

Abstract
Key points
Insulin enters the brain from the blood via a saturable transport system.
It is unclear how insulin is transported across the blood–brain barrier (BBB).
Using two models of the signalling‐related insulin receptor loss or inhibition, we show insulin transport can occur in vivo without the signalling‐related insulin receptor.
Insulin in the brain has multiple roles including acting as a metabolic regulator and improving memory.
Understanding how insulin is transported across the BBB will aid in developing therapeutics to further increase CNS concentrations.
Abstract
A saturable system transports insulin from blood across the blood–brain barrier (BBB) and into the central nervous system. Whether or not the classic or signalling‐related insulin receptor plays a role in mediating this transport in vivo is controversial. Here, we employed kinetics methods that distinguish between transport across the brain endothelial cell and reversible luminal surface receptor binding. Using a previously established line of mice with endothelial‐specific loss of the signalling‐related insulin receptor (EndoIRKO) or inhibiting the insulin receptor with the selective antagonist S961, we show insulin transport across the BBB is maintained. Rates of insulin transport were similar in all groups and transport was still saturable. Unlike transport, binding of insulin to the brain endothelial cell was decreased with the loss or inhibition of the signalling‐related insulin receptor. These findings demonstrate that the signalling‐related insulin receptor is not required for insulin transport across the BBB.
Keywords: blood‐brain barrier, insulin transport, insulin receptor
Key points
Insulin enters the brain from the blood via a saturable transport system.
It is unclear how insulin is transported across the blood–brain barrier (BBB).
Using two models of the signalling‐related insulin receptor loss or inhibition, we show insulin transport can occur in vivo without the signalling‐related insulin receptor.
Insulin in the brain has multiple roles including acting as a metabolic regulator and improving memory.
Understanding how insulin is transported across the BBB will aid in developing therapeutics to further increase CNS concentrations.
Introduction
Insulin in the central nervous system (CNS) is primarily derived from the blood (Schwartz et al. 1992; Banks, 2004). Thus, insulin must traverse the blood–brain barrier (BBB) to access the CNS. Imbalances in CNS insulin levels and sensitivity is implicated in many neurodegenerative diseases such as Alzheimer's and Parkinson's disease (Procaccini et al. 2016). In addition to influencing food intake and serum glucose (Porte et al. 2005), insulin in the CNS is neuroprotective, affecting synaptogenesis and nerve growth (Nelson et al. 2008; Banks et al. 2012) and has recently proved beneficial in improving cognition (Craft et al. 1996; Benedict et al. 2004; Reger et al. 2008; Salameh et al. 2015). Indeed, investigations into ways in which CNS insulin can be restored or increased in these CNS insulin‐resistant states are currently underway in clinical trials (Novak et al. 2014; Claxton et al. 2015). Further understanding the transport mechanism of insulin across the BBB and into the CNS could prove useful in developing therapeutics to overcome CNS insulin resistance.
The insulin binding sites present on brain endothelial cells (BECs) serve two functions: transport of insulin across the BBB and activation of classic receptors such as the signalling‐related insulin receptor and insulin‐like growth factor 1 (IGF‐1) receptor (van Houten & Posner, 1979; Jialal et al. 1985; Pardridge et al. 1985). Transport of insulin into the CNS has been investigated for over half a century (Elgee et al. 1954; Haugaard et al. 1954) and occurs in a saturable manner, demonstrating the insulin transport‐related protein is saturable (Baura et al. 1993; Banks et al. 1997a). Insulin transport across the BBB is highly regulated and altered in a number of states, including obesity, starvation, hyperglycaemia, Alzheimer's disease and diabetes (Banks, 2004). Insulin can be internalized by the signalling‐related insulin receptor present on endothelial cells (King & Johnson, 1985), a process termed receptor‐mediated endocytosis (Bar et al. 1983; King & Johnson, 1985; Duffy & Pardridge, 1987).
It has been an untested assumption that the signalling‐related insulin receptor also serves as the BBB transporter on BECs; that is, that insulin transport across the BBB occurs by receptor‐mediated transcytosis via the signalling‐related insulin receptor. Whereas receptor‐mediated endocytosis internalizes a vesicle that can return to the luminal surface, transcytosis incorporates the additional process of exocytosis at the abluminal side, and so is a mechanism of transport across the BBB. Transcytosis occurs in three major steps: (1) receptor‐mediated endocytosis at the luminal side of the BEC, (2) vesicle transfer across the endothelial cell, and (3) receptor‐mediated exocytosis at the abluminal side of the BEC.
With the recent advances in technology and accessibility to various genetic mouse models and inhibitors, we can better investigate the role of the signalling‐related insulin receptor located on endothelial cells. First, genetic mouse models lacking the signalling‐related insulin receptor in endothelial cells have been developed to study the role in vivo. Loss of the signalling‐related insulin receptor in the vascular endothelium has recently been investigated not only in the context of addressing the role of insulin action in endothelial function (Vicent et al. 2003) but also in determining the impact of the signalling‐related insulin receptor located in endothelial cells on downstream signalling in various tissues, including brain (Konishi et al. 2017; Wang et al. 2017). In describing the mouse model with the endothelial cell insulin receptor knockout used in this study (EndoIRKO mice) driven by Cdh5 Cre (VE‐Cadherin), Konishi et al. reported decreased levels of the signalling‐related insulin receptor in various endothelial populations, including aorta and lung (Konishi et al. 2017). In addition, the amount of fluorescein isothiocyanate (FITC)–insulin binding in the brain endothelium in EndoIRKO mice is significantly decreased suggesting a significant loss of the signalling‐related insulin receptor. While the loss of the insulin receptor in brain endothelium delayed insulin signalling in the hypothalamus, hippocampus and prefrontal cortex, direct transport of insulin across the BBB was not investigated. Second, selective inhibitors of the signalling‐related insulin receptor have been generated to further study the role of the insulin receptor in any cell type. The insulin receptor antagonist S961 has been used both in vitro and in vivo and has been shown to prevent insulin signalling in many cell types (Schaffer et al. 2008; Chan et al. 2016) and increase serum glucose levels (Vikram & Jena, 2010).
In order to determine if the signalling‐related insulin receptor is involved in BBB insulin transport, we used these two separate genetic and pharmacological models. We used EndoIRKO mice that have been previously defined and described (Konishi et al. 2017; Wang et al. 2017). We also tested our hypothesis in CD‐1 mice using the selective insulin receptor inhibitor S961 (Schaffer et al. 2008). Here, we report that transport of insulin across the BBB in vivo can occur independently of endothelial cell signalling‐related insulin receptors. We refer to this unidentified protein as the insulin transport‐related protein.
Methods
Ethical approval
The Institutional Animal Care and Use Committee at the Veterans Affairs Puget Sound (Seattle, WA, USA) approved all animal experimental protocols (0909) and all methods were carried out in accordance with the approved guidelines and regulations. The VA Puget Sound is a facility that is certified by the Association for Assessment and Accreditation of Laboratory Animal Care International. The protocols are in accordance with The Journal of Physiology’s guidelines on animal ethics.
Animals
The generation of EndoIRKO mice was described previously (Wang et al. 2017). Two‐month‐old EndoIRKO and wild type mice were used for the transport kinetics. Five‐month‐old mice were used for the remaining EndoIRKO studies. For the S961 inhibitor studies, male CD‐1 mice (2–3 months old) were purchased from Charles River Laboratories (Seattle, WA, USA). CD‐1 mice are an established model for BBB transport studies (Banks et al. 1997a, b ; Banks & Kastin, 1998). Mice had ad libitum access to food and water and were kept on a 12 h/12 h light/dark cycle. For all studies, mice were anaesthetized with an intraperitoneal injection of 0.1 ml of 40% urethane to minimize pain and distress. At the end of each study, mice were killed by decapitation while under anaesthesia.
Radioactive labelling of insulin, S961 and albumin
Ten micrograms of human insulin (Sigma‐Aldrich, St Louis, MO, USA) or S961 (Novo Nordisk, Copenhagen, Denmark) diluted in 0.25 m chloride‐free sodium phosphate buffer (PB), pH 7.5, was radioactively labelled with 0.5 mCi Na125I (Perkin Elmer, Waltham, MA, USA) by the chloramine‐T (Sigma‐Aldrich) method. Addition of 10 μg chloramine‐T in 0.25 m PB began the reaction. The reaction was terminated 1 min later with the addition of 100 μg sodium metabisulfite. The specific activity of 125I‐labelled insulin has previously been calculated to be 55 Ci/g (Banks et al. 1997a). Albumin (Sigma‐Aldrich) was radioactively labelled with 99mTc (GE Healthcare, Seattle, WA, USA). Briefly, 1 mg albumin was combined with 120 μg stannous tartrate and 20 μl 1 m HCl in 500 ml deionized water. One millicurie of 99mTc was added for the 20 min reaction. 125I‐insulin, 125I‐S961 and 99mTc‐albumin were purified on a column of Sephadex G‐10 (Sigma‐Aldrich). Protein labelling by 125I or by 99mTc isotopes was characterized by 15% trichloroacetic acid (TCA) precipitation. Greater than 90% radioactivity in the precipitated fraction was consistently observed for insulin, S961 and albumin.
Measurement of radioactive insulin transport across the BBB
Multiple‐time regression analysis was used as detailed previously (Blasberg et al. 1983; Patlak et al. 1983) and briefly described below to calculate the blood‐to‐brain steady‐state rate of unidirectional solute influx (K i) for insulin. The right jugular vein and left carotid artery were exposed for all studies. Mice were given an injection into the right jugular vein of 0.2 ml of lactated Ringer solution (LR) containing 1 × 106 c.p.m. of 125I‐insulin and 5 × 105 c.p.m. of 99mTc‐albumin for all studies unless specified. The amount of 125I‐insulin injected equates to approximately 8.26 ng of insulin, a dose that is about 15% of endogenous murine insulin serum levels (Banks et al. 1997c). 99mTc‐labelled albumin was co‐injected as a marker for vascular space (Blasberg et al. 1983). For all studies in the S961 group, mice received a co‐injection of PB (vehicle) or 1 μg S961/mouse with the intravenous radioactive injection. A separate group of mice was measured following a 6 h fast. The hypothalamus was dissected in this group to measure the influx of insulin into this specific brain region. Blood from the left carotid artery was collected between 1 and 10 min after intravenous injection. Mice were immediately decapitated and the whole brain removed and weighed. The arterial blood was centrifuged at 5400 g for 10 min at 4°C and serum collected. The levels of radioactivity in serum (50 μl) and brain were counted in a γ‐counter. The brain/serum (B/S) ratio (μl/g) of 125I‐insulin and 99mTc‐albumin in each gram of brain is calculated separately. Each B/S ratio of 125I‐insulin is corrected for vascular space by subtracting the paired B/S ratio of 99mTc‐albumin and defined as the delta. These values are plotted against the respective exposure time which represents the level of plasma arterial radioactivity integral divided by the final plasma radioactivity. Exposure time is calculated from the formula:
| (1) |
where Cp(t) is the level of radioactivity (c.p.m.) in serum at time (t). The influx of insulin is calculated from the following formula as originally described in eqn (6) of Blasberg et al. (1983):
| (2) |
where Am is level of radioactivity (c.p.m.) per gram of brain tissue at time t, Cpt is level of radioactivity (c.p.m.) per millilitre of arterial serum at time t, K i (μl/(g min)) is the steady‐state rate of unidirectional solute influx from blood to brain divided by the plasma concentration, and V i (μl/g) is the level of rapid and reversible binding for brain. The linear portion of the relation between the delta B/S ratio vs. exposure time is used to calculate the K i (μl/(g min)) and V i (μl/g) (Blasberg et al. 1983). The slope of the linearity measures K i and is reported with its standard error term. The current calculations have been adapted to modern graphical applications by using Prism to calculate the linear regression. The y‐intercept of the linearity measures V i, the initial volume of distribution in brain at t = 0. Multiple‐time linear regression analysis was also used to measure transport of 125I‐S961 across the BBB in the presence of either vehicle (PB), 1 μg S961 or 10 μg S961. The same methods and calculations were used as described above except that the B/S ratio was not corrected for levels of 99mTc‐albumin as it was not co‐injected.
Measurement of immunoactive insulin transport across the BBB
CD‐1 male mice fasted for 6 h were anaesthetized as described above and the right jugular vein exposed for an i.v. injection of 0.2 ml containing 1 μg human insulin ± 1 μg S961 in 1% bovine serum albumin (BSA)–LR and the solution allowed to circulate for a period of time (1–5 min). At the end of the time, blood was collected via the descending thoracic aorta. Brains were washed out with 20 ml ice‐cold LR to clear the vascular space, which also removes any reversible binding at the brain endothelium. Blood was centrifuged at 5400 g for 10 min and serum collected and stored at −80°C. Brains were snap frozen in liquid nitrogen and stored at −80°C. Brains were processed in radioimmunoprecipitation assay (RIPA) buffer (150 nm NaCl, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate, 20 mm Tris HCl, 0.5 m EDTA) containing a protease inhibitor cocktail (Sigma). Protein was measured and 5 μg/μl stock samples were generated. A human insulin Meso Scale Discovery (MSD) kit was used to measure human insulin levels (MSD, Rockville, MD, USA). A 125 μg quantity of protein from whole brain and serum diluted 100‐fold was measured for each sample. The B/S ratio was calculated and plotted against exposure time.
In vivo stability of insulin in serum
Blood was collected 10 min after the intravenous injection of 1 × 106 c.p.m. of 125I‐insulin in 0.2 ml LR. Blood was centrifuged at 5400 g for 10 min and 50 μl of the resulting serum was added to 250 μl of LR–BSA and vortexed. A 300 μl volume of 30% TCA was added, vortexed and centrifuged for 10 min at 5400 g. The resulting supernatant (S) and precipitate (P) were counted separately and the percentage (%Precip) of counts in the precipitate was calculated:
| (3) |
To correct for any degradation that might have occurred during the processing for acid precipitation, we added 125I‐insulin to non‐radioactive arterial whole blood and processed as above. Sample values were made relative to the processing controls. Since the degradation in serum was limited, there was no correction made for the pharmacokinetic data.
Transport saturability determined by intravenous injection
Blood was collected and the whole brain was removed and weighed 1–10 min after intravenous injection of the radiolabelled solution ± 1 μg/mouse of S961 ± 1 μg/mouse of non‐radioactive insulin, a dose about 120× that of the 125I‐insulin injected. Results were expressed as B/S ratio in units of μl/g after correcting for vascular space as described above. The influx for 125I‐insulin was calculated based on the equations above.
Capillary depletion
To verify the insulin detected in the brain was 125I‐insulin that had completely crossed through the brain endothelial cells, the capillary depletion method was used to separate cerebral capillaries and vascular components from brain parenchyma (Triguero et al. 1990). This method is a quality control to determine which fraction the radioactive insulin is present in with a very low standard error and therefore only requires an n = 2–3. Ten minutes after intravenous injection of the radiolabelled solution blood was collected from the carotid artery. The brains were removed, weighed and homogenized with 10 strokes of a glass homogenizer in 0.8 ml of physiological buffer (10 mm Hepes, 141 mm NaCl, 4 mm KCl, 2.8 mm CaCl2, 1 mm MgSO4, 1 mm NaH2PO4 and 10 mm d‐glucose adjusted to pH 7.4). A 40% dextran solution (1.6 ml) was added to the homogenate, vortexed and further homogenized with three strokes. Homogenates were centrifuged at 3200 g for 10 min at 4°C. The pellet, containing the capillaries, and the supernatant, representing the brain parenchymal/interstitial fluid space, were carefully separated. The ratio of radioactivity of 125I‐insulin in the supernatant (parenchyma) was corrected for vascular space by subtracting the ratio of 99mTc‐albumin in the supernatant.
Regional distribution
Five minutes after intravenous injection of the radiolabelled solution ± 1 μg S961, blood was collected and brains were removed. Brains were dissected on ice into 10 brain regions according to Glowinski & Iversen (1966). Radioactive values were corrected for vascular space as described above and made relative to the whole brain levels (μl/g) for each sample.
Statistics
Regression analysis and other statistical analyses were performed with the use of Prism 6.0 (GraphPad Software Inc., San Diego, CA, USA). Means are reported with their standard error terms and compared by one‐way analysis of variance (ANOVA) followed by Newman–Keuls post‐test. Two means were compared by Student's t test analysis. Linear regression lines were compared statistically with the Prism 6.0 software package (Zar, 1984). They are reported with their correlation coefficients (r) and P values with significance < 0.05.
Results
Insulin transport across the BBB attributable to loss of the endothelial insulin receptor
The transport of 125I‐insulin into the brain was measured over time. The plasma concentration of 125I‐insulin was logarithmic over the 10 min study period, varying by approximately 7% over this time (data not shown). The linear regression analysis showed a statistically significant relation between the delta B/S (brain/serum) ratio (y‐axis) of 125I‐insulin vs. exposure time (x‐axis) for both the wild type (r = 0.904, P = 0.013) and EndoIRKO mice (r = 0.903, P = 0.014; n = 6/group) (Table 1) and that these values were significantly different from zero. Therefore, the linear portion of the line was used to calculate the influx (K i) for insulin in each group. The blood‐to‐brain influx is 0.870 ± 0.21 μl/(g min) for 125I‐insulin in wild type mice and 0.545 ± 0.13 μl/(g min) in EndoIRKO mice (Fig. 1 A). Although the calculated estimate of the insulin influx for EndoIRKO mice is less than that for the wild type mice, the difference is not statistically different (P = 0.34).
Table 1.
Pharmacokinetics of insulin transport into the whole brain in fed mice
| Group | K i (μl/(g min)) | r | V i (μl/g) |
|---|---|---|---|
| Wild type | 0.870 ± 0.21 | 0.904 | 10.2 ± 1.02 |
| EndoIRKO | 0.545 ± 0.13 | 0.903 | 5.5 ± 1.38* |
| Vehicle | 0.738 ± 0.21 | 0.715 | 8.3 ± 1.39 |
| S961 | 1.21 ± 0.17 | 0.903 | −3.2 ± 1.79* |
Levels of 125I‐insulin present in whole brain were measured 1–10 min after injection and plotted in Fig. 1. Multiple‐time linear regression analysis was performed to determine the influx of insulin (K i) and vascular binding (Vi). Data are presented as means ± SEM. * P < 0.05 vs. respective control group (Wild type or Vehicle).
Figure 1. Transport of radioactive insulin across the BBB with loss or inhibition of the insulin receptor.
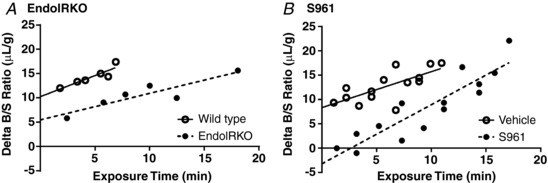
A, level of 125I‐insulin in whole brain was measured in wild type (n = 6, open circles) and EndoIRKO (n = 6, filled circles) mice. B, level of 125I‐insulin in whole brain was measured in vehicle (n = 14, open circles) and mice co‐injected with 1 μg S961 (n = 14, filled circles). Values are corrected for vascular space and expressed as the Delta B/S ratio (μl/g). Linear regression analysis was included (straight or dashed line) to depict the influx.
Similar results were observed with insulin receptor inhibition (Fig. 1 B). Linear regression analysis showed a statistically significant relation between the delta B/S ratio vs. exposure time regardless of vehicle (r = 0.715, P = 0.004) or S961 co‐injection (S961 r = 0.903, P < 0.0001; n = 14/group). The K i in vehicle‐treated mice was 0.738 ± 0.21 μl/(g min) for 125I‐insulin and 1.214 ± 0.17 μl/(g min) for S961‐treated mice (Fig. 1 B). In this case, S961 increased the influx but the estimated influxes were not statistically different (P = 0.11). These results suggest the signalling‐related insulin receptor is not completely necessary for insulin transport across the BBB.
In contrast to transport, the initial volumes of distribution (V i, y‐intercept) for the two groups in each model were significantly different (Table 1). The V i reflects the rapidly reversible component of the vasculature and when corrected for vascular space, as we did here, reflects primarily receptor binding. As expected, EndoIRKO had a lower V i (5.5 ± 1.38 μl/g) compared to wild type mice (10.2 ± 1.02 μl/g, P < 0.0001). The nearly 50% reduction in V i indicates a decrease in the amount of insulin binding to the endothelial surface in EndoIRKO mice and is similar to recently published data showing decreased insulin binding in the frontal cortex (Konishi et al. 2017). In addition, the V i for radioactive insulin was inhibited by S961 in CD‐1 mice (Vehicle V i = 8.3 ± 1.39 μl/g vs. S961 V i = −3.2 ± 1.79 μl/g, P < 0.0001), indicating a decrease in the amount of insulin binding to the endothelial surface.
There was no correlation between the B/S ratio of 99mTc‐albumin and exposure time in any of the groups as expected, suggesting lack of uptake, and no change in the vascular space (data not shown).
Insulin BBB transport following fasting
To determine if fasting altered the level of BBB insulin transport, we fasted CD‐1 mice for 6 h before the radioactive insulin transport study. Similar to the study in Fig. 1 B, we co‐injected one cohort of mice with 1 μg S961. As shown in Fig. 2, insulin transport occurs whether the insulin receptor is inhibited or not under fasted conditions (Vehicle K i = 1.051 ± 0.13 μl/(g min) vs. S961 K i = 0.782 ± 0.21 μl/(g min), P = 0.282). The amount of reversible binding to the endothelium (y‐intercept) is significantly different between the two groups (Vehicle V i = 4.4 ± 1.02 μl/g vs. S961 V i = 1.1 ± 1.68 μl/g, P < 0.0001; Table 2) similar to the data shown in Fig. 1 B, suggestive of successful inhibition of the insulin receptor. The half‐life for the radiolabelled insulin in the fasted mice was not different between the two groups and was approximately 3–4 min. In addition, we included a control group that had not been fasted (Fed). The results suggest that a 6 h fast does not significantly change insulin transport across the BBB compared to Fed animals (K i = 1.048 ± 0.1 μl/(g min), P = 0.45).
Figure 2. Effect of insulin receptor inhibition on the transport of radioactive insulin across the BBB following fasting.
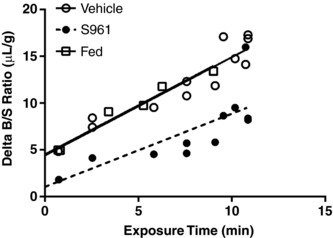
Level of 125I‐insulin in whole brain was measured in mice co‐injected with vehicle (n = 12, open circles) or 1 μg S961 (n = 12, filled circles), and fed mice (n = 6, open squares). Values are corrected for vascular space and expressed as the Delta B/S ratio (μl/g). Linear regression analysis was included (straight or dashed line) to depict the influx.
Table 2.
Pharmacokinetics of insulin transport into the whole brain following a 6 h fast
| Group | K i (μl/(g min)) | r | V i (μl/g) |
|---|---|---|---|
| Vehicle | 1.051 ± 0.13 | 0.935 | 4.4 ± 1.02 |
| S961 | 0.782 ± 0.21 | 0.766 | 1.1 ± 1.68* |
| Fed | 1.048 ± 0.10 | 0.982 | 4.5 ± 0.516 |
Levels of 125I‐insulin present in whole brain were measured 1–10 min after injection and plotted in Fig. 2. Multiple‐time linear regression analysis was performed to determine the influx of insulin (K i) and vascular binding (V i). Data are presented as means ± SEM. * P < 0.05 vs. Vehicle.
Immunoactive insulin BBB transport following insulin receptor inhibition
In order to corroborate our findings based on radioactive insulin, we repeated the study in CD‐1 mice using exogenous human insulin and measured immunoactive levels of insulin present in the brain and serum. We did so using an immunoassay specific for human insulin, so that any insulin detected in brain clearly crossed the BBB. We saw a similar lack of effect of S961 on the transport of human insulin across the BBB (Fig. 3) as our radioactive study (Fig. 1 B).
Figure 3. Transport of immunoactive insulin across the BBB with inhibition of the insulin receptor.
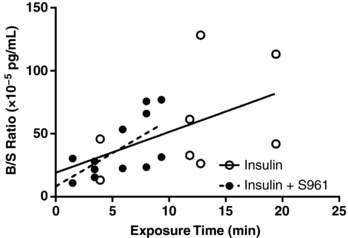
Level of human insulin in whole brain was measured in mice co‐injected with 1 μg insulin plus vehicle (n = 8, open circles) or 1 μg S961 (n = 12, filled circles). Values are expressed as the Delta B/S ratio (10−5 pg/ml). Linear regression analysis was included (straight or dashed line) to depict the influx.
Transport of S961 across the BBB
To determine whether S961 only bound to BECs or was also transported across the BBB, we studied its brain pharmacokinetics as well. There was no correlation between the B/S ratio of 125I‐S961 and exposure time (r = 0.231, P = 0.62), indicative of lack of transport (Fig. 4). However, the average level of 125I‐S961 present at the brain endothelium was very high (68.3 ± 5.17 μl/g). Co‐injection of unlabelled S961 at two doses, 1 and 10 μg, significantly decreased this binding (P < 0.0001), with no effect of dose (15.6 ± 0.95 vs. 11.2 ± 0.71 μl/g, respectively). These data suggest S961 binds to the BEC but is not transported across the BBB.
Figure 4. Transport of S961 across the BBB.
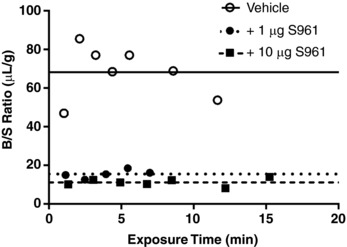
Levels of 125I‐S961 in whole brain were measured in mice co‐injected with vehicle (n = 7, open circles), 1 μg S961 (n = 5, filled circles) or 10 μg S961 (n = 7, filled squares). Values are expressed as the B/S ratio (μl/g). As slope of the linear regression was not significantly different from zero, the average level of 125I‐S961 present in each group was depicted by a straight, dotted or dashed line.
Effect of loss of the endothelial insulin receptor on insulin penetration of the BEC (capillary depletion)
As a control in order to verify that the 125I‐insulin detected in our studies was due to 125I‐insulin present in the brain, rather than sequestration by the BECs, the method of capillary depletion was used to measure the distribution of 125I‐insulin between capillaries and brain parenchyma after 10 min of circulation (Fig. 5). The majority of the 125I‐insulin was present in the parenchyma compared to the capillary fraction in all groups. The amount of 125I‐insulin present in the parenchyma of wild type mice was 77.4 ± 0.5% compared to only 22.6 ± 0.5% in the capillary fraction (n = 2/group). The amount present in the parenchyma of EndoIRKO mice was 83.3 ± 0.1% (n = 2/group). When the receptor was inhibited with S961, 70.3 ± 1.3% of 125I‐insulin was present in the parenchyma while 77.4 ± 2.3% was present in the vehicle treated group (n = 3/group). These data demonstrate that insulin transport across the BEC is not affected by loss or inhibition of the insulin endothelial receptor.
Figure 5. Transfer of 125I‐insulin across the brain endothelial cell.
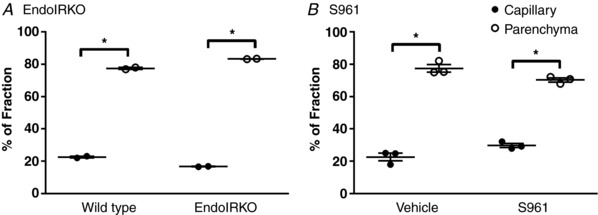
After separating the capillaries (filled circles) from the remaining brain parenchyma (open circles), the percentage of 125I‐insulin present in the parenchyma was significantly greater than in the capillary fraction in both wild type and EndoIRKO mice (A; n = 2) and in mice co‐injected with vehicle or S961 (B; n = 3). All data are expressed as means ± SEM (* P < 0.05, t test).
Effect of loss of the endothelial insulin receptor on insulin stability in serum
To verify that the majority of the 125I‐insulin detected in serum was still intact, protein was precipitated and radioactivity measured in the pellet and supernatant. Ten minutes after circulation, the majority of 125I precipitated with the protein pellet indicating that insulin was intact. There was no significant difference in stability of 125I‐insulin between wild type (73.7 ± 8.8%) and EndoIRKO mice (66.2 ± 2.9%) suggesting the loss of the insulin receptor does not affect insulin degradation (n = 2/group). However, addition of S961 did increase the stability of 125I‐insulin in serum (70 ± 3.8%) vs. vehicle (51 ± 2.6%, P = 0.016, n = 3). These results suggest that deletion of the insulin receptor on endothelial cells does not affect stability of insulin in serum but that addition of the inhibitor S961 does enhance the stability of serum insulin.
Effect of loss of the endothelial insulin receptor on saturability of insulin transport across the BBB
Addition of 1 μg unlabelled insulin inhibited the transport of 125I‐insulin into the brain in mice co‐injected with vehicle or 1 μg S961 (Fig. 6, Table 3). Saturability of a transport system is defined by self‐inhibition (Kastin & Pan, 2008). These results are in line with previous reports supporting blood‐to‐brain insulin transport as saturable (Duffy & Pardridge, 1987; Schwartz et al. 1991; Baura et al. 1993; Banks et al. 1997a), and the protein that is responsible for insulin transport still acts as a saturable transporter.
Figure 6. Saturability of insulin transport into the whole brain.
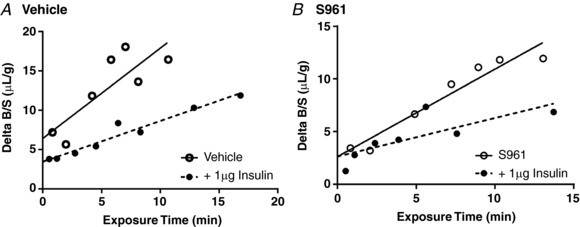
A, levels of 125I‐insulin present in whole brain was measured after injection ± 1 μg unlabelled insulin. B, to evaluate the impact of S961 on the saturability of 125I‐insulin transport, separate mice were co‐injected with 1 μg S961 ± 1 μg unlabelled insulin. Values are corrected for vascular space and expressed as the Delta B/S ratio (μl/g). Linear regression analysis was included (straight or dashed line) to depict the influx.
Table 3.
Saturability of insulin transport into the whole brain
| Group | K i (μl/(g min)) | P | r | V i (μl/g) |
|---|---|---|---|---|
| Vehicle | 1.149 ± 0.35 | 0.83 | 6.41 ± 2.20 | |
| Vehicle + 1 μg insulin | 0.518 ± 0.05* | 0.043 | 0.97 | 3.45 ± 0.42 |
| S961 | 0.830 ± 0.10 | 0.97 | 2.64 ± 0.79 | |
| S961 + 1 μg insulin | 0.365 ± 0.13* | 0.018 | 0.78 | 2.65 ± 0.85 |
Pharmacokinetics of insulin transport into the whole brain when co‐administered with or without unlabelled insulin. Levels of 125I‐insulin present in whole brain were measured 1–10 min after co‐injection with either Vehicle or 1 μg S961 ± 1 μg unlabelled insulin and linear data plotted in Fig. 7. Multiple‐time regression analysis was performed to determine the influx of insulin (K i) and vascular binding (V i). Data are presented as means ± SEM. * P < 0.05 vs. respective control group (Vehicle or S961).
Effect of inhibition of the endothelial insulin receptor on regional distribution of insulin
To determine if insulin distribution throughout the brain was different with inhibition of the insulin receptor, we co‐injected vehicle or 1 μg S961 with 125I‐insulin and 99mTc‐albumin and measured the amount of 125I‐insulin present in the brain regions 5 min after intravenous injection (Fig. 7 A). There was no difference in the 125I‐insulin level in any brain region except in the hypothalamus, in which S961 resulted in a 56% reduction. The regional difference in insulin levels suggests insulin transport and/or insulin receptor binding in the hypothalamus could occur via different mechanisms compared to other brain regions. To investigate which contributing factor (transport or binding) occurs in the hypothalamus, we measured the kinetics of 125I‐insulin transport into the hypothalamus of CD‐1 mice with or without 1 μg S961 (Fig. 7 B). We found S961 decreased the binding (S961 V i = −1.76 ± 5.4 μl/g vs. Vehicle V i = 11.42 ± 3.8 μl/g, P < 0.001) in this brain region rather than the influx (S961 K i = 1.66 ± 0.64 μl/(g min) vs. Vehicle K i = 1.27 ± 0.46 μl/(g min), P = 0.62).
Figure 7. Brain distribution of insulin with inhibition of the insulin receptor.
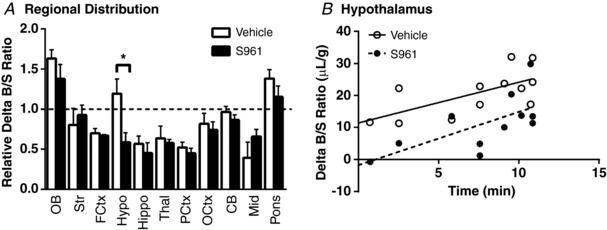
A, levels of 125I‐insulin were measured in brain regions collected after 5 min in mice co‐injected with vehicle (n = 4, open bars) or 1 μg S961 (n = 6, filled bars). Each brain region is corrected for vascular space and expressed relative to the whole brain Delta B/S ratio. All data are expressed as means ± SEM (* P < 0.05, t test). B, level of 125I‐insulin in hypothalamus was measured in mice co‐injected with vehicle (n = 12, open circles) or 1 μg S961 (n = 11, filled circles). Values are corrected for vascular space and expressed as the Delta B/S ratio (μl/g). Linear regression analysis was included (straight or dashed line) to depict the influx. OB, olfactory bulb; Str, striatum; FCtx, frontal cortex; Hypo, hypothalamus; Hippo, hippocampus; Thal, thalamus; PCtx, parietal cortex; OCtx, occipital cortex; CB, cerebellum; Mid, midbrain.
Discussion
The results presented here suggest that insulin transport across the BBB can occur independently of the signalling‐related insulin receptor. By using two separate approaches to inhibit signalling‐related insulin receptor binding at the BBB, a genetic model (EndoIRKO mice) and a pharmacological inhibitor (S961), we investigated the pharmacokinetics (radioactive and immunoactive) of insulin transport across the BBB. We report the influx (K i) of insulin is not significantly altered, although the amount of insulin binding the endothelium (V i) is decreased, with loss of the signalling receptor. Insulin stability in serum was increased with the use of S961. In addition, inhibiting the binding of insulin to the signalling‐related insulin receptor did not block the ability of insulin to fully cross the BBB or affect saturability of insulin transport across the BBB.
BECs contain many insulin‐binding sites (Frank et al. 1985; Miller et al. 1994), but which sites are transporters vs. signalling receptors is unknown. To investigate whether insulin transport into the brain in vivo was dependent on the signalling‐related insulin receptor, we first used mice lacking this insulin receptor on endothelial cells through Cre recombination driven by the Cdh5 (VE‐cadherin) promoter (EndoIRKO mice) (Konishi et al. 2017; Wang et al. 2017). Our results, as well as others (Konishi et al. 2017), suggest there is not complete loss of the signalling‐related insulin receptor in BECs. Since the reported V i in our study for insulin in the EndoIRKO mice is not zero, it is likely there is insulin binding to the residual signalling‐related insulin receptor. We compare the results obtained from the genetic deletion of the signalling‐related insulin receptor in endothelial cells to the use of the selective signalling‐related insulin receptor antagonist S961 (Schaffer et al. 2008). Our data using S961 show that it fully inhibits insulin binding yet does not affect insulin transport across the BBB into whole brain.
Insulin stability in serum was increased with addition of S961. Since the inhibitor is similar to the structure of insulin, it is possible enzymes responsible for insulin degradation targeted the inhibitor rather than the radioactive insulin. In addition, since the inhibitor blocks the ability of insulin to bind to the insulin receptor, there could be a concurrent increase in the availability of 125I‐labelled insulin in the circulation. As our kinetic studies correct for the amount of 125I‐insulin present in the circulation, the increased level of intact radiolabelled insulin does not alter our results.
Insulin is labelled by attaching the radioactive iodide to tyrosine (Banks et al. 1997a). Radiolabelled insulin has been used since at least the 1980s to investigate insulin binding to the insulin receptor (Frank et al. 1985, 1986). The only radioactive non‐peptide fragments generated could be iodide and iodotyrosine. Iodide transport across the BBB is minimal (Banks et al. 1997a). In addition, there is a brain‐to‐blood efflux transport system in place for iodide (Cserr & Berman, 1978), while there is no efflux system for insulin (Cashion et al. 1996). This combination would limit any net accumulation of radioactive fragments in the brain. Insulin that has been inactivated by freezing and radioactively labelled is not transported across the BBB. This suggests insulin must be in the biologically active conformation for transport (Urayama & Banks, 2008). In addition, our current results and previous studies have found immunoactive levels of human insulin after peripheral administration in brain that align with the radioactive studies (Banks et al. 1997c). Also, in our study, unlabelled insulin inhibited the radioactive insulin transport. These data support the idea that the radioactivity recovered from the brain after intravenous injection is intact 125I‐insulin. Interestingly, the measures of variance were much greater for the kinetics parameters when determined with immunoactive than with radioactive insulin. While the linear regression was not significant for the insulin‐only group (r = 0.459, P = 0.252), the S961 group did have a significant linear regression (r = 0.651, P = 0.0218). With the low levels of insulin entry into the CNS, this experiment shows how much more sensitive the radioactive studies can be. This is presumably because of the greater sensitivity and reproducibility that is achievable with radioactivity. Similar to previous studies (Banks et al. 1999), our capillary depletion data showed 125I‐insulin was not being sequestered by BECs. These experiments allow us to better interpret the data obtained from the kinetic experiments.
To determine the influx of insulin into the brain, we used the highly sensitive multiple‐time regression analysis technique (Patlak et al. 1983) adapted to modern graphical applications by using Prism to calculate the linear regression. Multiple‐time regression analysis shows that the influx of insulin across the BBB in fed wild type mice (0.870 ± 0.21 μl/(g min)) is similar to the mean of previously reported influxes, 0.75 μl/(g min) (Banks et al. 1997a). When we investigated insulin transport in EndoIRKO mice or mice with S961, the influx was not significantly affected compared to their controls despite the slight variance. The correlation coefficients (r) for each linear regression were very significant, suggesting adequate power to detect significant differences. The S961 result is in contrast to published data stating the major pathway for brain microvascular insulin uptake is via insulin receptor‐mediated transfer when also using a high dose of S961 (Meijer et al. 2016). However, only a single time point sampling of brain radioactive insulin levels was taken and so transport vs. receptor binding could not be distinguished. In addition, the dose of S961 used to define insulin transport in Sprague–Dawley rats was about 60× greater than the highest dose used in the current study. It may be at very high levels that S961 does interfere with transport of insulin across the BBB. We also verified pre‐treating mice with S961 had no effect by administering S961 30 min prior to measuring 125I‐insulin BBB transport. Even with pre‐treatment, we saw no differences in the influx and insulin binding was still significantly decreased (data not shown). While a paper by the same group also showed S961 prevented insulin uptake in an in vitro BEC model (Gray et al. 2017), a more recently published paper investigating the role of S961 in an in vitro BEC model by a separate group showed insulin transport across the BEC can occur independently of the insulin receptor (Hersom et al. 2018). These discrepancies should be taken into consideration when working with in vitro models as the culturing conditions (high levels of insulin in the media required) and tightness of the BEC barrier can be quite different.
In a seminal paper by King & Johnson (1985), it was shown in vitro that insulin was transported across aortic endothelial cells and transport was saturable, similar to our in vivo results. Yet, using an antibody derived from patients with insulin resistance, insulin binding to the endothelial cell and transport across the endothelium was inhibited in a dose‐dependent manner. Perhaps it is possible the antibody, being polyclonal, also recognized the protein responsible for insulin transport. However, a recent paper showed that insulin transport across the muscle endothelium was similarly independent of the insulin receptor using S961 (Williams et al. 2018). Our results show for the first time in vivo that insulin transport into the brain still occurs with loss of the endothelial signalling‐related insulin receptor.
We show that differences in the amount of insulin binding to the brain endothelium is significantly altered with loss or inhibition of the signalling‐related receptor. This is expected as the receptor serves as a primary binding site for insulin and it has been shown insulin can bind to brain capillaries (Schwartz et al. 1990). Using the EndoIRKO mice and S961 inhibitor, we experimentally reduced or removed the main receptor binding site for insulin on endothelial cells, as confirmed by the decrease in V i. S961 inhibits the insulin receptor on any cell type. However, EndoIRKO mice do not have complete insulin receptor knockout in all BECs and, therefore, the binding of insulin to residual endothelial cell insulin receptors or on other cells types (such as epithelial cells in the choroid plexus) likely contributes to the V i observed here. The decrease in V i also serves as a control for successful conditional knockout and inhibition of the receptor in endothelial cells. Together, these data indicate that the signalling‐related insulin receptor does bind circulating insulin at the lumen of BECs, but is not the sole protein responsible for transporting insulin across the BBB into the CNS interstitium.
It has previously been reported that fed EndoIRKO serum insulin levels are approximately two times greater than their wild type littermates (Konishi et al. 2017). In addition, when S961 is given i.v., serum insulin levels increase approximately threefold (Vikram & Jena, 2010). We observed the same increase in serum insulin level in fed EndoIRKO mice and 6 h‐fasted CD‐1 mice 5 min after 1 μg S961 i.v. injection (data not shown). However, if anything, the increased level of serum insulin would decrease the influx of insulin transport across the BBB since the system is saturable. The transporter for insulin has been shown to be totally saturable at serum concentrations of 10 ng/ml in mice (Banks et al. 1997c). In addition, the data from our fed vs. fasted mice suggest the change in serum insulin level that occurs with short‐term fasting is not enough to alter transport across the BBB.
Previous studies have shown the olfactory bulb, pons–medulla and hypothalamus have the greatest levels of insulin transport (Banks & Kastin, 1998; Banks et al. 1999). We observed similar regional findings in our study. Interestingly, the level of insulin present in the hypothalamus relative to whole brain levels was decreased with S961. We followed up this study to measure the pharmacokinetics of insulin transport into the hypothalamus in the presence of S961. We found that while the influx was not altered with insulin receptor inhibition, receptor binding was significantly decreased.
A small percentage of insulin can be transported by the separate transport system for IGF‐1 (Yu et al. 2006). Insulin can bind to the IGF‐1 receptor but with much lower affinity (Wilcox, 2005). However, there is no compensation of the IGF‐1 receptor protein levels in the endothelium of EndoIRKO mice as measured in the aorta (Konishi et al. 2017). In addition, it is not likely the IGF‐1 receptor compensates completely to transport insulin with acute inhibition of the insulin receptor with S961. The affinity of S961 for the insulin receptor is four magnitudes greater than for IGF‐1, and has an even greater affinity for the insulin receptor than insulin itself (Schaffer et al. 2008). Lastly, as the transport system for insulin has a much higher capacity than that for IGF‐1 (Yu et al. 2006), we would still expect to observe a decrease in insulin transport with loss of the receptor if transport were dependent on the signalling‐related insulin receptor. Therefore, it is not likely the IGF‐1 receptor is responsible for insulin transport across the BBB. Additionally, megalin (or LRP‐2) has been suggested to bind and internalize insulin (Orlando et al. 1998), in addition to many other hormones, including leptin and IGF‐1. However, the transport systems for leptin and insulin have been shown to be independent of one another (Banks et al. 1996). We did test whether an inhibitor for megalin using the receptor‐associated protein (RAP) inhibited insulin transport across the BBB and found no difference (data not shown).
The question of whether or not the signalling‐related insulin receptor is also the transporter for insulin has prevailed for at least 30 years (Banks, 2004). The data presented here show that the signalling‐related insulin receptor is not absolutely required for transport of insulin across the BBB. To compare, there are substances that are transported across the BBB by their receptor, such as tumour necrosis factor α (TNFα) (Pan & Kastin, 2002). However, in most cases, the receptor for a particular peptide is not also responsible for the transport of the peptide across the BBB. For example, the transporter for PACAP27, a pluripotent neuropeptide, is peptide transport system‐6, and differs from the canonical signalling‐related receptors PAC1, VPAC1 and VPAC2 (Dogrukol‐Ak et al. 2009). In addition, epidermal growth factor (Pan & Kastin, 1999) transporters differ from their signalling‐related receptor.
Determining that the signalling‐related insulin receptor is not the BBB insulin transport‐related protein has many important ramifications. It provides a mechanism by which insulin resistance at peripheral tissues or in the brain does not necessarily involve insulin transport. By extension, discovering the protein(s) responsible for transport of insulin may provide insight on the marked variations in insulin BBB transport due to the physiological state (Banks & Kastin, 1998). It is not surprising there are multiple proteins involved in insulin signalling and transport due to the many regulations it has within the CNS. In addition, identifying the transporter for insulin would contribute to understanding various diseases due to a deficiency of insulin transport, including disruptions in the CNS/periphery connection in the metabolic syndrome and changes in cognition due to decreased levels of CNS insulin. Regarding drug delivery, the insulin receptor has long been targeted by Trojan horse approaches because of the assumption that BBB insulin transport is insulin receptor mediated. In retrospect, these approaches are likely inducing adsorptive endocytosis/transcytosis mechanisms, routing the cargo primarily to lysosomes.
As the role for insulin in various neurodegenerative diseases is becoming more apparent, understanding the regulation of insulin transport into the brain will aid in treatments. Indeed, insulin levels and receptors in the CNS are also decreased with Alzheimer's disease and age (Craft et al. 1998; Frolich et al. 1998) which could be a result of transporter dysfunction. Of course, the metabolic regulation of CNS insulin will be greatly impacted by the discovery of the insulin transport‐related protein. Knowing how to enhance the transport of insulin to the CNS will aid in many therapeutic conditions. In conclusion, this is the first in vivo evidence that the signalling‐related insulin receptor may not be solely responsible for transport of insulin across the BEC and into the brain.
Additional information
Competing interests
None declared.
Author contributions
The experiments were carried out at the VA Puget Sound in Seattle, WA, USA. E.M.R. and W.A.B. designed the experiments. E.M.R. executed the experiments. E.M.R., C.R.‐M. and W.A.B. interpreted the data. E.M.R., C.R.‐M. and W.A.B. wrote and reviewed the manuscript. All authors approved the final version of the manuscript and agree to be accountable to the accuracy and integrity of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by the National Institutes of Health (T32AG000057 to E.M.R., R21CA185196 to C.R.‐M. and RO1AG046619 to W.A.B.) and by the Veterans Affairs Puget Sound Health Care System Research & Development.
Acknowledgements
We would like to thank Dr Lauge Schäffer from Novo Nordisk (Denmark) for providing the S961 peptide.
Biography
Elizabeth M. Rhea's current research interests focus on the role of insulin in the brain and ways to increase CNS insulin. She believes this paper has a huge impact on fields in which CNS insulin is implicated: obesity, diabetes, Alzheimer's. Many programmes are targeting the insulin receptor to deliver drugs and therapies to the CNS with minimal success. We know little about how substrates are actually transported across the BBB. Her ultimate research goal is to characterize this transport mechanism in order to target the endogenous insulin transport system, treat CNS insulin resistance, and offer an alternative route for drug delivery.

Edited by Laura Bennet & Weifang Rong
This is an Editor's Choice article from the 1 October 2018 issue.
References
- Banks WA (2004). The source of cerebral insulin. Eur J Pharmacol 490, 5–12. [DOI] [PubMed] [Google Scholar]
- Banks WA, Jaspan JB, Huang W & Kastin AJ (1997a). Transport of insulin across the blood‐brain barrier: saturability at euglycemic doses of insulin. Peptides 18, 1423–1429. [DOI] [PubMed] [Google Scholar]
- Banks WA, Jaspan JB & Kastin AJ (1997b). Effect of diabetes mellitus on the permeability of the blood‐brain barrier to insulin. Peptides 18, 1577–1584. [DOI] [PubMed] [Google Scholar]
- Banks WA, Jaspan JB & Kastin AJ (1997c). Selective, physiological transport of insulin across the blood‐brain barrier: novel demonstration by species‐specific radioimmunoassays. Peptides 18, 1257–1262. [DOI] [PubMed] [Google Scholar]
- Banks WA & Kastin AJ (1998). Differential permeability of the blood‐brain barrier to two pancreatic peptides: insulin and amylin. Peptides 19, 883–889. [DOI] [PubMed] [Google Scholar]
- Banks WA, Kastin AJ, Huang W, Jaspan JB & Maness LM (1996). Leptin enters the brain by a saturable system independent of insulin. Peptides 17, 305–311. [DOI] [PubMed] [Google Scholar]
- Banks WA, Kastin AJ & Pan W (1999). Uptake and degradation of blood‐borne insulin by the olfactory bulb. Peptides 20, 373–378. [DOI] [PubMed] [Google Scholar]
- Banks WA, Owen JB & Erickson MA (2012). Insulin in the brain: there and back again. Pharmacol Ther 136, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar RS, DeRose A, Sandra A, Peacock ML & Owen WG (1983). Insulin binding to microvascular endothelium of intact heart: a kinetic and morphometric analysis. Am J Physiol 244, E447–E452. [DOI] [PubMed] [Google Scholar]
- Baura GD, Foster DM, Porte D Jr, Kahn SE, Bergman RN, Cobelli C & Schwartz MW (1993). Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J Clin Invest 92, 1824–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J & Kern W (2004). Intranasal insulin improves memory in humans. Psychoneuroendocrinology 29, 1326–1334. [DOI] [PubMed] [Google Scholar]
- Blasberg RG, Fenstermacher JD & Patlak CS (1983). Transport of α‐aminoisobutyric acid across brain capillary and cellular membranes. J Cereb Blood Flow Metab 3, 8–32. [DOI] [PubMed] [Google Scholar]
- Cashion MF, Banks WA & Kastin AJ (1996). Sequestration of centrally administered insulin by the brain: effects of starvation, aluminum, and TNF‐α. Horm Behav 30, 280–286. [DOI] [PubMed] [Google Scholar]
- Chan JY, LaPara K & Yee D (2016). Disruption of insulin receptor function inhibits proliferation in endocrine‐resistant breast cancer cells. Oncogene 35, 4235–4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A, Callaghan M, Arbuckle M, Behl C & Craft S (2015). Long acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early‐stage Alzheimer's disease dementia. J Alzheimers Dis 45, 1269–1270. [DOI] [PubMed] [Google Scholar]
- Craft S, Newcomer J, Kanne S, Dagogo‐Jack S, Cryer P, Sheline Y, Luby J, Dagogo‐Jack A & Alderson A (1996). Memory improvement following induced hyperinsulinemia in Alzheimer's disease. Neurobiol Aging 17, 123–130. [DOI] [PubMed] [Google Scholar]
- Craft S, Peskind E, Schwartz MW, Schellenberg GD, Raskind M & Porte D Jr (1998). Cerebrospinal fluid and plasma insulin levels in Alzheimer's disease: relationship to severity of dementia and apolipoprotein E genotype. Neurology 50, 164–168. [DOI] [PubMed] [Google Scholar]
- Cserr HF & Berman BJ (1978). Iodide and thiocyanate efflux from brain following injection into rat caudate nucleus. Am J Physiol 235, F331–F337. [DOI] [PubMed] [Google Scholar]
- Dogrukol‐Ak D, Kumar VB, Ryerse JS, Farr SA, Verma S, Nonaka N, Nakamachi T, Ohtaki H, Niehoff ML, Edwards JC, Shioda S, Morley JE & Banks WA (2009). Isolation of peptide transport system‐6 from brain endothelial cells: therapeutic effects with antisense inhibition in Alzheimer and stroke models. J Cereb Blood Flow Metab 29, 411–422. [DOI] [PubMed] [Google Scholar]
- Duffy KR & Pardridge WM (1987). Blood‐brain‐barrier transcytosis of insulin in developing rabbits. Brain Res 420, 32–38. [DOI] [PubMed] [Google Scholar]
- Elgee NJ, Williams RH & Lee ND (1954). Distribution and degradation studies with insulin I131 . J Clin Invest 33, 1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank HJ, Jankovic‐Vokes T, Pardridge WM & Morris WL (1985). Enhanced insulin binding to blood‐brain barrier in vivo and to brain microvessels in vitro in newborn rabbits. Diabetes 34, 728–733. [DOI] [PubMed] [Google Scholar]
- Frank HJ, Pardridge WM, Morris WL, Rosenfeld RG & Choi TB (1986). Binding and internalization of insulin and insulin‐like growth factors by isolated brain microvessels. Diabetes 35, 654–661. [DOI] [PubMed] [Google Scholar]
- Frolich L, Blum‐Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A, Hoyer S, Zochling R, Boissl KW, Jellinger K & Riederer P (1998). Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm (Vienna) 105, 423–438. [DOI] [PubMed] [Google Scholar]
- Glowinski J & Iversen LL (1966). Regional studies of catecholamines in the rat brain. I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J Neurochem 13, 655–669. [DOI] [PubMed] [Google Scholar]
- Gray SM, Aylor KW & Barrett EJ (2017). Unravelling the regulation of insulin transport across the brain endothelial cell. Diabetologia 60, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugaard N, Vaughan M, Haugaard ES & Stadie WC (1954). Studies of radioactive injected labeled insulin. J Biol Chem 208, 549–563. [PubMed] [Google Scholar]
- Hersom M, Helms HC, Schmalz C, Pedersen TA, Buckley ST & Brodin B (2018). The insulin receptor is expressed and functional in cultured blood‐brain barrier endothelial cells, but does not mediate insulin entry from blood‐to‐brain. Am J Physiol Endocrinol Metab (in press; 10.1152/ajpendo.00350.2016). [DOI] [PubMed] [Google Scholar]
- Jialal I, Crettaz M, Hachiya HL, Kahn CR, Moses AC, Buzney SM & King GL (1985). Characterization of the receptors for insulin and the insulin‐like growth factors on micro‐ and macrovascular tissues. Endocrinology 117, 1222–1229. [DOI] [PubMed] [Google Scholar]
- Kastin AJ & Pan W (2008). Blood‐brain barrier and feeding: regulatory roles of saturable transport systems for ingestive peptides. Curr Pharm Des 14, 1615–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GL & Johnson SM (1985). Receptor‐mediated transport of insulin across endothelial cells. Science 227, 1583–1586. [DOI] [PubMed] [Google Scholar]
- Konishi M, Sakaguchi M, Lockhart SM, Cai W, Li ME, Homan EP, Rask‐Madsen C & Kahn CR (2017). Endothelial insulin receptors differentially control insulin signalling kinetics in peripheral tissues and brain of mice. Proc Natl Acad Sci U S A 114, E8478–E8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer RI, Gray SM, Aylor KW & Barrett EJ (2016). Pathways for insulin access to the brain: the role of the microvascular endothelial cell. Am J Physiol Heart Circ Physiol 311, H1132–H1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DW, Keller BT & Borchardt RT (1994). Identification and distribution of insulin receptors on cultured bovine brain microvessel endothelial cells: possible function in insulin processing in the blood‐brain barrier. J Cell Physiol 161, 333–341. [DOI] [PubMed] [Google Scholar]
- Nelson TJ, Sun MK, Hongpaisan J & Alkon DL (2008). Insulin, PKC signalling pathways and synaptic remodelling during memory storage and neuronal repair. Eur J Pharmacol 585, 76–87. [DOI] [PubMed] [Google Scholar]
- Novak V, Milberg W, Hao Y, Munshi M, Novak P, Galica A, Manor B, Roberson P, Craft S & Abduljalil A (2014). Enhancement of vasoreactivity and cognition by intranasal insulin in type 2 diabetes. Diabetes Care 37, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando RA, Rader K, Authier F, Yamazaki H, Posner BI, Bergeron JJ & Farquhar MG (1998). Megalin is an endocytic receptor for insulin. J Am Soc Nephrol 9, 1759–1766. [DOI] [PubMed] [Google Scholar]
- Pan W & Kastin AJ (1999). Entry of EGF into brain is rapid and saturable. Peptides 20, 1091–1098. [DOI] [PubMed] [Google Scholar]
- Pan W & Kastin AJ (2002). TNFα transport across the blood‐brain barrier is abolished in receptor knockout mice. Exp Neurol 174, 193–200. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, Eisenberg J & Yang J (1985). Human blood‐brain barrier insulin receptor. J Neurochem 44, 1771–1778. [DOI] [PubMed] [Google Scholar]
- Patlak CS, Blasberg RG & Fenstermacher JD (1983). Graphical evaluation of blood‐to‐brain transfer constants from multiple‐time uptake data. J Cereb Blood Flow Metab 3, 1–7. [DOI] [PubMed] [Google Scholar]
- Porte D Jr, Baskin DG & Schwartz MW (2005). Insulin signalling in the central nervous system: a critical role in metabolic homeostasis and disease from C. elegans to humans. Diabetes 54, 1264–1276. [DOI] [PubMed] [Google Scholar]
- Procaccini C, Santopaolo M, Faicchia D, Colamatteo A, Formisano L, de Candia P, Galgani M, De Rosa V & Matarese G (2016). Role of metabolism in neurodegenerative disorders. Metabolism 65, 1376–1390. [DOI] [PubMed] [Google Scholar]
- Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, Fishel MA, Plymate SR, Breitner JC, DeGroodt W, Mehta P & Craft S (2008). Intranasal insulin improves cognition and modulates β‐amyloid in early AD. Neurology 70, 440–448. [DOI] [PubMed] [Google Scholar]
- Salameh TS, Bullock KM, Hujoel IA, Niehoff ML, Wolden‐Hanson T, Kim J, Morley JE, Farr SA & Banks WA (2015). Central nervous system delivery of intranasal insulin: mechanisms of uptake and effects on cognition. J Alzheimers Dis 47, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer L, Brand CL, Hansen BF, Ribel U, Shaw AC, Slaaby R & Sturis J (2008). A novel high‐affinity peptide antagonist to the insulin receptor. Biochem Biophys Res Commun 376, 380–383. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Bergman RN, Kahn SE, Taborsky GJ Jr, Fisher LD, Sipols AJ, Woods SC, Steil GM & Porte D Jr (1991). Evidence for entry of plasma insulin into cerebrospinal fluid through an intermediate compartment in dogs. Quantitative aspects and implications for transport. J Clin Invest 88, 1272–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Figlewicz DF, Kahn SE, Baskin DG, Greenwood MR & Porte D Jr (1990). Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic Zucker rats. Peptides 11, 467–472. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Figlewicz DP, Baskin DG, Woods SC & Porte D Jr (1992). Insulin in the brain: a hormonal regulator of energy balance. Endocr Rev 13, 387–414. [DOI] [PubMed] [Google Scholar]
- Triguero D, Buciak J & Pardridge WM (1990). Capillary depletion method for quantification of blood‐brain barrier transport of circulating peptides and plasma proteins. J Neurochem 54, 1882–1888. [DOI] [PubMed] [Google Scholar]
- Urayama A & Banks WA (2008). Starvation and triglycerides reverse the obesity‐induced impairment of insulin transport at the blood‐brain barrier. Endocrinology 149, 3592–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Houten M & Posner BI (1979). Insulin binds to brain blood vessels in vivo. Nature 282, 623–625. [DOI] [PubMed] [Google Scholar]
- Vicent D, Ilany J, Kondo T, Naruse K, Fisher SJ, Kisanuki YY, Bursell S, Yanagisawa M, King GL & Kahn CR (2003). The role of endothelial insulin signalling in the regulation of vascular tone and insulin resistance. J Clin Invest 111, 1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikram A & Jena G (2010). S961, an insulin receptor antagonist causes hyperinsulinemia, insulin‐resistance and depletion of energy stores in rats. Biochem Biophys Res Commun 398, 260–265. [DOI] [PubMed] [Google Scholar]
- Wang X, Haring MF, Rathjen T, Lockhart SM, Sorensen D, Ussar S, Rasmussen LM, Bertagnolli MM, Kahn CR & Rask‐Madsen C (2017). Insulin resistance in vascular endothelial cells promotes intestinal tumour formation. Oncogene 36, 4987–4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox G (2005). Insulin and insulin resistance. Clin Biochem Rev 26, 19–39. [PMC free article] [PubMed] [Google Scholar]
- Williams IM, Valenzuela FA, Kahl SD, Ramkrishna D, Mezo AR, Young JD, Wells KS & Wasserman DH (2018). Insulin exits skeletal muscle capillaries by fluid‐phase transport. J Clin Invest 128, 699–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Kastin AJ & Pan W (2006). Reciprocal interactions of insulin and insulin‐like growth factor I in receptor‐mediated transport across the blood‐brain barrier. Endocrinology 147, 2611–2615. [DOI] [PubMed] [Google Scholar]
- Zar JH (1984). Biostatistical Analysis. Prentice‐Hall, Englewood Cliffs, NJ, USA. [Google Scholar]