]
- Reynolds JN, Wickens JR. Dopamine-dependent plasticity of corticostriatal synapses. Neural Netw. 2002;15:507–521. doi: 10.1016/s0893-6080(02)00045-x. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci. 2004;7:583–584. doi: 10.1038/nn1244. [DOI] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci. 2004;24:1673–1679. doi: 10.1523/JNEUROSCI.5214-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi J, Lovinger DM. Induction of striatal long-term synaptic depression by moderate frequency activation of cortical afferents in rat. J Physiol. 2005;562:245–256. doi: 10.1113/jphysiol.2004.068460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas AG, Davis MI, Lovinger DM, Mateo Y. Dopamine dynamics and cocaine sensitivity differ between striosome and matrix compartments of the striatum. Neuropharmacology. 2016;108:275–283. doi: 10.1016/j.neuropharm.2016.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo D, Sinha R. The neurobiology of alcohol craving and relapse. Handb Clin Neurol. 2014;125:355–368. doi: 10.1016/B978-0-444-62619-6.00021-5. [DOI] [PubMed] [Google Scholar]
- Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD. Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron. 2006;50:765–779. doi: 10.1016/j.neuron.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Shan Q, Ge M, Christie MJ, Balleine BW. The acquisition of goal-directed actions generates opposing plasticity in direct and indirect pathways in dorsomedial striatum. J Neurosci. 2014;34:9196–9201. doi: 10.1523/JNEUROSCI.0313-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hamilton SE, Nathanson NM, Surmeier DJ. Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci. 2005;25:7449–7458. doi: 10.1523/JNEUROSCI.1381-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Moussawi K, Zhou W, Toda S, Kalivas PW. Heroin relapse requires long-term potentiation-like plasticity mediated by NMDA2b-containing receptors. Proc Natl Acad Sci USA. 2011;108:19407–19412. doi: 10.1073/pnas.1112052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiflett MW, Balleine BW. Contributions of ERK signaling in the striatum to instrumental learning and performance. Behav Brain Res. 2011;218:240–247. doi: 10.1016/j.bbr.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindou T, Ochi-Shindou M, Wickens JR. A Ca(2+) threshold for induction of spike-timing-dependent depression in the mouse striatum. J Neurosci. 2011;31:13015–13022. doi: 10.1523/JNEUROSCI.3206-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Wang X, Rose KL, Ramikie TS, Cavener VS, Rentz T, Baucum AJ, Jalan-Sakrikar N, Mackie K, Winder DG, Patel S, Colbran RJ. CaMKII regulates diacylglycerol lipase-alpha and striatal endocannabinoid signaling. Nat Neurosci. 2013;16:456–463. doi: 10.1038/nn.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Winder DG, Patel S, Colbran RJ. The initiation of synaptic 2-AG mobilization requires both an increased supply of diacylglycerol precursor and increased postsynaptic calcium. Neuropharmacology. 2015;91:57–62. doi: 10.1016/j.neuropharm.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano CA, Locke JL, Mathews TA, Lopez MF, Becker HC, Jones SR. Dopamine synthesis in alcohol drinking-prone and -resistant mouse strains. Alcohol. 2017;58:25–32. doi: 10.1016/j.alcohol.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KS, Graybiel AM. Habit formation coincides with shifts in reinforcement representations in the sensorimotor striatum. J Neurophysiol. 2016;115:1487–1498. doi: 10.1152/jn.00925.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JP, Murphy KP. Activation of cyclic AMP-dependent protein kinase is required for long-term enhancement at corticostriatal synapses in rats. Neurosci Lett. 2002;329:217–221. doi: 10.1016/s0304-3940(02)00659-6. [DOI] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Tillo SE, Xiong WH, Takahashi M, Miao S, Andrade AL, Fortin DA, Yang G, Qin M, Smoody BF, Stork PJS, Zhong H. Liberated PKA Catalytic Subunits Associate with the Membrane via Myristoylation to Preferentially Phosphorylate Membrane Substrates. Cell Rep. 2017;19:617–629. doi: 10.1016/j.celrep.2017.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4:a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci. 2007;27:3663–3676. doi: 10.1523/JNEUROSCI.0448-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corbille AG, Bertran-Gonzalez J, Herve D, Girault JA. Inhibition of ERK pathway or protein synthesis during reexposure to drugs of abuse erases previously learned place preference. Proc Natl Acad Sci USA. 2006;103:2932–2937. doi: 10.1073/pnas.0511030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- Walker-Gray R, Stengel F, Gold MG. Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches. Proc Natl Acad Sci USA. 2017;114:10414–10419. doi: 10.1073/pnas.1701782114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lanfranco MF, Gibb SL, Yowell QV, Carnicella S, Ron D. Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. J Neurosci. 2010;30:10187–10198. doi: 10.1523/JNEUROSCI.2268-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Xia JX, Li J, Zhou R, Zhang XH, Ge YB, Ru YX. Alterations of rat corticostriatal synaptic plasticity after chronic ethanol exposure and withdrawal. Alcohol Clin Exp Res. 2006;30:819–824. doi: 10.1111/j.1530-0277.2006.00095.x. [DOI] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ, Balleine BW. Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning. Eur J Neurosci. 2004;19:181–189. doi: 10.1111/j.1460-9568.2004.03095.x. [DOI] [PubMed] [Google Scholar]
- Yin HH, Mulcare SP, Hilario MR, Clouse E, Holloway T, Davis MI, Hansson AC, Lovinger DM, Costa RM. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat Neurosci. 2009;12:333–341. doi: 10.1038/nn.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason JT, Espana RA, Jones SR. Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods. 2011;202:158–164. doi: 10.1016/j.jneumeth.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sulzer D. Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci. 2004;7:581–582. doi: 10.1038/nn1243. [DOI] [PubMed] [Google Scholar]