

Abstract
Protein‐losing enteropathy (PLE) due to leakage of lymph into the gut sometimes occurs in young patients after Fontan palliation but is very rarely reported with other aetiologies of chronic heart failure (HF). PLE leads to severe hypoalbuminemia and immunodeficiency and is associated with poor prognosis. The mechanisms and the predispositions to PLE are poorly understood. Here, we report an adult patient with advanced HF due to non‐ischaemic non‐dilated hypocontractile cardiomyopathy who developed severe PLE, probably owing to increased ventricular stiffness and constraint by atypically placed epicardial electrode encircling both ventricles. Importantly, both PLE and immunodeficiency completely resolved after heart transplantation.
Keywords: Protein‐losing enteropathy, Heart failure, Cardiac transplantation
Introduction
Protein‐losing enteropathy (PLE) may develop in patients with primary intestinal disease or abdominal lymphatic obstruction or less frequently in some patients with cardiac disease. PLE may occur as a late complication of Fontan procedure1, 2, 3, 4 or in patients with constrictive pericarditis4, 5 or rheumatic tricuspid regurgitation,6 but it is probably very rare in other conditions that lead to biventricular heart failure (HF). The mechanisms and reasons responsible for PLE development are still poorly understood. Besides chronic elevation of central venous pressure,2 PLE development may involve lymphatic vessel rupture1 or intestinal wall inflammation.7 Chronic loss of lymphatic fluid rich in immunoglobulin and lymphocytes can lead to secondary immune deficiency. Because PLE has adverse prognostic consequences, reversibility of PLE in HF patients is critically important when considering radical HF therapy, such as cardiac transplantation. PLE has been shown to reverse after transplant in young patients with failed Fontan circulation,8 but its reversibility is not established in other conditions and in adults. Here, we report a patient in whom PLE with immune deficiency developed as a complication of hypokinetic non‐dilated cardiomyopathy, probably due to combination of increased ventricular stiffness and encircling epicardial electrode,9 that completely resolved after heart transplantation.
Case report
A 56‐year‐old man was referred to our centre for advanced HF management. He had cardiac hypertrophy and murmur since childhood and was diagnosed with hypertrophic cardiomyopathy. He noticed shortness of breath and exercise intolerance 16 years ago, coinciding with transition of hypertrophic cardiomyopathy into non‐dilated hypokinetic phenotype.10 Ten years ago, he underwent mitral valve repair with annuloplasty and epicardial lead implantation on left ventricular (LV) lateral wall for the purpose of cardiac resynchronization owing to the presence of LV bundle branch block. He had cardiac resynchronization therapy defibrillator device implanted and connected to LV epicardial and right ventricular (RV) endovasal leads. The surgery and the device implantation were performed in his country of origin, and there were no further details available. Several years later, he noticed oedema formation and began to regularly use loop diuretics. Four years ago, he developed reccurent chest infections and was diagnosed with common variable immunodeficiency with low immunoglobulin (Ig) G1–3 and lymphopenia but normal levels of IgM and IgG4. Several months prior to the presentation, he experienced progressive exercise dyspnoea, enhanced oedema formation, and poor tolerance of flat supine position. His bowel frequency was normal, and he did not report diarrhoea. At presentation to us, he was in New York Heart Association Class III, with peripheral oedemas and jugular venous distention. Cardiac imaging showed normal LV thickness, reduced LV ejection fraction (30%), increased LV diastolic diameter (60 mm, 29 mm/m2), dilated non‐collapsing inferior vena cava (30 mm), mild–moderate RV dysfunction, and mild tricuspid regurgitation (1–2/4) but no pulmonary hypertension or pericardial thickening. Endomyocardial biopsy of the right ventricle showed cardiomyocyte hypertrophy and extensive myocardial fibrosis, but no amyloid. Coronary angiography showed patent coronary arteries. On cardiac computed tomography (CT), we noticed long epicardial LV electrode almost encircling the heart at mid‐ventricular level, including the right ventricle (Figure 1). Right heart catheterization (after diuresis) showed central venous pressure 14 mmHg, pulmonary artery (PA) pressure 37/20/28 mmHg, PA wedge 19 mmHg, and cardiac output of 4.5 L/min. Pressure tracings at rest did not show evidence of constriction. DNA sequencing confirmed the presence of mutation myosin heavy chain gene (MYH7 c.2156G>A; p.Arg719Gln) that is pathogenic for familiar hypertrophic cardiomyopathy; an identical mutation was also found in a similarly affected first‐degree relative.
Figure 1.
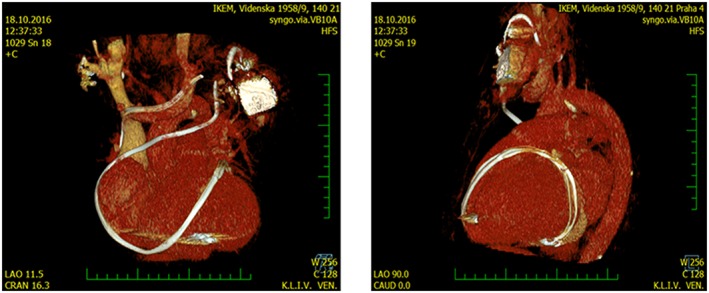
Computed tomography reconstruction (left, antero‐posterior aspect; right, lateral aspect) of atypical course of long left ventricular pacing electrode that encircled and adhered to the epicardium of the failing heart, which might be implicated in the development of protein‐losing enteropathy (see text).
Biochemistry showed BNP of 300 ng/mL and profound reduction of albumin, total serum protein, and IgG (Figure 2 A). Liver and kidney morphology and function were normal, there was no proteinuria. Despite having no gastrointestinal symptoms, the patient had elevated α1‐antitrypsin (A1AT) in spot stool sample (743 μg/L, 2.6 times above the upper limit), indicating possibility of PLE. Gastroscopy, colonoscopy, and CT small‐bowel study showed normal gastrointestinal and retroperitoneal morphology. Whole‐body positron emission tomography–CT excluded malignancy. Deep duodenoscopy with small‐bowel biopsy showed dilatation of venules and lymphatic vessels of intestinal mucosa, but no evidence of parasites, intestinal lymphoma, coeliac, or inflammatory bowel disease. Per exclusion, chronic HF became the only possible reason for excessive gastrointestinal protein loss. To put our case into the context of advanced HF population, we show data from consecutive advanced HF patients evaluated in our institution. The degree of hypoalbuminemia was more profound than other advanced HF patients evaluated in our centre (Figure 2 B). Our patient had far lower blood albumin and but only moderate elevation of RA pressure than had other patients in advanced HF population undergoing invasive haemodynamic study (Figure 2 C).
Figure 2.
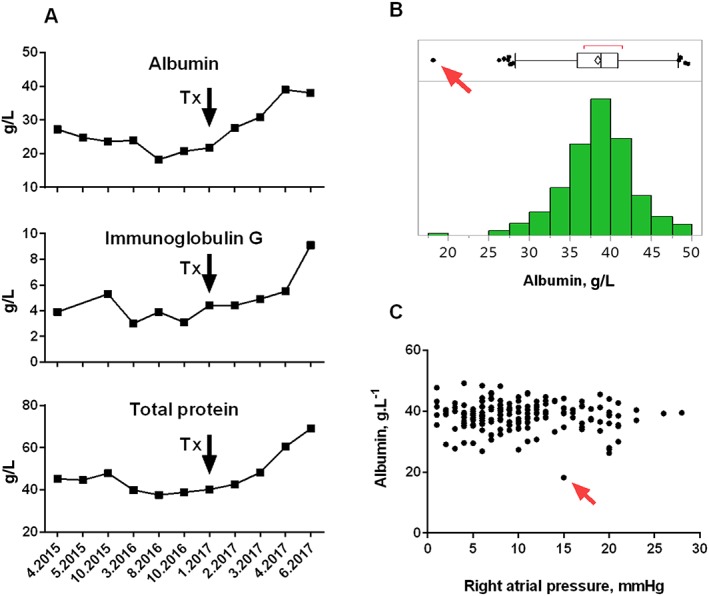
(A) The time course of plasma concentration of albumin, immunoglobulin G, and total protein in our patient. The arrow indicates the time point of heart transplantation (Tx). Note the time lag between Tx and recovery of protein levels to normal. (B) The frequency distribution histogram (bottom) and box and whisker plot (top) of plasma albumin levels in 409 consecutive patients with advanced HF evaluated for heart transplant candidacy at our centre (mean albumin level 38 ± 4.2 g/L). The PLE case is labelled by the red arrow. (C) The relationship between right atrial pressure and plasma albumin in 181 consecutive patients with advanced HF who underwent right heart catheterization and biochemical analysis. The PLE case is labelled by the red arrow. Absence of right atrial hypertension does not rule out hypoalbuminemia due to PLE. HF, heart failure; PLE, protein‐losing enteropathy.
The patient started high‐protein diet, intermittent albumin and immunoglobulin replacement therapy, and i.v. diuretics, but his haemodynamics gradually deteriorated, and he became dependent on inotropic therapy. After discussions with the patient and experts from other transplant centres, we decided to proceed with heart transplantation. The patient underwent uncomplicated transplantation in January 2017. During the procedure, the surgeon noticed that excessively long epicardial LV electrode firmly adhered to epicardial surface and almost encircled the heart. After an uneventful transplantation, the patient was started on regular triple immunosuppressant regime (prednisone, mycophenolate, and tacrolimus). He needed substitutions of albumin and IgG, but the need was decreasing, and after 4 months, plasma albumin, immunoglobulin, and stool A1AT levels returned to normal (Figure 2 A). Therefore, PLE, including humoral immunodeficiency, completely resolved. Further clinical course was uncomplicated, with return of excellent quality of life.
Discussion
In patients with advanced HF, mild hypoalbuminemia is common owing to the presence of haemodilution, cachexia, malnutrition, impaired liver function, and malabsorption. More rarely, altered integrity of intestinal lymphatic system may contribute to massive protein loss, leading to PLE with severe hypoalbuminemia. Because in vivo albumin tracing studies are not routinely available, the diagnosis of cardiac PLE is based on increased faecal content of A1AT (protein not degraded by intestinal bacteria and serving as a marker of increased intestinal protein loss) and on excluding other reasons for PLE.2 Studies that would systematically examine prevalence of PLE in advanced HF population using objective diagnostic tools (faecal A1AT concentration) are missing. The distribution frequency of plasma albumin levels in advanced HF patients evaluated in our centre (Figure 2 B) does not show leftward skewness, suggesting that severe hypoalbuminemia due to PLE is probably rare in this population. Besides the reported case, none of our advanced HF patients had plasma albumin concentration ˂ 25 g/L.
The consequences and the mechanisms responsible for cardiac PLE are incompletely understood. It has been hypothesized that lymphatic over‐distention due to transmission of elevated central venous pressure in predisposed patients can lead to the rupture of intestinal lymphatic vessels and to ensuing leakage of protein‐rich lymph into the lumen of the small intestine.1 Despite that increased central venous pressure is considered to be the prerequisite for cardiac disease‐related PLE, our results illustrate that degree of PLE poorly correlates with haemodynamics, indicating that other factors are causally involved.2 One explanation could be an existence of developmental abnormalities of hepato‐duodenal lymphatic vessels that may be prone to rupture if exposed to increased pressure, leading to focal leakage of the lymph into the duodenal lumen, as recently demonstrated.1 Instantaneous peaks of central venous pressure may be more relevant for lymphatic damage rather than value of central venous pressure at rest. Moreover, increased inflammatory cytokines may increase intestinal epithelial permeability and lead to higher flux of albumin across intestinal mucosa, explaining why some patients with post‐Fontan PLE respond to oral corticosteroids.2, 7 Our case indicates that if integrity of intestinal lymphatic system is breached, the leak of albumin‐rich lymph can continue even at mildly elevated RA pressure. It is clinically important to realize (Figure 2 C) that the absence of severe RA pressure elevation does not exclude the presence of severe PLE.
Our case illustrates that PLE with ensuing immunodeficiency may occur not only in patients with pericardial constriction3, 4 or post‐Fontan surgery1 but also in patients with HF due to non‐ischaemic cardiomyopathy with non‐dilated hypocontractile phenotype. Why this particular patient developed PLE is not known. It can be speculated that his RV diastolic compliance was reduced owing to the restrictive nature of his burned‐out hypertrophic cardiomyopathy.11 Diastolic compliance could be also externally restrained by ingrown, excessively long, and atypically bended epicardial pacing electrode encircling the heart (Figure 2 A), perhaps leading to more steep RV pressure–volume relation with increased preload,9 causing episodes of markedly elevated central venous pressure, pressure build‐up, and rupture of anatomically predisposed lymphatic vessels.1 A case of PLE due to ‘occult’ pericardial constriction from pre‐existing cardiac surgery was previously described.3 We did not perform complete left–right haemodynamic study with volume challenge to unmask the presence of constrictive physiology in a normovolemic patient. However, owing to advanced LV systolic and diastolic dysfunction, the transplantation was indeed a better option than pericardiectomy in this patient.
The second important observation is slow but complete reversibility of PLE (including humoral immunodeficiency) after cardiac transplantation. It is plausible to speculate that protracted and profound reduction of right atrial pressure after transplantation led to redirection of lymphatic flow from the gut into the liver and ductus thoracicus.1 Such unloading may even lead to natural ‘sealing’ of perforated lymphatic channels. It is also possible that immunosuppressive drugs can inhibit inflammation and improve the barrier function of the gut.7
In summary, our case illustrates that PLE may occur in patients with non‐ischaemic cardiomyopathy and that it can lead to humoral immunodeficiency and is reversible by cardiac transplantation. PLE should not be considered a contraindication to heart transplantation, even in non‐Fontan adult HF patients.
Funding
The authors are supported by research grants from the Ministry of Health of the Czech Republic (MZ 15‐27682A, NV16‐27496A), Czech Science Foundation (GA 15‐14200S), and Grant Agency for Czech Health Research Council (AZV 17‐28784A).
Melenovsky, V. , Kubanek, M. , and Kacer, P. (2018) Protein‐losing enteropathy in an adult with non‐ischaemic cardiomyopathy: complete reversal by heart transplantation. ESC Heart Failure, 5: 842–845. 10.1002/ehf2.12342.
References
- 1. Itkin M, Piccoli DA, Nadolski G, Rychik J, DeWitt A, Pinto E, Rome J, Dori Y. Protein‐losing enteropathy in patients with congenital heart disease. J Am Coll Cardiol 2017; 69: 2929–2937. [DOI] [PubMed] [Google Scholar]
- 2. Umar SB, DiBaise JK. Protein‐losing enteropathy: case illustrations and clinical review. Am J Gastroenterol 2010; 105: 43–49. [DOI] [PubMed] [Google Scholar]
- 3. Muller C, Globits S, Glogar D, Klepetko W, Knoflach P. Constrictive pericarditis without typical haemodynamic changes as a cause of oedema formation due to protein‐losing enteropathy. Eur Heart J 1991; 12: 1140–1143. [DOI] [PubMed] [Google Scholar]
- 4. Wilkinson P, Pinto B, Senior JR. Reversible protein‐losing enteropathy with intestinal lymphangiectasia secondary to chronic constrictive pericarditis. N Engl J Med 1965; 273: 1178–1181. [DOI] [PubMed] [Google Scholar]
- 5. Davidson JD, Waldmann TA, Goodman DS, Gordon RS Jr. Protein‐losing gastroenteropathy in congestive heart‐failure. Lancet 1961; 1: 899–902. [DOI] [PubMed] [Google Scholar]
- 6. Strober W, Cohen LS, Waldmann TA, Braunwald E. Tricuspid regurgitation. A newly recognized cause of protein‐losing enteropathy, lymphocytopenia and immunologic deficiency. Am J Med 1968; 44: 842–850. [DOI] [PubMed] [Google Scholar]
- 7. Thacker D, Patel A, Dodds K, Goldberg DJ, Semeao E, Rychik J. Use of oral budesonide in the management of protein‐losing enteropathy after the Fontan operation. Ann Thorac Surg 2010; 89: 837–842. [DOI] [PubMed] [Google Scholar]
- 8. Bernstein D, Naftel D, Chin C, Addonizio LJ, Gamberg P, Blume ED, Hsu D, Canter CE, Kirklin JK, Morrow WR. Outcome of listing for cardiac transplantation for failed Fontan: a multi‐institutional study. Circulation 2006; 114: 273–280. [DOI] [PubMed] [Google Scholar]
- 9. Peters RW, Scheinman MM, Raskin S, Thomas AN. Unusual complications of epicardial pacemakers. Recurrent pericarditis, cardiac tamponade and pericardial constriction. Am J Cardiol 1980; 45: 1088–1094. [DOI] [PubMed] [Google Scholar]
- 10. Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Bohm M, Duboc D, Gimeno J, de Groote P, Imazio M, Heymans S, Klingel K, Komajda M, Limongelli G, Linhart A, Mogensen J, Moon J, Pieper PG, Seferovic PM, Schueler S, Zamorano JL, Caforio AL, Charron P. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non‐dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2016; 37: 1850–1858. [DOI] [PubMed] [Google Scholar]
- 11. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 2017; 121: 749–770. [DOI] [PMC free article] [PubMed] [Google Scholar]