

Abstract
Key points
Synaptic transmission relies on the recruitment of neurotransmitter‐filled vesicles to presynaptic release sites. Increased intracellular calcium buffering slows the recovery from synaptic depression, suggesting that vesicle recruitment is a calcium‐dependent process.
However, the molecular mechanisms of vesicle recruitment have only been investigated at some synapses.
We investigate the role of calcium in vesicle recruitment at the cerebellar mossy fibre to granule cell synapse. We find that increased intracellular calcium buffering slows the recovery from depression following physiological stimulation. However, the recovery is largely resistant to perturbation of the molecular pathways previously shown to mediate calcium‐dependent vesicle recruitment.
Furthermore, we find two pools of vesicles with different recruitment speeds and show that models incorporating two pools of vesicles with different calcium‐independent recruitment rates can explain our data. In this framework, increased calcium buffering prevents the release of intrinsically fast‐recruited vesicles but does not change the vesicle recruitment rates themselves.
Abstract
During sustained synaptic transmission, recruitment of new transmitter‐filled vesicles to the release site counteracts vesicle depletion and thus synaptic depression. An elevated intracellular Ca2+ concentration has been proposed to accelerate the rate of vesicle recruitment at many synapses. This conclusion is often based on the finding that increased intracellular Ca2+ buffering slows the recovery from synaptic depression. However, the molecular mechanisms of the activity‐dependent acceleration of vesicle recruitment have only been analysed at some synapses. Using physiological stimulation patterns in postsynaptic recordings and step depolarizations in presynaptic bouton recordings, we investigate vesicle recruitment at cerebellar mossy fibre boutons. We show that increased intracellular Ca2+ buffering slows recovery from depression dramatically. However, pharmacological and genetic interference with calmodulin or the calmodulin–Munc13 pathway, which has been proposed to mediate Ca2+‐dependence of vesicle recruitment, barely affects vesicle recovery from depression. Furthermore, we show that cerebellar mossy fibre boutons have two pools of vesicles: rapidly fusing vesicles that recover slowly and slowly fusing vesicles that recover rapidly. Finally, models adopting such two pools of vesicles with Ca2+‐independent recruitment rates can explain the slowed recovery from depression upon increased Ca2+ buffering. Our data do not rule out the involvement of the calmodulin–Munc13 pathway during stronger stimuli or other molecular pathways mediating Ca2+‐dependent vesicle recruitment at cerebellar mossy fibre boutons. However, we show that well‐established two‐pool models predict an apparent Ca2+‐dependence of vesicle recruitment. Thus, previous conclusions of Ca2+‐dependent vesicle recruitment based solely on increased intracellular Ca2+ buffering should be considered with caution.
Keywords: Synapse, short‐term plasticity, vesicle recruitment, calcium buffering
Key points
Synaptic transmission relies on the recruitment of neurotransmitter‐filled vesicles to presynaptic release sites. Increased intracellular calcium buffering slows the recovery from synaptic depression, suggesting that vesicle recruitment is a calcium‐dependent process.
However, the molecular mechanisms of vesicle recruitment have only been investigated at some synapses.
We investigate the role of calcium in vesicle recruitment at the cerebellar mossy fibre to granule cell synapse. We find that increased intracellular calcium buffering slows the recovery from depression following physiological stimulation. However, the recovery is largely resistant to perturbation of the molecular pathways previously shown to mediate calcium‐dependent vesicle recruitment.
Furthermore, we find two pools of vesicles with different recruitment speeds and show that models incorporating two pools of vesicles with different calcium‐independent recruitment rates can explain our data. In this framework, increased calcium buffering prevents the release of intrinsically fast‐recruited vesicles but does not change the vesicle recruitment rates themselves.
Introduction
During short bursts of activity, some synapses exhibit short‐term depression (Zucker & Regehr, 2002). One major contribution to depression is the depletion of rapidly releasable presynaptic vesicles (Schneggenburger et al. 2002; Neher, 2015). Vesicle consumption is counterbalanced by the simultaneous recruitment of new vesicles to vacant presynaptic release sites. At a variety of synapses, evidence for an acceleration of vesicle recruitment has been observed during and following high‐frequency synaptic activity. This acceleration has been hypothesized to arise from an accumulation of calcium (Ca2+) within the presynaptic terminal and was often probed by the application of membrane‐permeable Ca2+ buffers (Zucker, 1989; Wang & Kaczmarek, 1998). Particularly, a slowing of the recovery from synaptic depression upon buffer application was taken as evidence for the hypothesis that Ca2+ accelerates vesicle recruitment (Dittman & Regehr, 1998; Stevens & Wesseling, 1998; von Gersdorff et al. 1998; Wang & Kaczmarek, 1998; Gomis et al. 1999; Wang & Manis, 2008; Babai et al. 2010), although see also Wu & Borst (1999) or Mennerick & Matthews (1996). However, the molecular mechanisms mediating accelerated vesicle recruitment are poorly understood and have only been investigated in more detail at a few types of synapses. At the calyx of Held, an extensively studied auditory brain stem presynaptic terminal (Forsythe, 1994; Borst & Soria van Hoeve, 2012), direct presynaptic inhibition of calmodulin demonstrated the Ca2+‐dependence of vesicle recruitment (Sakaba & Neher, 2001a). Subsequently, calmodulin was found to bind Munc13‐1 at a conserved binding domain, and binding could be disrupted by a single point mutation (Munc13‐1W464R; Junge et al. 2004). In cultured hippocampal synapses and in the calyx of Held, Munc13‐1W464R shows an enhanced synaptic depression and a slower recovery from depression (Junge et al. 2004; Lipstein et al. 2013).
At the cerebellar mossy fibre boutons (cMFB) to granule cell (GC) synapse, previous studies explained the time‐course of short‐term plasticity with models assuming Ca2+‐independent vesicle recruitment (Saviane & Silver, 2006; Hallermann et al. 2010). Furthermore, release kinetics of cMFBs during prolonged depolarization in the presence of EGTA provides circumstantial evidence for Ca2+‐independent vesicle recruitment (Ritzau‐Jost et al. 2014). However, Ca2+‐dependence of vesicle recruitment and the Ca2+‐calmodulin–Munc13‐1 pathway have not been studied at the cMFB to GC synapse. Therefore, we investigated the mechanisms of vesicle recruitment at cMFB to GC synapses, which are ideally suited for investigating these mechanisms because cMFBs reliably transmit high frequencies (van Kan et al. 1993; Jörntell & Ekerot, 2006; Rancz et al. 2007) and glutamate release is mediated by a few release sites with rapid vesicle recruitment (Hallermann & Silver, 2013).
Methods
Ethical approval
All experiments were approved in advance by the Institutional Ethics Committees and animals were treated in accordance with the European (EU Directive 2010/63/EU, Annex IV for animal experiments), National and Leipzig, as well as Göttingen University, guidelines. All investigators understand the Journal of Physiology’s ethical principles and their work complies with the animal ethics checklist.
Brain slice preparation
Experiments were performed in mature mice (≥ postnatal day 21) of either sex and of the following genetic backgrounds: C57BL/6 (referred to as wild‐type) (Figs 1, 2 and 4) or Munc13‐1 mutant mice (Munc13‐1W464R/W464R referred to as Munc13‐1W464R compared to their homozygous wild‐type allele littermates, Munc13‐1WT/WT, termed ‘control’) (Fig. 3). Animals for the experiments shown in Figs 1, 2 and 4 were bred at the Medical Experimental Center of the Faculty of Medicine, Leipzig University (Leipzig, Germany) and animals for the experiments shown in Fig. 3 were bred at the Max Planck Institute of Experimental Medicine (Göttingen, Germany). The experimenter had no prior information on the animal genotypes and individual experiments were analysed before the experimenter was unblinded. Munc13‐1 mutant and control mice were kindly provided by Nils Brose (Max‐Planck‐Institute of Experimental Medicine, Göttingen, Germany) and they were genotyped before and after the experiments by PCR. All animals were fed ad libitum. Mice were anaesthetized with isoflurane and thereafter rapidly killed by decapitation. Cerebella were sliced parasagittal into 300 μm thin slices (VT1200 vibratome; Leica Microsystems, Wetzlar, Germany) and subsequently kept in artificial cerebrospinal fluid (aCSF; for composition, see ‘Solutions’ below) at 37°C for 30 min before being transferred to aCSF at room temperature until the experiments were performed.
Figure 1. EGTA‐AM slows recovery from synaptic depression.
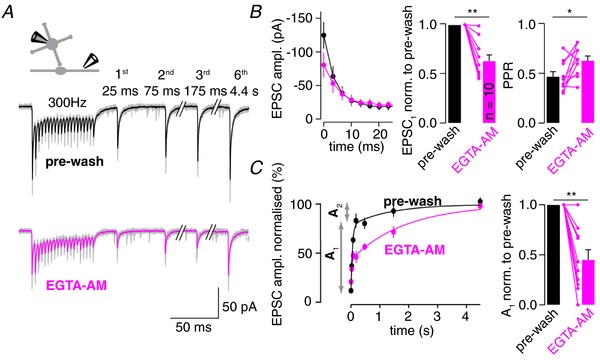
A, example EPSC trains elicited by 20 axonal stimulations at 300 Hz before (pre‐wash) and after wash‐in of EGTA‐AM (100 μm) via the bath perfusion in a single experiment (individual traces in grey, average before and after wash‐in in black and magenta, respectively). Trains were followed by stimuli at different time points addressing recovery from synaptic depression. Inset: illustration of a whole‐cell patch clamp recording from a GC and extracellular stimulation of a mossy fibre axon. B, Left: average time‐course of the first eight EPSC amplitudes within the train before and after EGTA‐AM wash‐in. Middle: average amplitude of initial train EPSC (EPSC1) after EGTA‐AM application normalized to the pre‐wash amplitude. Right: PPR before and after EGTA‐AM wash‐in. C, Left: average recovery of EPSC amplitudes following 300 Hz stimulation before and after EGTA‐AM wash‐in superimposed with bi‐exponential fits. The fast initial and slower second recovery components of the pre‐wash data are indicated by labelled arrows (A 1 and A 2). Right: average reduction of the amplitude of A 1 by EGTA‐AM normalized to the pre‐wash A 1. Colour code in (B) and (C) as in (A); dots in (B) and (C), as well as all bar graphs, denote the mean ± SEM; lines and dots in bar graphs depict individual experiments; Wilcoxon signed‐rank test, * P < 0.05, ** P < 0.01; n = 10 for (B) and (C), where n indicates the number of recordings from different cells.
Figure 2. A membrane‐permeable calmodulin blocker does not alter synaptic recovery.
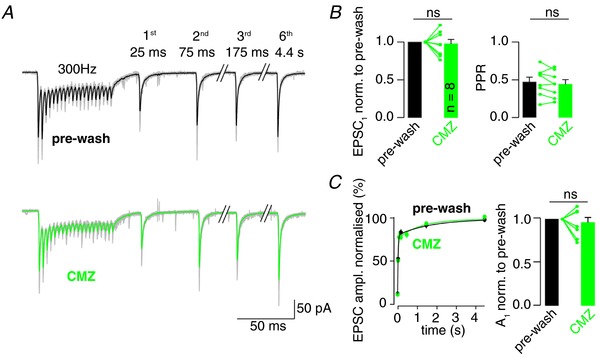
A, example 300 Hz EPSC trains and recovery EPSCs before (pre‐wash) and after bath application of CMZ (10 μm) in a single experiment (individual traces in grey, average for pre‐wash and CMZ in black and green, respectively). B, Left: average amplitude of EPSC1 after CMZ application normalized to the pre‐wash amplitude. Right: PPR before and after CMZ wash‐in. C, Left: average recovery of EPSC amplitudes following 300 Hz stimulation before and after CMZ wash‐in superimposed with bi‐exponential fits. Right: average reduction of the fast component of the bi‐exponential fit (A 1) by CMZ normalized to the corresponding value before wash‐in. Colour code in (B) and (C) as in (A); dots in (C), as well as all bar graphs, denote the mean ± SEM; lines and dots in bar graphs depict effect in single experiments; Wilcoxon signed‐rank test, ns indicates P > 0.05; n = 8 for (B) and (C), where n indicates the number of recordings from different cells.
Figure 4. Paired pre‐ and postsynaptic recordings reveal two pools of vesicle with different, calmodulin‐independent recruitment speeds.

A, example voltage command for cMFB (V m), Ca2+ currents in cMFB (I Ca), EPSCs in GC (I post) and cumulative release rate (N ves) during paired 3 ms depolarizations with an interstimulus interval (ISI) of 30 ms. Left inset: illustration of a simultaneous whole‐cell patch clamp recording from GC and cMFB. Inset in N ves: superposition of N ves during the first and second 3 ms depolarization (scale bar = 500 μs). B, superposition of N ves during the first (left) and second (right) 3 ms depolarization for different ISIs as indicated by colour code. C, average Ca2+ current amplitude (I Ca), average total number of released vesicles (N) and average number of rapidly released vesicles (N 1) plotted vs. ISIs for control peptide (20 μm, black) and MLCK peptide (20 μm, green). N and N 1 are normalized to the values of the first 3 ms depolarization; n = 7 and 5 for recordings with the control and MLCK peptide, respectively, where n is the number of paired recordings.
Figure 3. Interference with the calmodulin‐Munc13‐1 pathway does not alter synaptic recovery.
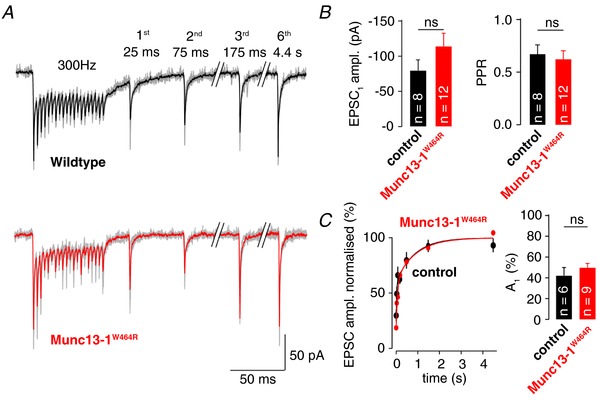
A, example EPSC trains evoked at 300 Hz with recovery EPSCs in control and Munc13‐1W464R mice (single experiments, individual traces in grey, mean traces for wild‐type and Munc13 mutants in black and red, respectively). B, Left: average amplitude of EPSC1 in control and Munc13‐1W464R mice. Right: PPR in control and Munc13‐1W464R mice. C, Left: average recovery of EPSC amplitudes following 300 Hz train stimulation and bi‐exponential fits to the average recovery time courses in control and Munc13‐1W464R mice. Right: average fractional amplitude of the fast component of the bi‐exponential fit (A 1) for control and Munc13‐1W464R mice. Colour code in (B) and (C) as in (A); dots in (C), as well as all bar graphs, denote the mean ± SEM; Mann–Whitney U test; ns indicates P > 0.05; n = 8 and 12 for control and Munc13‐1W464R mice, where n indicates the number of recordings from different cells; for analysis of A 1 in (C), two recordings for control and three for Munc13‐1W464R mice were excluded because the recovery time course was mono‐exponential.
Axonal mossy fibre train stimulation
Postsynaptic whole‐cell patch clamp recordings from GCs were performed using borosilicate glass pipettes (Science Products, Hofheim, Germany) pulled to a pipette resistance (R pip) of 5–7 MΩ (DMZ Universal Electrode Puller; Zeitz Instruments, Martinsried, Germany). Patch pipettes were filled with a potassium gluconate‐based solution, whereas, for extracellular mossy fibre stimulations, borosilicate pipettes (R pip ∼ 10 MΩ) were loaded with aCSF. The experiments were performed at room temperature unless noted otherwise. All current clamp and voltage clamp recordings from cMFBs and GCs were performed with an EPC10/2 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). Pre‐ and postsynaptic recordings were corrected for liquid junction potentials by 4.2 and 12 mV, respectively. For the stimulation of mossy fibre axons, the surface of the brain slice surrounding the recorded GC was screened with a second electrode until EPSCs in GCs were evoked reliably. Only those experiments in which stimulation evoked EPSCs with a rapid monophasic rising, a single peak, and a similar amplitude (for single stimuli), as well as constant initial and steady‐state EPSC amplitudes (for trains), were interpreted as resulting from solitary mossy fibre stimulation and included in the analysis.
Train stimulation of mossy fibre axons was performed as described in Hallermann et al. (2010) and Ritzau‐Jost et al. (2014). In short, after identification of solitary mossy fibre inputs, train stimulations were repeated every 30 s throughout the individual experiments and EGTA‐AM (100 μm) or calmidazolium (CMZ) (10 μm), both dissolved in 0.1% DMSO, was added to the bath perfusion after 5 min of stable stimulation (‘pre‐wash’ in Figs 1 and 2). After EPSCs reached steady‐state amplitudes following wash‐in (at least 5 min following wash‐in granted in each experiment), EPSCs were assigned to the EGTA‐AM/CMZ subgroup (Figs 1 and 2). For control experiments, DMSO alone was added to the bath perfusion (0.1% final concentration) but did not show any effect on synaptic transmission (n = 8; data not shown). Presynaptic train stimulation (20× at 300 Hz) was followed by recovery pulses 25, 75, 175, 475, 1475 and 4475 ms after the last train EPSC to quantify recovery from synaptic depression. EPSC amplitudes were analysed as the phasic component of the EPCS as described previously (Hallermann et al. 2010). EPSC amplitudes during recovery (normalized to the amplitude of the first EPSC in the train) were fitted with bi‐exponential fits revealing time constants τ1 and τ2 with amplitudes A 1 and A 2, respectively. The paired‐pulse ratio (PPR) was calculated as the ratio of the second over the first phasic EPSC amplitude during the train. Data were analysed using Igor Pro, version 6.32A (WaveMetrics, Lake Oswego, OR, USA), extended by Patcher's Power Tool, version 2.19 (http://www3.mpibpc.mpg.de/groups/neher/index.php?page=aboutppt) and NeuroMatic, version 2.00 (Rothman & Silver, 2018, http://www.neuromatic.thinkrandom.com/) plug‐ins, as well as self‐written analysis routines.
Paired recordings between GCs and cMFBs
Recordings were performed at 35–37°C essentially as described previously (Ritzau‐Jost et al. 2014). In short, GC patch solution contained a fluorescent dye (20 μm Atto 488 carboxy or 20 μm Atto 594 carboxy; ATTO‐TEC, Siegen, Germany) and neighbouring cMFBs were identified by means of infrared differential interference contrast optics (Eclipse FN‐1 microscope; Nikon, Tokyo, Japan) with a 100× objective (NA 1.1). After establishing a whole cell presynaptic patch clamp configuration, a paired recording was assumed if repetitive brief presynaptic depolarizations reliably evoked immediate fast rising GC EPSCs. Dual voltage pulses of 3 ms duration each were applied presynaptically at different interstimulus intervals (10, 30, 100, 300, 1000 and 3000 ms; defined as the end of the first depolarization until initiation of the second depolarization). Simultaneously, presynaptic Ca2+ currents and postsynaptic EPSCs were recorded. Ca2+ current amplitudes were calculated from the steady‐state current at the end of the depolarization preceding the tail current. EPSCs were used to calculate presynaptic vesicular release rates by deconvolution. Only experiments with uncompensated pre‐ and postsynaptic series resistances below 35 and 25 MΩ, respectively, were considered for the analysis. Series resistances were electronically compensated by 50–80% (10 or 100 μs).
Deconvolution
Deconvolution of postsynaptic currents (Fig. 4) was performed as described by Neher & Sakaba (2001) and was identical to the reported implementation of this method at the cMFB to GC synapse (Ritzau‐Jost et al. 2014). The postsynaptic current (I post) was then used to calculate the release rate (not shown) and the cumulative release rate (N ves). Cumulative release rates during both 3 ms pulses were fitted with the sum of two exponential functions. Time constants were constrained to the average time constants determined previously (426 μs and 5.27 ms for τ1 and τ2, respectively; Ritzau‐Jost et al. 2014) and the size of the fast‐releasing vesicle pool (N 1) was determined by the amplitude of the first component of this fit.
Modelling
The simple two‐pool model (Fig. 5 A) was based on model 3 of Hallermann et al. (2010) and run with the C++ compiler of XCode 9 on macOSX 10.13 (Apple Inc., Cupertino, CA, USA). In short, a pool of high‐ and low‐p r vesicles was considered. The number of low‐p r vesicles was four times higher and the release probability was two times lower compared to high‐p r vesicles. High‐p r vesicles were slowly recruited from the pool of low‐p r vesicles (rate constant ∼0.5 s−1). Low‐p r vesicles were rapidly recruited from the pool of supply vesicles (rate constant ∼33 s−1). The number of supply vesicles was 300 per high‐p r vesicle (Saviane & Silver, 2006), resulting in almost no reduction in the number of supply vesicles during our train of 20 action potentials. All rate constants were Ca2+‐independent. Synaptic facilitation was implemented according to Markram et al. (1998). Similar results were obtained when another model of facilitation was used (data not shown; Trommershäuser et al. 2003; for parameters, see Hallermann et al. 2010). The best‐fit release probability of the high‐ and low‐p r vesicles was 0.88 and 0.44, respectively, for control and 0.76 and 0.24, respectively, for EGTA‐AM data. This corresponds to a reduction of 13% and 44% for high‐ and low‐p r vesicles, respectively. All other parameters were identical to the values in Hallermann et al. (2010) with minor adjustments of the fast and slow rate constant of vesicle recruitment (50.1 and 0.50 s–1, respectively) to fit the data in the present study (identical for control and EGTA‐AM data). We also tested a model in which EGTA reduces the release probability and blocks synaptic facilitation.
Figure 5. A simple two‐pool model predicts an apparent Ca2+‐dependence of vesicle recruitment.

A, illustration of simple model assuming two pools of vesicles (N low pr and N high pr) displaying high and low release probability (p r). N high pr vesicles are slowly recruited from N low pr vesicles and N low pr are rapidly recruited from a supply pool (not shown). Increasing presynaptic Ca2+ buffering (EGTA) reduces the release probability of N low pr more than that of N high pr vesicles. In some models, EGTA blocks facilitation of release probabilities. B, release probabilities of low‐ and high‐p r vesicles (top) and EPSC amplitudes normalized to the first EPSC amplitude (bottom, dots indicate recorded data adopted from Fig. 1) during 300 Hz stimulation. Predictions of the model described in (A) are shown as lines (solid black line for model with pre‐wash; solid magenta line for a model including EGTA and facilitation unaffected by EGTA; dashed magenta line for a model with EGTA fully blocking facilitation). C, average experimental (Fig. 1) and simulated reduction of EPSC1 amplitude (left) and PPR (right). D, release probabilities of low‐ and high‐p r vesicles (top) and normalized EPSC amplitudes (bottom) recorded during the end of the train and the initial recovery (left) and during the entire recovery (right) superimposed with the predictions of the models described in (A) (colour code as in B). E, average reduction of the fast recovery component (A 1) recorded (Fig. 1) and predicted by the models described in (A). Dots in (B) and (D), as well as all bar graphs, denote the mean ± SEM; solid bars depict recorded data, model prediction as open bars; * P < 0.05, ** P < 0.01 as in Fig. 1.
The 3‐D model (Fig. 6 A) was based on Delvendahl et al. (2015) and run with CalC 6.9.1 (Matveev et al. 2002) and post‐processed with Mathematica 10 (Wolfram Research, Champaign, IL, USA). The five‐site kinetic model of Schneggenburger & Neher (2000) was used with the parameters of Wang et al. (2008) obtained for the mature calyx of Held. Rates of the scheme were adjusted by a Q 10 value of 2.5 and by slight adjustment of the affinity with a scaling factor of 1.065 as described in Delvendahl et al. (2015) resulting in k on = 4.06 × 108 s–1·m −1, k off = 4.42 × 104 s−1, vesicle fusion rate (γ) = 1.80 × 104 s–1 and b = 0.25. Two types of vesicles were added to the model with distances of 6.5 and 15 nm to the nearest Ca2+ channel, resulting in a release probability of 0.88 and 0.45, respectively. The number of remote vesicles was four times higher than the number of closely‐coupled vesicles. The addition of 20 mm EGTA with constrained binding and unbinding rates for Ca2+ (Smith et al. 1984; Delvendahl et al. 2015) resulted in a release probability of 0.77 and 0.24, respectively. The rate constants of vesicle recruitment were identical to the simple two‐pool model and the supply pool was assumed to be infinite. The default grid size was 50 × 50 × 30 in x–y–z directions and the default CalC‐accuracy parameter was 10−5. Varying the grid size between 40 × 40 × 30 and 100 × 100 × 60 in x–y–z directions and the CalC‐accuracy parameter between 10−4 and 10−6 provided almost identical results (e.g. <2% difference in release probability).
Figure 6. A 3‐D model also predicts an apparent Ca2+‐dependent recruitment.

A, illustration of the 3‐D model with two types of vesicles and Ca2+ channel to vesicle distances of 6.5 and 15 nm, respectively. The vesicles close to Ca2+ channels experience higher local Ca2+ transients and therefore have a higher p r. The recruitment rates are, as in the simple two‐pool model, Ca2+‐independent, slow for close vesicles and fast for remote vesicles. To simulate the EGTA‐AM data, EGTA at a concentration of 20 mm with realistic Ca2+ binding and unbinding rates was added to the simulation. B, release probabilities of low‐ and high‐p r vesicles (top) and EPSC amplitudes normalized to the first EPSC amplitude (bottom) during 300 Hz stimulation superimposed with the predictions of the 3‐D model described in (A) (dots indicate recorded data adopted from Fig. 1; black solid line for model with pre‐wash condition; solid magenta line for EGTA condition). C, experimental and simulated reduction of EPSC1 amplitude (left) and PPR (right). D, release probabilities of low‐ and high‐p r vesicles (top) and normalized EPSC amplitudes (bottom) during the end of the train and the initial recovery (left) and the entire recovery (right) superimposed with the predictions of the 3‐D model (colour code as in B). E, recorded average reduction of the fast recovery component (A 1) and prediction of the 3‐D model described in (A). Dots in (B) and (D), as well as all bar graphs, denote the mean ± SEM; recorded data are depicted as solid bars, model prediction as open bars, * P < 0.05, ** P < 0.01.
Another 3‐D model with two pools differing not in distance to Ca2+ channels but instead with respect to their molecular priming (i.e. sensitivity to Ca2+) was identical to the model shown in Fig. 6, but the coupling distance was 15 nm for both pools of vesicles. The k on and k off rates of the Ca2+ sensor for fusion were multiplied by 1.3 and 0.7, respectively, for high‐p r vesicles, and by 0.75 and 1.3, respectively, for low‐p r vesicles. This resulted in a release probability of 0.88 and 0.45 for high‐ and low‐p r vesicles, respectively. The addition of 20 mm EGTA with constrained binding and unbinding rates for Ca2+ (Smith et al. 1984; Delvendahl et al. 2015) decreased the release probability to 0.77 and 0.30, respectively (data not shown). The code to reproduce the data is available (see ‘Code’ section below).
Solutions
The extracellular solution (aCSF) during mossy fibre stimulation contained (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 20 glucose, 2 CaCl2 and 1 MgCl2. For wash‐in experiment, EGTA‐AM or CMZ were added to the bath perfusion in individual experiments (bath concentration of 100 and 10 μm for EGTA‐AM and CMZ, respectively; both dissolved in 0.1% DMSO. The intracellular solution used for postsynaptic recordings contained (in mm): 150 K‐gluconate, 3 Mg‐ATP, 0.3 Na‐GTP, 10 K‐Hepes, 10 NaCl and 0.05 or 0.2 EGTA. For the EPSC recordings shown in Figs 1, 2, 3, 4, 10 μm dl‐2‐amino‐5‐phospho‐novalerate (DL‐APV) was added to the extracellular solution.
For paired recordings (Fig. 4), 0.001 mm TTX, 5 μm 4‐AP and 20 μm TEA‐Cl were added to the aCSF. 100 μm cyclothiazide and 1 mm kynurenic acid were added to the aCSF to prevent postsynaptic AMPA receptor desensitization and saturation, respectively. This approach is based on Neher & Sakaba (2001) and the high concentration of kynurenic acid is intended to counterbalance the increased AMPA receptor affinity induced by cyclothiazide (Fucile et al. 2006).
The intracellular presynaptic patch solution contained (in mm): 135 CsCl, 20 TEA‐Cl, 4 MgATP, 0.3 NaGTP, 5 Na2‐phosphocreatine, 10 Hepes and 0.2 EGTA. Furthermore, either 20 μm of the myosin light chain kinase (MLCK) peptide (alias calmodulin inhibitory peptide; Calbiochem, San Diego, CA, USA) or 20 μm of a calmodulin inhibitory control peptide (alias control peptide; Calbiochem) were added to the presynaptic pipette solution. Chemicals were obtained from Sigma‐Aldrich (St Louis, MO, USA) unless otherwise stated.
Statistical analysis
If not noted otherwise, data are expressed as the mean ± SEM. Sample sizes for each group (n) are provided as recordings from individual GCs or as individual paired recordings. The total number of animals used was 10 for EGTA‐AM, six for CMZ, six and eight for Munc13W464R and Munc13WT/WT, and six and five for each control condition and CaM inhibitory peptide treatment, respectively, in paired recordings. Results were excluded from further analysis if uncompensated pre‐ or postsynaptic series resistance increased above 35 or 25 MΩ, respectively, holding currents steadily exceeded −50 pA, experiments lasted less than 10 min following wash‐in (Figs 1 and 2), experiments had less than six traces suitable for averaging and experiments included solitary synaptic inputs that were not stimulated reliably (see ‘Axonal mossy fibre train stimulation’ above). For dependent (Figs 1 and 2) and independent samples (Figs 3 and 4), the Wilcoxon signed rank and the Mann–Whitney U tests were coducted, respectively, with Igor Pro.
Code
The C++ code to reproduce the two‐pool model results and the CalC scripts and Mathematica code to reproduce the 3‐D model results are available at: https://github.com/HallermannLab/2018_JP_ApparentCa.
Results
EGTA‐AM slows recovery from synaptic depression
To investigate the recovery from synaptic depression at the cMFB to GC synapse, we recorded EPSCs in GCs evoked by high‐frequency extracellular stimulation of mossy fibre axons (20 stimuli at 300 Hz followed by stimuli at increasing intervals of 25 ms to 5 s). Simulations were repetitively performed before and after bath application of the membrane‐permeable Ca2+ buffer EGTA‐AM (Fig. 1 A). EGTA‐AM application induced a reduction in the initial EPSC amplitude to 63 ± 6% of the pre‐wash amplitude (P = 0.002; n = 10; Wilcoxon signed‐rank test) and a slight increase in the PPR from 0.47 ± 0.05 to 0.63 ± 0.05 (P = 0.048; n = 10; Wilcoxon signed‐rank test) (Fig. 1 B). Before EGTA‐AM application, the time course of recovery from synaptic depression was bi‐exponential with time constants of τ1 = 53 ± 10 ms and τ2 = 2060 ± 710 ms and a relative amplitude of the fast component (A 1) of 69 ± 7% (n = 10). Following EGTA‐AM application, the recovery was profoundly slowed with time constants similar to pre‐wash values (τ1 = 46 ± 14 ms and τ2 = 2530 ± 430 ms) but with a strong reduction in the amplitude of the fast component to 45 ± 10% of the pre‐wash value (P = 0.004; n = 10; Wilcoxon signed‐rank test) (Fig. 1 C). These data show that increased Ca2+ buffering strongly inhibits rapid recovery of synaptic transmission from depression at cMFBs.
A membrane‐permeable calmodulin blocker does not alter synaptic recovery
Because a slowing of synaptic recovery by Ca2+ buffers would be consistent with the findings of a Ca2+‐dependent acceleration of vesicle recruitment (Wang & Kaczmarek, 1998), we next investigated the Ca2+‐calmodulin–Munc13‐1 pathway, which is a well‐established molecular interaction mediating Ca2+‐dependent vesicle recruitment. Therefore, we repeated the experiments shown in Fig. 1 but washed‐in CMZ, a membrane‐permeable calmodulin blocker (Fig. 2 A). Bath application of CMZ (10 μm) had no significant effect on initial EPSC amplitude or PPR (P = 0.64 and 0.55, respectively; n = 8; Wilcoxon signed‐rank test) (Fig. 2 B). Furthermore, the bi‐exponential recovery time course of EPSC amplitudes was resistant to CMZ (τ1 = 30 ± 9 ms and τ2 = 2330 ± 520 ms before wash‐in, after CMZ application τ1 = 34 ± 11 ms and τ2 = 2690 ± 1360 ms; A 1 = 79 ± 4% and 76 ± 6% before and after wash‐in of CMZ, respectively; P = 0.25; n = 8; Wilcoxon signed‐rank test) (Fig. 2 C). These findings suggest that calmodulin does not significantly contribute to vesicle recruitment at the cMFB to GC synapse during physiologically short, high‐frequency activity.
Interfering with the calmodulin–Munc13‐1 pathway does not alter synaptic recovery
CMZ is applied extracellularly and we cannot exclude a scenario in which it does not sufficiently enter the presynaptic terminal. Therefore, we next investigated knock‐in mice expressing Munc13‐1 with a point mutation that precisely interferes with the calmodulin–Munc13‐1 interaction (Munc13‐1W464R) (Lipstein et al. 2013). We again applied 300 Hz train stimulations and compared responses elicited in GCs of Munc13‐1W464R mice and their control littermates (Fig. 3 A). Initial EPSC amplitudes were not significantly different between mutant and control mice (80 ± 15 pA and 114 ± 19 pA for control and Munc13‐1W464R, respectively; n = 8 and 12; P = 0.25, Mann–Whitney U test) (Fig. 3 B) and tended towards slightly larger amplitudes in mutant mice in accordance with previous observations (Junge et al. 2004; Lipstein et al. 2013). Furthermore, mutant mice did not show differences in PPR (0.67 ± 0.09 and 0.58 ± 0.06 with n = 8 and 12 for control and Munc13‐1W464R; P = 0.59, Mann–Whitney U test) or recovery following a 300 Hz stimulation (τ1 = 56 ± 21 ms and τ2 = 2260 ± 550 ms for control; τ1 = 83 ± 23 ms and τ2 = 1460 ± 410 ms for Munc13‐1W464R; n = 6 and 9 respectively; P = 0.16; Mann–Whitney U test) (Fig. 3 C). These data show that the calmodulin–Munc13‐1 pathway has a marginal impact on the time course of recovery from synaptic depression following brief high‐frequency transmission at cMFB to GC synapses.
Paired pre‐ and postsynaptic recordings reveal two pools of vesicles with different, calmodulin ‐independent recruitment speeds
Interference with the Ca2+‐calmodulin–Munc13‐1 pathway was so far achieved by extracellular application of CMZ, as well as a specific mutation abolishing the calmodulin–Munc13‐1 interaction. To ensure successful direct and acute blockade of the presynaptic pathway, we next performed simultaneous paired voltage clamp recordings from presynaptic cMFBs and the postsynaptic granule cell (Fig. 4 A). To limit exocytosis to the amount observed during physiological 300 Hz train stimulation, voltage clamp stimuli of 3 ms to 0 mV were applied (Ritzau‐Jost et al. 2014). Evoked presynaptic Ca2+ currents and postsynaptic EPSCs were simultaneously recorded and the cumulative release (N ves) was calculated based on deconvolution methods (Fig. 4 A). To monitor recovery from the first stimulus, a second stimulus (3 ms to 0 mV) was applied to the presynaptic terminal with varying interstimulus intervals (10–3000 ms) (Fig. 4 B). The number of vesicles released during the 3 ms depolarization (N) recovered with a bi‐exponential time course (Fig. 4 C, middle). To dissect the recovery of fast and slowly releasing vesicles and to relate these findings to action potential–evoked release, the cumulative release was fitted bi‐exponentially, with the fast and slow component representing the fast (N 1) and slowly (N 2) released vesicle, respectively (Ritzau‐Jost et al. 2014). A slow recovery of N 1 was observed, indicating that fast releasable vesicles recovered slowly with a time constant of 2.3 s. The rapid component of recovery in N thus corresponds to slowly releasing vesicles (N 2). These data indicate that two pools of synaptic vesicles exist at cMFBs and that the fast releasing vesicles recover slowly, whereas the slow releasing vesicles recover rapidly.
To test the role of calmodulin in vesicle recruitment, MLCK peptide (20 μm), a potent inhibitor of calmodulin, was included in the presynaptic pipette solution (Sakaba & Neher, 2001a). The inclusion of MLCK peptide affected neither presynaptic Ca2+ current amplitudes, nor the vesicular release time course (Fig. 4 C). Particularly, the recovery time course of fast releasable vesicles was unchanged in the presence of MLCK peptide (Fig. 4 C). Consistent with our previous findings with CMZ and mutant mice, these experiments indicate a limited contribution of calmodulin to vesicle recruitment upon short stimulation at cMFB to GC synapses.
A simple two‐pool model predicts an apparent Ca2+‐dependence of vesicle recruitment
To investigate whether the two pools of vesicles with different recruitment speeds can explain the observed effect of EGTA‐AM, we first used a simple kinetic state model adopting two pools of vesicles with different release probabilities (p r) and different Ca2+ ‐independent recruitment rates, which has previously been established for this synapse (Hallermann et al. 2010). The parameters were only marginally adjusted to obtain the best‐fit results for the pre‐wash data. Next, the EGTA‐AM data were fit to the model allowing the release probability of both pools to change to a different extent, whereas all other parameters of the model were kept constant (i.e. only two free parameters). The best‐fit to the EGTA‐AM data was obtained by a 13% and 44% reduction in release probability for high‐p r and low‐p r vesicles, respectively. Surprisingly, the model precisely predicted the initial time course of EPSC depression (Fig. 5 B), first EPSC amplitude and the PPR (Fig. 5 C), steady‐state depression and time course of recovery (Fig. 5 D), and also the amplitude of the rapid component of recovery (Fig. 5 E) for both the pre‐wash and the EGTA‐AM condition.
To investigate the impact of facilitation, we also tested a model in which synaptic facilitation is fully blocked by EGTA (Fig. 5, dashed line). The best‐fit to the EGTA‐AM data revealed a 48% reduction in release probability of low‐p r vesicles. The reduction in release probability of high‐p r was constrained to 13%. This model reproduced the recovery well, but not the depression, arguing against a complete block of facilitation by EGTA, as also reported previously (Atluri & Regehr, 1998; Turecek & Regehr, 2018). Independent of the blocking effect of EGTA on facilitation, both models predict a slowing of the recovery from depression upon EGTA application. Thus, simple models assuming two heterogeneous vesicle pools with Ca2+‐independent recruitment rates can explain our data.
A 3‐D model also predicts an apparent Ca2+‐dependent vesicle recruitment
In addition to the simple models described above, we used a more realistic 3‐D model of the active zone previously established for the cMFB to GC synapse (Delvendahl et al. 2015). In this model, we also implemented two pools of vesicles with different Ca2+‐independent recruitment rates (Fig. 6 A) and adjusted the vesicle to Ca2+ channel coupling distance to 6.5 and 15 nm, respectively, to obtain a best‐fit to the control data. Note that, in this model, the facilitation is not a free parameter but, instead, results from the interplay of Ca2+ diffusion, buffering, and the Ca2+‐binding and ‐unbinding rates of the release sensors. EGTA‐AM data were simulated by adding 20 mm EGTA with previously defined Ca2+‐binding and ‐unbinding rate constants (Smith et al. 1984; Delvendahl et al. 2015). Despite the highly constrained parameters of the model, the experimental data were captured well. Particularly, the effect of EGTA on the fast component of recovery (Fig. 6 E) was reproduced. Thus, a more realistic 3‐D model of active zones featuring two pools of vesicles with different recruitment speeds and coupling distance to Ca2+ channels corroborates the results of the simple two‐pool models and demonstrates the apparent Ca2+‐dependence of vesicle recruitment. In this framework, EGTA decelerates the recovery from depression simply by preferentially blocking release sites that have an intrinsically rapid recruitment speed.
Discussion
We show that, following short bursts of high‐frequency transmission, EGTA profoundly slows the recovery from depression (Fig. 1), although calmodulin and the Ca2+‐calmodulin–Munc13‐1 pathway do not contribute significantly at cMFB to GC synapses (Figs 2, 3, 4). Furthermore, we show that cMFBs contain two pools of releasable vesicles with different release probabilities and recruitment speeds (Fig. 4) and this finding by itself predicts a strong effect of EGTA on the speed of recovery from depression (Figs 5 and 6). We conclude that application of EGTA, a widely‐used tool to probe the role of Ca2+, is not sufficient to confirm Ca2+‐dependence of vesicle recruitment at synapses.
Apparent Ca2+‐dependence of vesicle recruitment
The main finding of the present study is that two pools of vesicles with different coupling distances to Ca2+ channels and different recruitment speeds predict an apparent Ca2+‐dependence of vesicle recruitment. Our simulations demonstrate that EGTA can have a strong effect on the speed of recovery from synaptic depression even when the individual rates of vesicle recruitment are Ca2+ ‐independent. Intuitively, this can be easily understood because (i) EGTA preferentially blocks the release of vesicles remote from Ca2+ channels and (ii) it is a straightforward assumption that these vesicles are intrinsically recruited faster compared to vesicles that have to closely colocalize to Ca2+ channels via a time‐consuming process. In this framework, the activity‐dependent acceleration of vesicle recruitment is mediated not by a Ca2+‐dependent recruitment rate but, instead, by the intrinsically fast‐recruited remote vesicles, for which the release probability exhibits activity‐depended facilitation (Wu & Borst, 1999).
In our experiments, EGTA significantly reduced the amplitude of the first EPSC in the train stimulation (Fig. 1 B). In some studies at other synapses, EGTA did not reduce the basal EPSC amplitude but still slowed recovery from depression (Yang & Xu‐Friedman, 2008; Cho et al. 2011; Johnson et al. 2017). If the basal amplitude is not reduced, the two‐pool mechanism described in the present study cannot provide an explanation for the slowed recovery upon EGTA application. Thus, the present study suggests the need to carefully investigate the effect of EGTA on release probability (i.e. the EPSC amplitude). If EGTA has no effect on the basal EPSC amplitude, the conclusion of a Ca2+‐dependent vesicle recruitment based on a slowed recovery appears to be more justified.
We cannot rule out the possibility that other molecular pathways could contribute to the effect of Ca2+ on recovery speed observed in the present study at the cMFB. Particularly, Ca2+‐dependent pathways involving the interaction of diacylgylcerol/phospholipase C or Ca2+/phospholipids with Munc13s (Rhee et al. 2002; Lou et al. 2008; Shin et al. 2010; Lee et al. 2013), Ca2+ channel facilitation and/or inactivation (Forsythe et al. 1998; Taschenberger et al. 2002; Xu & Wu, 2005; Lin et al. 2012) and Ca2+‐calmodulin‐dependent kinases (Sun et al. 2006; Lee et al. 2008; Srinivasan et al. 2008) could be obstructed by EGTA. However, our data clearly demonstrate that EGTA is a non‐sufficient tool for analysing the Ca2+‐dependence of vesicle recruitment at synapses. Instead, interference with a specific molecular pathway is required to reveal the role of Ca2+ in vesicle recruitment (Sakaba & Neher, 2001a; Junge et al. 2004; Wang & Manis, 2008; Lee et al. 2012; Lipstein et al. 2013; Van Hook & Thoreson, 2014).
The Ca2+‐calmodulin–Munc13‐1 pathway
We observed a minor role of the Ca2+‐calmodulin–Munc13‐1 pathway in vesicle recruitment at cMFBs (Figs 2, 3, 4). However, several caveats should be emphasixed with respect to the possible roles of the pathway. First, our results do not rule out a significant contribution of the Ca2+‐calmodulin–Munc13‐1 pathway during stronger types of stimulation at the cMFB. It has previously been shown that the Ca2+‐calmodulin–Munc13‐1 pathway is not involved in vesicle recruitment triggered by weaker stimuli, such as 5 ms depolarization at the calyx of Held (Sakaba & Neher, 2001a) or during the initial phase of train stimulation (Lee et al. 2012). Thus, our data appear to be consistent with previous studies at other synapses and stronger stimuli might well trigger vesicle recruitment mediated by the Ca2+‐calmodulin–Munc13‐1 pathway at cMFBs. However, this is not the focus of the present study. By contrast, the novelty of the present study is the demonstration of a strong effect of EGTA on the recovery from depression, which is not mediated by calmodulin and the Ca2+‐calmodulin–Munc13‐1 pathway. We show that this can be explained by a two‐pool mechanism. This option has previously been considered (Wu & Borst, 1999) but, to our knowledge, the apparent Ca2+‐dependence of vesicle recruitment has not been demonstrated before.
The second caveat concerns indications suggesting that the importance of Ca2+ and calmodulin in vesicle recruitment might change developmentally. Although it was shown that blocking calmodulin by CMZ slows vesicle recruitment during 100 Hz stimulation in young animals (Lee et al. 2012), recruitment after stimulation with up to 200 Hz was resistant to EGTA‐AM and CMZ in older animals (Lee et al. 2012). Furthermore, the involvement of calmodulin in other processes, such as Ca2+ current inactivation and vesicle endocytosis, was shown to decrease during development (Nakamura et al. 2008; Yamashita et al. 2010). However, it should also be noted that a developmental decrease was not observed when vesicle recruitment was compared for Munc13‐1W464R mice at between P9–12 and P14–17 days (Lipstein et al. 2013). Thus, our data do not exclude an involvement of the Ca2+‐calmodulin–Munc13‐1 pathway in vesicle recruitment at cMFBs of mice that are younger than those investigated in the present study. Finally, a subset of our recordings was performed at room temperature (postsynaptic train recordings in Figs. 1, 2, 3), although it was recently shown that, for example, fast endocytosis strongly depends on temperature (Delvendahl et al. 2016).
Two pools of vesicles
Two pools of release ready vesicles have probably been described best at the calyx of Held: fast‐ and slow‐releasing vesicle pools (FRP and SRP) (Sakaba & Neher, 2001a, b). However, the SRP vesicles appear to contribute little to synchronous action potential–evoked release at the calyx of Held (Sakaba, 2006). Therefore, the two pools of vesicles used in our models to describe synchronous action potential–evoked release at cMFBs most probably do not correspond to FRP and SRP vesicles at the calyx of Held (Delvendahl & Hallermann, 2016). It was recently reported that FRP vesicles at the calyx of Held can be subdivided further into FRP1 (normally primed) and FRP2 (super‐primed) vesicles (Lee et al. 2013; Taschenberger et al. 2016). Both pools can be released upon action potential stimulation and FRP2 exhibited higher release probabilities than FRP1. The two pools used in our models (Figs 5 and 6) therefore most probably correspond to FRP1 and FRP2 vesicles at the calyx of Held. Based on this interpretation, the rapid Ca2+ ‐independent recruitment of our low‐p r vesicles corresponds to the previously described rapid Ca2+ ‐independent transition of SRP to FRP vesicles at the calyx (referred to as SRP‐dependent recovery) (Lee et al. 2012). Consistently, super‐priming is considered to be a slow process occurring over the time scale of seconds (Neher, 2017), as was the rate constant found in the present study for the recruitment of high‐p r vesicles. Two pools of release‐ready vesicles have also been described at many other types of synapses and their molecular distinctions are currently being revealed (Böhme et al. 2016; Datta et al. 2017; Wentzel et al. 2018).
Molecular or positional priming
The apparent Ca2+‐dependent vesicle recruitment demonstrated in the present study relies on a differential effect of EGTA on the two pools of vesicles. This is predicted if both pools have different coupling distances to the Ca2+ channels (Fig. 6). In addition to the model with different coupling distances to Ca2+ channels, we have also tested a model assuming two pools of vesicles with same distance to Ca2+ channels but with different molecular priming (i.e. sensitivity to Ca2+; data not shown). Despite the same distance of the vesicles to the Ca2+ channels, EGTA reduced the release probability of the vesicles with low Ca2+ sensitivity slightly more compared to the vesicles with high Ca2+ sensitivity. Therefore, this model also predicted the apparent Ca2+‐dependence of vesicle recruitment, although the effect was smaller. Thus, a significant differential effect of EGTA on the two pools of vesicles requires that the two pools differ in their coupling distance to Ca2+ channels. This assumption appears to be reasonable (time consuming migration of vesicles towards Ca2+ channels) but has not been tested at cMFBs. At the calyx of Held, FRP1 and FRP2 have been suggested to differ in their degree of molecular priming (Taschenberger et al. 2016) and the phospholipase C‐diacylglycerol pathway appears to be involved in the transition from FRP1 to FPR2 (Lee et al. 2013), although differential positional priming of FRP1 and FRP2 has not been studied to our knowledge. Interestingly, long‐term plasticity at hippocampal mossy fibre boutons, which might be mediated by increased super‐priming, involves a shortening of the coupling distance of Ca2+ sensor to Ca2+ channels (Midorikawa & Sakaba, 2017). Furthermore, at parallel fibre to Purkinje cell synapses, Munc13‐3 mediates super‐priming (Ishiyama et al. 2014), most probably by shortening the coupling distance (Kusch et al. 2018).
The strong effect of EGTA on the initial EPSC amplitude in the present study indicates a significant contribution of remote vesicles to action potential–evoked release at cMFBs (Keller et al. 2015). This appears to be in contrast to our previous conclusions of nanodomain coupling (Delvendahl & Hallermann, 2016) based on the small effect of 5 mm EGTA on the fast component of release during step depolarizations (Ritzau‐Jost et al. 2014). However, our model predicts that 5 mm EGTA slows the rise time of the initial release component during a step depolarization to 0 mV by only 3% (data not shown). This is consistent with findings at the calyx, where 5 mm EGTA also has a small effect on the initial release kinetics of vesicles with a coupling distance as remote as ∼25 nm during step depolarizations (Chen et al. 2015; Nakamura et al. 2018). Thus, our data suggest that both closely (6.5 nm) and more remotely (15 nm) coupled vesicles mediate action potential–evoked release at the cMFBs. However, the exact coupling distances critically depend on the assumed release scheme and intra‐bouton EGTA concentration upon EGTA‐AM application. Based on a recent definition of nanodomain as <100 nm (Eggermann et al. 2011), both coupling distances would be classified as part of the nanodomain.
In conclusion, we demonstrate that EGTA, but not interference with the Ca2+‐calmodulin–Munc13‐1 pathway, slows recovery from synaptic depression after high‐frequency transmission at cMFB to GC synapses. Furthermore, we provide evidence for two pools of vesicles with different recruitment kinetics. Finally, models with two pools of vesicles predict a slowed recovery upon EGTA application, although all vesicle recruitment rates of the models are Ca2+ ‐independent. In this framework, EGTA slows recovery from depression simply by preferentially blocking the release of intrinsically fast‐recruited vesicles. Thus, the application of Ca2+ buffers by itself does not provide a sufficient means to test the Ca2+‐dependence of vesicle recruitment.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
Experiments were performed at the Carl‐Ludwig‐Institute for Physiology of the Faculty of Medicine, Leipzig University, and at the European Neuroscience Institute, Göttingen. SH and ARJ contributed to the conception and design of the work. Munc13 mutant and control mice were generated and genotyped by NL. ARJ, JV, NL, JE and SH were responsible for data acquisition, analysis and interpretation. LJ and SH performed the modelling. ARJ and SH drafted the manuscript. All authors have critically revised and approved the final copy of the manuscript submitted for publication. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work is supported by the Heisenberg‐Program of the German Research Foundation to S.H. (HA 6386/2‐1, 3‐1).
Acknowledgements
We thank Erwin Neher for critically reading the manuscript and Nils Brose for feedback and the provision of the mutant mouse line.
Biography
Andreas Ritzau‐Jost received his medical training at Leipzig University, where, early on, he became fascinated in molecular neuroscience. Parallel to his medical studies, he earned his doctorate degree (MD) in the laboratory of Stefan Hallermann for investigating mechanisms that enable kilohertz synaptic transmission in the cerebellum. After graduating in 2016, he continued studying mechanisms of synaptic transmission in cerebellar brain slices, as well as cortical cell culture, by applying cutting‐edge electrophysiological and optical techniques.

Edited by: Ian Forsythe & Michael Shipston
Linked articles This article is highlighted in a Perspectives article by Jackman and von Gersdorff. To read this article, visit http://doi.org/10.1113/JP276673.
References
- Atluri PP & Regehr WG (1998). Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci 18, 8214–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babai N, Bartoletti TM & Thoreson WB (2010). Calcium regulates vesicle replenishment at the cone ribbon synapse. J Neurosci 30, 15866–15877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhme MA, Beis C, Reddy‐Alla S, Reynolds E, Mampell MM, Grasskamp AT, Lützkendorf J, Bergeron DD, Driller JH, Babikir H, Göttfert F, Robinson IM, O'Kane CJ, Hell SW, Wahl MC, Stelzl U, Loll B, Walter AM & Sigrist SJ (2016). Active zone scaffolds differentially accumulate Unc13 isoforms to tune Ca2+ channel‐vesicle coupling. Nat Neurosci 19, 1311–1320. [DOI] [PubMed] [Google Scholar]
- Borst JG & Soria van Hoeve J (2012). The calyx of Held synapse: from model synapse to auditory relay. Annu Rev Physiol 74, 199–224. [DOI] [PubMed] [Google Scholar]
- Chen Z, Das B, Nakamura Y, DiGregorio DA & Young SM, Jr (2015). Ca2+ channel to synaptic vesicle distance accounts for the readily releasable pool kinetics at a functionally mature auditory synapse. J Neurosci 35, 2083–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Li GL & von Gersdorff H (2011). Recovery from short‐term depression and facilitation is ultrafast and Ca2+ dependent at auditory hair cell synapses. J Neurosci 31, 5682–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta P, Gilliam J, Thoreson WB, Janz R & Heidelberger R (2017). Two pools of vesicles associated with synaptic ribbons are molecularly prepared for release. Biophys J 113, 2281–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvendahl I & Hallermann S (2016). The cerebellar mossy fiber synapse as a model for high‐frequency transmission in the mammalian CNS. Trends Neurosci 39, 722–737. [DOI] [PubMed] [Google Scholar]
- Delvendahl I, Jablonski L, Baade C, Matveev V, Neher E & Hallermann S (2015). Reduced endogenous Ca2+ buffering speeds active zone Ca2+ signaling. Proc Natl Acad Sci U S A 112, E3075–E3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvendahl I, Vyleta NP, von Gersdorff H & Hallermann S (2016). Fast, temperature‐sensitive and clathrin‐independent endocytosis at central synapses. Neuron 90, 492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS & Regehr WG (1998). Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci 18, 6147–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP & Jonas P (2011). Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci 13, 7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID (1994). Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro . J Physiol 479, 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes‐Davies M, Cuttle MF & Takahashi T (1998). Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron 20, 797–807. [DOI] [PubMed] [Google Scholar]
- Fucile S, Miledi R & Eusebi F (2006). Effects of cyclothiazide on GluR1/AMPA receptors. Proc Natl Acad Sci U S A 103, 2943–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis A, Burrone J & Lagnado L (1999). Two actions of calcium regulate the supply of releasable vesicles at the ribbon synapse of retinal bipolar cells. J Neurosci 19, 6309–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallermann S, Fejtova A, Schmidt H, Weyhersmüller A, Silver RA, Gundelfinger E & Eilers J (2010). Bassoon speeds vesicle reloading at a central excitatory synapse. Neuron 18, 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallermann S & Silver RA (2013). Sustaining rapid vesicular release at active zones: potential roles for vesicle tethering. Trends Neurosci 36, 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama S, Schmidt H, Cooper BH, Brose N & Eilers J (2014). Munc13‐3 superprimes synaptic vesicles at granule cell‐to‐basket cell synapses in the mouse cerebellum. J Neurosci 34, 14687–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Olt J, Cho S, von Gersdorff H & Marcotti W (2017). The coupling between Ca2+ channels and the exocytotic Ca2+ sensor at hair cell ribbon synapses varies tonotopically along the mature cochlea. J Neurosci 37, 2471–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörntell H & Ekerot CF (2006). Properties of somatosensory synaptic integration in cerebellar granule cells in vivo . J Neurosci 26, 11786–11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge HJ, Rhee J‐S, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C & Brose N (2004). Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short‐term synaptic plasticity. Cell 118, 389–401. [DOI] [PubMed] [Google Scholar]
- Keller D, Babai N, Kochubey O, Han Y, Markram H, Schurmann F & Schneggenburger R (2015). An exclusion zone for Ca2+ channels around docked vesicles explains release control by multiple channels at a CNS synapse. PLoS Comput Biol 11, e1004253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusch V, Bornschein G, Loreth D, Bank J, Jordan J, Baur D, Watanabe M, Kulik A, Heckmann M, Eilers J & Schmidt H (2018). Munc13‐3 is required for the developmental localization of calcium channels to presynaptic active zones and the nanopositioning of Cav2.1 channels near the release sensor. Cell Reports 22, 1965–1973. [DOI] [PubMed] [Google Scholar]
- Lee JS, Ho WK & Lee SH (2012). Actin‐dependent rapid recruitment of reluctant synaptic vesicles into a fast‐releasing vesicle pool. Proc Natl Acad Sci U S A 109, E765–E774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Ho WK, Neher E & Lee SH (2013). Superpriming of synaptic vesicles after their recruitment to the readily releasable pool. Proc Natl Acad Sci U S A 110, 15079–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Kim MH, Ho WK & Lee SH (2008). Presynaptic release probability and readily releasable pool size are regulated by two independent mechanisms during posttetanic potentiation at the calyx of Held synapse. J Neurosci 28, 7945–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KH, Erazo‐Fischer E & Taschenberger H (2012). Similar intracellular Ca2+ requirements for inactivation and facilitation of voltage‐gated Ca2+ channels in a glutamatergic mammalian nerve terminal. J Neurosci 32, 1261–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipstein N, Sakaba T, Cooper BH, Lin KH, Strenzke N, Ashery U, Rhee JS, Taschenberger H, Neher E & Brose N (2013). Dynamic control of synaptic vesicle replenishment and short‐term plasticity by Ca2+‐calmodulin‐Munc13‐1 signaling. Neuron 79, 82–96. [DOI] [PubMed] [Google Scholar]
- Lou X, Korogod N, Brose N & Schneggenburger R (2008). Phorbol esters modulate spontaneous and Ca2+‐evoked transmitter release via acting on both Munc13 and protein kinase C. J Neurosci 28, 8257–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Wang Y & Tsodyks M (1998). Differential signaling via the same axon of neocortical pyramidal neurons. Proc Natl Acad Sci U S A 95, 5323–5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveev V, Sherman A & Zucker RS (2002). New and corrected simulations of synaptic facilitation. Biophys J 83, 1368–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S & Matthews G (1996). Ultrafast exocytosis elicited by calcium current in synaptic terminals of retinal bipolar neurons. Neuron 17, 1241–1249. [DOI] [PubMed] [Google Scholar]
- Midorikawa M & Sakaba T (2017). Kinetics of releasable synaptic vesicles and their plastic changes at hippocampal mossy fiber synapses. Neuron 96, 1033–1040. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Yamashita T, Saitoh N & Takahashi T (2008). Developmental changes in calcium/calmodulin‐dependent inactivation of calcium currents at the rat calyx of Held. J Physiol 586, 2253–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Reva M & DiGregorio DA (2018). Variations in Ca2+ influx can alter chelator‐based estimates of Ca2+ channel‐synaptic vesicle coupling distance. J Neurosci 38, 3971–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E (2015). Merits and limitations of vesicle pool models in view of heterogeneous populations of synaptic vesicles. Neuron 87, 1131–1142. [DOI] [PubMed] [Google Scholar]
- Neher E (2017). Some subtle lessons from the calyx of Held synapse. Biophys J 112, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E & Sakaba T (2001). Combining deconvolution and noise analysis for the estimation of transmitter release rates at the calyx of Held. J Neurosci 21, 444–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancz EA, Ishikawa T, Duguid I, Chadderton P, Mahon S & Häusser M (2007). High‐fidelity transmission of sensory information by single cerebellar mossy fibre boutons. Nature 450, 1245–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Südhof TC, Takahashi M, Rosenmund C & Brose N (2002). Beta phorbol ester‐ and diacylglycerol‐induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 108, 121–133. [DOI] [PubMed] [Google Scholar]
- Ritzau‐Jost A, Delvendahl I, Rings A, Byczkowicz N, Harada H, Shigemoto R, Hirrlinger J, Eilers J & Hallermann S (2014). Ultrafast action potentials mediate kilohertz signaling at a central synapse. Neuron 84, 152–163. [DOI] [PubMed] [Google Scholar]
- Rothman JS & Silver RA (2018). NeuroMatic: an integrated open‐source software toolkit for acquisition, analysis and simulation of electrophysiological data. Front Neuroinform 12, 14, 10.3389/fninf.2018.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T (2006). Roles of the fast‐releasing and the slowly releasing vesicles in synaptic transmission at the calyx of Held. J Neurosci 26, 5863–5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T & Neher E (2001a). Calmodulin mediates rapid recruitment of fast‐releasing synaptic vesicles at a calyx‐type synapse. Neuron 32, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Sakaba T & Neher E (2001b). Quantitative relationship between transmitter release and calcium current at the calyx of Held synapse. J Neurosci 21, 462–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C & Silver RA (2006). Fast vesicle reloading and a large pool sustain high bandwidth transmission at a central synapse. Nature 439, 983–987. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R & Neher E (2000). Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406, 889–893. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T & Neher E (2002). Vesicle pools and short‐term synaptic depression: lessons from a large synapse. Trends Neurosci 25, 206–212. [DOI] [PubMed] [Google Scholar]
- Shin OH, Lu J, Rhee JS, Tomchick DR, Pang ZP, Wojcik SM, Camacho‐Perez M, Brose N, Machius M, Rizo J, Rosenmund C & Südhof TC (2010). Munc13 C2B domain is an activity‐dependent Ca2+ regulator of synaptic exocytosis. Nat Struct Mol Biol 17, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Liesegang GW, Berger RL, Czerlinski G & Podolsky RJ (1984). A stopped‐flow investigation of calcium ion binding by ethylene glycol bis(beta‐aminoethyl ether)‐N,N′‐tetraacetic acid. Anal Biochem 143, 188–195. [DOI] [PubMed] [Google Scholar]
- Srinivasan G, Kim JH & von Gersdorff H (2008). The pool of fast releasing vesicles is augmented by myosin light chain kinase inhibition at the calyx of Held synapse. J Neurophysiol 99, 1810–1824. [DOI] [PubMed] [Google Scholar]
- Stevens CF & Wesseling JF (1998). Activity‐dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron 21, 415–424. [DOI] [PubMed] [Google Scholar]
- Sun J, Bronk P, Liu X, Han W & Südhof TC (2006). Synapsins regulate use‐dependent synaptic plasticity in the calyx of Held by a Ca2+/calmodulin‐dependent pathway. Proc Natl Acad Sci U S A 103, 2880–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, Leão RM, Rowland KC, Spirou GA & von Gersdorff H (2002). Optimizing synaptic architecture and efficiency for high‐frequency transmission. Neuron 36, 1127–1143. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Woehler A & Neher E (2016). Superpriming of synaptic vesicles as a common basis for intersynapse variability and modulation of synaptic strength. Proc Natl Acad Sci U S A 113, E4548–E4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trommershäuser J, Schneggenburger R, Zippelius A & Neher E (2003). Heterogeneous presynaptic release probabilities: functional relevance for short‐term plasticity. Biophys J 84, 1563–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek J & Regehr WG (2018). Synaptotagmin 7 mediates both facilitation and asynchronous release at granule cell synapses. J Neurosci 38, 3240–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hook MJ & Thoreson WB (2014). Endogenous calcium buffering at photoreceptor synaptic terminals in salamander retina. Synapse 68, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kan PL, Gibson AR & Houk JC (1993). Movement‐related inputs to intermediate cerebellum of the monkey. J Neurophysiol 69, 74–94. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Sakaba T, Berglund K & Tachibana M (1998). Submillisecond kinetics of glutamate release from a sensory synapse. Neuron 21, 1177–1188. [DOI] [PubMed] [Google Scholar]
- Wang LY & Kaczmarek LK (1998). High‐frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394, 384–388. [DOI] [PubMed] [Google Scholar]
- Wang LY, Neher E & Taschenberger H (2008). Synaptic vesicles in mature calyx of Held synapses sense higher nanodomain calcium concentrations during action potential‐evoked glutamate release. J Neurosci 28, 14450–14458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y & Manis PB (2008). Short‐term synaptic depression and recovery at the mature mammalian endbulb of Held synapse in mice. J Neurophysiol 100, 1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzel C, Delvendahl I, Sydlik S, Georgiev O & Müller M (2018). Dysbindin links presynaptic proteasome function to homeostatic recruitment of low release probability vesicles. Nat Commun 9, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG & Borst JG (1999). The reduced release probability of releasable vesicles during recovery from short‐term synaptic depression. Neuron 23, 821–832. [DOI] [PubMed] [Google Scholar]
- Xu J & Wu L (2005). The decrease in the presynaptic calcium current is a major cause of short‐term depression at a calyx‐type synapse. Neuron 46, 633–645. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Eguchi K, Saitoh N, von Gersdorff H & Takahashi T (2010). Developmental shift to a mechanism of synaptic vesicle endocytosis requiring nanodomain Ca2+ . Nat Neurosci 13, 838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H & Xu‐Friedman MA (2008). Relative roles of different mechanisms of depression at the mouse endbulb of Held. J Neurophysiol 99, 2510–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS (1989). Short‐term synaptic plasticity. Annu Rev Neurosci 12, 13–31. [DOI] [PubMed] [Google Scholar]
- Zucker RS & Regehr WG (2002). Short‐term synaptic plasticity. Annu Rev Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]