

Abstract
Background
The five‐year survival rate of non‐small cell lung cancer (NSCLC) patients is very low. MiR‐873 is involved in the growth, metastasis, and differentiation of tumors. Herein, we determined the target gene and influence of miR‐873 in NSCLC.
Methods
MiRanda and Targetscan websites were used to predict the target gene of miR‐873 in NSCLC. Luciferase activity was examined using a dual luciferase reporter gene assay kit. The viability, tube formation, and proliferation of cells were analyzed by cell counting kit‐8, angiogenic analysis, and flow cytometry, respectively. The levels of miR‐873 and GLI1 were evaluated using quantitative real‐time PCR and Western blot assays.
Results
Low levels of GLI1 and high levels of miR‐873 were observed in an NSCLC cell line (PC9) highly sensitive to EGFR‐tyrosine kinase inhibitors. There was a negative correlation between miR‐873 and GLI1 expression in PC9 and PC9/GR cells. The inhibition of miR‐873 enhanced GLI1 levels. MiR‐873 expression was inhibited by gefitinib. Gefitinib markedly reduced the viability, tube formation, and cell number in PC9 cells. However, suppression of miR‐873 enhanced the resistance and knockdown of GLI1 enhanced the sensitivity of PC9 cells to gefitinib.
Conclusions
GLI1 is a target gene of miR‐873 in NSCLC. The inhibition of miR‐873 increased gefitinib resistance of NSCLC cells via the upregulation of GLI1. These results indicate that miR‐873‐GLI1 signaling is involved in gefitinib resistance in NSCLC.
Keywords: Angiogenesis, GLI1, miR‐873, non‐small cell lung cancer
Introduction
Lung cancer (LC) is one of the most common malignant tumors in the world, and the leading cause of cancer‐related death in China. Non‐small cell lung cancer (NSCLC) accounts for approximately 85% of LC cases. At the time of diagnosis, approximately 75% of patients are in middle and late stages. The five‐year survival rate is very low.1, 2 Platinum‐based doublet chemotherapy is the first‐line standard treatment for late‐stage NSCLC.3, 4, 5 The emergence of molecular targeted drugs has brought great promise for the treatment of LC.6, 7, 8 In patients with EGFR sensitive mutations, the efficacy rate of EGFR‐tyrosine kinase inhibitors (TKI) is 71.2%, thus EGFR‐TKIs have become first‐line drugs for patients with EGFR sensitive mutant advanced LC. However, almost all patients eventually suffer from drug resistance. The primary or secondary drug resistance of EGFR‐TKIs greatly limits their clinical application9, 10, 11 therefore, finding a mechanism to alter drug resistance is of significant importance.
MicroRNA (miRNAs) are non‐coding RNAs, mainly mediating gene expression at posttranscriptional levels.12, 13 Hundreds of miRNAs have been found. In the human genome, miRNAs regulate protein encoding genes at a rate of approximately 1:3, controlling cell apoptosis, proliferation, differentiation, metabolism, individual development and tumorigenesis, and drug resistance.14, 15, 16 Target gene expression is regulated by two mechanisms: (i) binding to the untranslated region (3'UTR) of the target messenger RNA (mRNA) 3' end, inhibiting its translation; and (ii) like small interfering RNA (siRNA), miRNA binds to the target and degrades target mRNA.17, 18, 19 Recent studies have found that miRNAs regulate drug resistance by mediating their targeting genes in various cancers.20, 21, 22, 23, 24 In recent years, abnormal expression of miR‐873 has been found in many kinds of tumors, such as breast and ovarian cancers, and glioma.25, 26, 27 MiR‐873 plays a role in promoting cancer or anticancer by regulating tumor cell invasion, migration, proliferation, apoptosis, and sensitivity to chemotherapeutic drugs.28, 29, 30, 31 Nevertheless, the role of miR‐873 in drug resistance in NSCLC is still unknown.
We detected mRNA levels of miR‐873 in normal human lung epithelial cells and highly sensitive EGFR‐TKI NSCLC cells by quantitative real‐time (qRT)‐PCR. According to data from the miRanda and Targetscan websites, we predicted the binding sites of miR‐873 to related target genes. The influence of miR‐873 on NSCLC repressed by gefitinib was explored.
Methods
Cell culture
Normal human lung epithelial (BEAS‐2B), EGFR‐TKI highly sensitive NSCLC (PC9), EGFR‐TKI resistant (PC9/GR), and human embryonic kidney (HEK293T) cell lines were provided by Shanghai Mingjing Biology Co., Ltd. (Shanghai, China). BEAS‐2B and HEK293T cells were cultured in Dulbecco's modified Eagle medium (Solarbio, Shanghai, China), including 10% fetal bovine serum (Gibco, Grand Island, NY, USA), 100 U/mL penicillin, and 100 ug/mL streptomycin (Thermo Fisher Scientific, Shanghai, China), and incubated at 37°C with 5% CO2 (Thermo Electron, Marietta, OH, USA).
Cell transfection
The miR‐873 inhibitor and empty vector (mock) were obtained from Shanghai Tuoran Biology Co., Ltd (Shanghai, China). The negative control siRNA (MBS 8241404) and GLI1 siRNA (MBS8208749) were purchased from MyBio Source (San Diego, CA, USA). PC9 cells were transfected with mimics or siRNA (50 pmol) using Lipofectamine 2000 (Solarbio) for 48 hours according to a standardized method.
Using data from the miRanda and Targetscan websites, we predicted the binding site of the GLI1 gene to miR‐873. The 3'UTR of GLI1 with affinity for miR‐873 and a mutant reporter were cloned to the downstream of firefly luciferase of psiCHECK‐2 vector (Hibio, Hangzhou, China). MiR‐873 was then co‐transfected into HEK293T cells using Lipofectamine 2000. After transfection, luciferase activity analysis was performed.
Quantitative real‐time PCR assay
Total RNA was harvested by TRIzol (Yeasen, Shanghai, China). One microgram of RNA was applied to synthesize complementary DNA (cDNA) using a TIANScript cDNA Synthesis kit (Tiangen, Beijing, China). The reaction conditions were: 85°C for 15 minutes and 4°C for five minutes. The cDNA was amplified using a SYBR Premix ExTaq II kit (Thermo Fisher Scientific). The reaction conditions were: 95°C for five minutes, 92°C for 15 seconds and 60°C for 35 seconds for 30 cycles, and 72°C for 35 seconds. The primers are listed in Table 1. U6 and
Table 1.
Primer sequences
| Primer name | Sequence (5′‐3′) | Product size (bp) |
|---|---|---|
| miR‐873‐forward | CTGCACTCCCCCACCTG | |
| miR‐873‐reverse | GTGCAGGGTCCGAGGT | 220 |
| GLI1‐forward | TACTCACGCCTCGAAAACCT | |
| GLI1‐reverse | AGGACCATGCACTGTCTTGA | 235 |
| U6‐forward | ACACCAAGCAGTCCGAAGAG | |
| U6‐reverse | ACAAAATTTCTCACGCCGGT | 220 |
| GAPDH‐forward | CCATCTTCCAGGAGCGAGAT | |
| GAPDH‐reverse | TGCTGATGATCTTGAGGCTG | 222 |
glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) were used as internal controls. The 2‐ΔΔCT formula was used to assess gene expression.
Western blot
Total protein was harvested by radioimmunoprecipitation assay (high) (Beyotime, Shanghai, China). A BCA protein assay kit (Jining Shiye, Shanghai, China) was used to detect protein content. Protein was separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membrane (Tengxiang, Yueqing, Wenzhou, Zhejiang, China). The membranes were sealed using tris‐buffered saline plus tween 20 containing 5% non‐fat milk at room temperature for 90 minutes, and hybridized to anti‐GLI1 (ab134906) and anti‐GAPDH (ab157156) (dilution: 1:1200; Abcam, Cambridge, UK) at 4°C overnight. Corresponding secondary antibodies (rabbit anti‐mouse immunoglobulin G [IgG], Cell Signaling Technology, Danvers, MA, USA; #58802, 1:7000; goat anti‐mouse IgG, ab6785, 1:7000, Abcam) were added to bind to the membranes at 37°C for 60 minutes. The blots were exposed by a WFH‐101B gel imaging analysis system (Jinke, Shanghai, China).
Luciferase activity analysis
A dual luciferase reporter gene assay kit (Yeasen) was used to measure luciferase activity following the standard method. HEK293T cells were lysed using lysate. Cell suspension was transferred to a black enzyme plate. The cells were then resuspended using medium. The supernatant was seeded in a 96‐well plate, then firefly and sea kidney luciferase reaction solutions were added to the plates. The data were measured using a multimode reader (SuPerMax‐3100, Shanpu, Shanghai, China).
Cell counting kit‐8 analysis
Cell counting kit‐8 (Beyotime, Shanghai, China) was used to assess the viability of PC9 cells. PC9 cells were cultured in 96‐well plates (2 × 103 cells/well) in the incubator for 24 hours. Cells were then exposed to miR‐873 inhibitor, empty vector (mock), and 40 nM gefitinib for 48, 72, and 144 hours. After treatment, the CCK‐8 reagent was dripped to the well, and cells were cultured in the incubator for four hours. Absorbance was assessed using a multimode reader.
Angiogenic analysis
PC9 cells were cultured in 6‐well plates (3 × 104 cells/well) in the incubator for 24 hours. Cells were exposed to miR‐873 inhibitor, empty vector (mock), and 40 nM gefitinib for 48 hours. The medium from each group was collected for the following experiments. Matrigel Basement Membrane Matrix (BD Biosciences, San Jose, CA, USA) was added into a 96‐well plate and allowed to polymerize at 37°C for 30 minutes. The human umbilical vein endothelial cells (2 × 104/well) were plated in a 96‐well plate containing the pre‐described condition medium and maintained for 20 hours at 37°C, 5% CO2. Finally, the endothelial cell tube was observed under an inverted microscope (TL3000 Ergo; Leica, Microsystems, Wetzlar, Germany). Tube formation was defined as: 1000 × total area of connected tubes/total image area.
Flow cytometry
The proliferation of PC9 cells was detected by carboxyfluorescein succinimidyl amino ester (CFSE). After cell treatment, cell suspension was prepared. CFSE solution was added into the cell suspension and shaken up. Cell proliferation was evaluated using flow cytometry (Attune NxT, Thermo Fisher Scientific).
Statistical analysis
The data were presented as mean ± standard deviation. One‐way analysis of variance was performed to evaluate the differences between groups using SPSS version 20 (IBM Corp., Armonk, NY, USA). P < 0.05 was considered statistically significant. Each experiment was repeated at least three times.
Results
MiR‐873 expression was negatively correlated with GLI1 expression in PC9 cells
The mRNA levels of miR‐873 and GLI1 were detected by qRT‐PCR assay, which revealed a significant decrease of GLI1 mRNA, but an increase of miR‐873 mRNA in PC9 cells compared to BEAS‐2B cells (Fig 1a,b) (P < 0.05) (Fig 1a,b). MiR‐873 expression was negatively correlated with GLI1 expression in PC9 cells (Fig 1c,d). As shown in Figure 1e, the half‐maximal inhibitory concentration (IC50) of gefitinib was < 20 nM in PC9 cells; however, the IC50 of gefitinib reached > 40 nM when the cells were treated with miR‐873 inhibitors.
Figure 1.
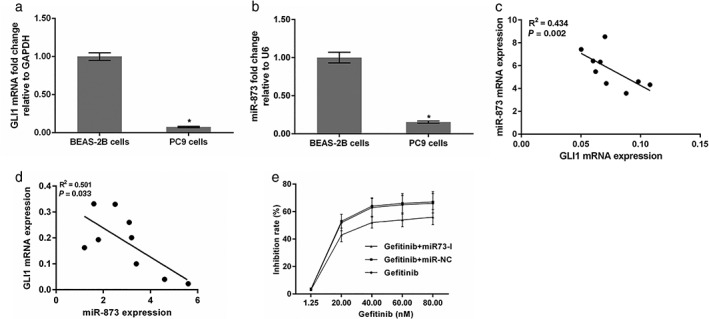
miR‐873 expression was negatively correlated with GLI1 expression in PC9 cells. (a,b) GLI1 and miR‐873 expression were assessed by quantitative real‐time PCR. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) and U6 were used as internal controls. *P < 0.01 vs. BEAS‐2B cells. The correlation between between GLI1 and miR‐873 expression was determined in (c) PC‐9 and (d) PC‐9/GR cells. (e) The inhibition rate of gefitinib in the presence of miR‐873 I (miR‐873 inhibitor). mRNA, messenger RNA.
GLI1 is a target gene of miR‐873
The qRT‐PCR data revealed that miR‐873 expression was obviously decreased when PC9 cells were transfected with the miR‐873 inhibitor (P < 0.05) (Fig 2a). GLI1 mRNA and protein expression were obviously upregulated when PC9 cells were administered an miR‐873 inhibitor (P < 0.05) (Fig 2b,c). Data from the miRanda and Targetscan websites predicted the binding site of miR‐873 on the GLI1 gene. There is a potential binding site in the 3’UTR region of the GLI1 gene (Fig 2d). Additionally, luciferase activity report assay findings showed that the relative luciferase activity of GLI1–3’UTR was markedly decreased in the presence of miR‐873. However, the relative luciferase activity of GLI1–3’UTR mut was not altered by miR‐873 (P < 0.05) (Fig 2e).
Figure 2.
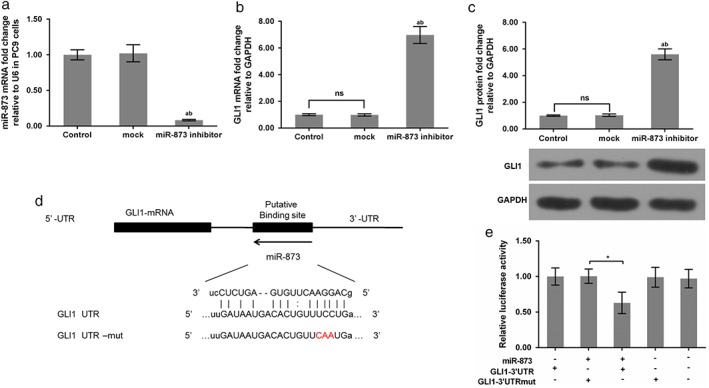
GLI1 was a potential target gene of miR‐873. (a) miR‐873 messenger RNA (mRNA) expression was evaluated by quantitative real‐time (qRT)‐PCR. qRT‐PCR and Western blot assays were performed to assess GLI1 (b) mRNA and (c) protein levels. *P < 0.01 versus control; **P < 0.01 versus mock. (d) The binding site of GLI1 to miR‐873 was predicted. The GLI1 3’ untranslated region (UTR)‐mutant (mut) is presented. (e) The 3’UTR of GLI1 with affinity for miR‐873 and a mutant reporter were cloned to the downstream of firefly luciferase of psiCHECK‐2 vector. miR‐873 was transfected into HEK293T cells. Luciferase activity was examined. *P < 0.01 versus GLI1 3’UTR; **P < 0.01 miR‐873 + GLI1–3’UTR + mut. NS, not significant.
Effect of miR‐873 on cell viability, proliferation, and angiogenesis
The expression of miR‐873 was decreased in cells treated with gefitinib or miR‐873 inhibitors. The combination of gefitinib and miR‐873 inhibitors further decreased the expression of miR‐873 (Fig 3). As CCK‐8 data shows, gefitinib predominantly decreased cell viability at 72 and 144 hours. Cell viability was notably increased in cells treated with miR‐873 inhibitor and gefitinib compared to gefitinib or mock (P < 0.05) (Fig 4a). Angiogenic analysis revealed that gefitinib reduced tube formation. Tube formation was drastically augmented by the miR‐873 inhibitor (P < 0.05) (Fig 4b,c). Meanwhile, flow cytometry data revealed a decrease in cell numbers in cells administered gefitinib, but an increase in cell numbers in cells exposed to miR‐873 inhibitor and gefitinib (P < 0.05) (Fig 4d).
Figure 3.
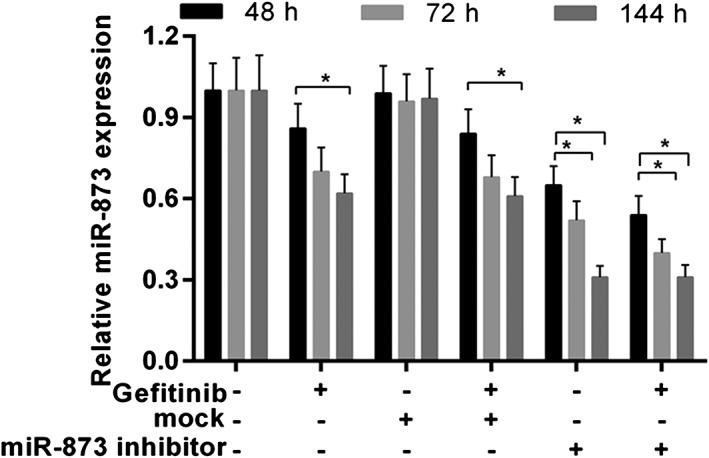
MiR‐873 expression was detected by quantitative real‐time (qRT)‐PCR. PC9 cells were exposed to miR‐873 inhibitor, empty vector (mock), and 40 nM gefitinib for 48, 72, and 144 hours. *P < 0.05.
Figure 4.
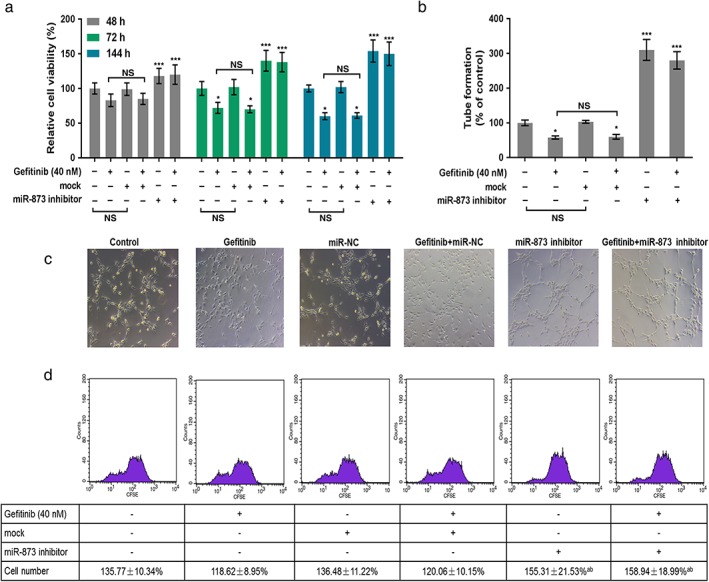
MiR‐873 inhibition increased the proliferation and angiogenesis of PC9 cells inhibited by gefitinib. (a) PC9 cells were exposed to miR‐873 inhibitor, empty vector (mock), and 40 nM gefitinib for 48, 72, and 144 hours. Cell viability was evaluated by cell counting kit‐8. (b,c) Tube formation of PC9 cells was analyzed using angiogenic analysis. (d) Cell proliferation was determined by flow cytometry. *P < 0.01 versus control. **P < 0.01 gefitinib or mock. NS, not significant.
Repression of miR‐873 enhanced GLI1 expression in PC9 cells
Western blot and qRT‐PCR were used to determine the molecular mechanism of miR‐873 on PC9 cells. As qRT‐PCR shows, gefitinib conspicuously upregulated the mRNA level of GLI1. When cells were exposed to the miR‐873 inhibitor and gefitinib, the GLI1 mRNA level was enhanced compared to gefitinib or mock (P < 0.05) (Fig 5a). The protein trend of GLI1 was also increased by miR‐873 inhibitor and gefitinib compared to gefitinib or mock (P < 0.05) (Fig 5b,c). GLI1 knockdown increased the sensitivity of PC9 to gefitinib. Our data also revealed that downregulation of GLI1 promoted apoptosis in PC9 cells compared to gefitinib treatment alone (P < 0.05) (Fig 6), indicating that inhibition of GLI1 improved the effectiveness of gefitinib in PC9 cells.
Figure 5.
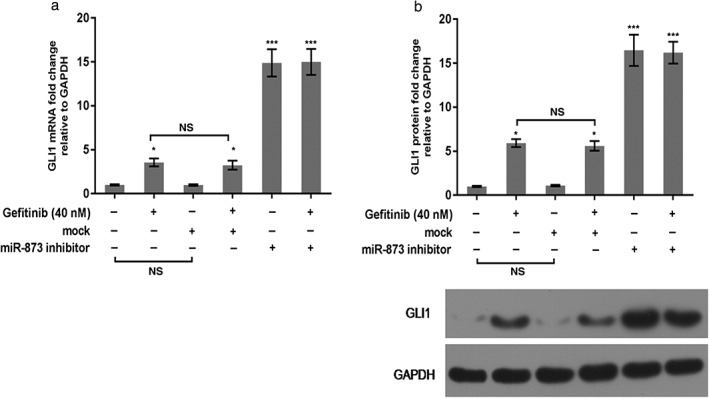
miR‐873 inhibition enhanced GLI1 expression in PC9 cells. (a,b) Messenger RNA (mRNA) and protein levels of GLI1 were identified using (a) quantitative real‐time PCR and (b) Western blot assays. *P < 0.01 versus control; **P < 0.01 gefitinib or mock. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; NS, not significant.
Figure 6.
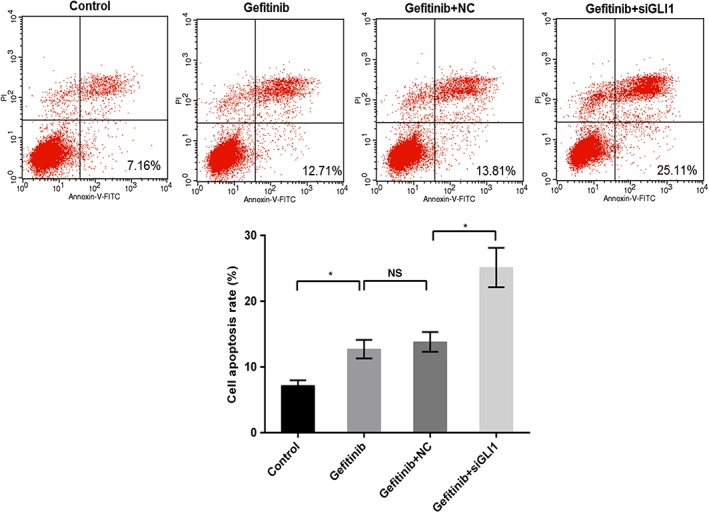
Downregulation of GLI1 enhanced cell sensitivity to gefitinib. PC9 cells were exposed to 40 nM gefitinib, gefitinib + small interfering (si)RNA negative control (NC), and gefitinib+siGLI1; untreated cells were considered the control. Cell apoptosis was determined by flow cytometry assay. *P < 0.05.
Discussion
In the present study, GLI1 was identified as a potential target gene for miR‐873 in NSCLC according to data from the miRanda and Targetscan websites. MiR‐873 and GLI1 expression has a negative correlation in NSCLC cells (PC9) highly sensitive to EGFR‐TKIs. Relative luciferase activity was markedly enhanced in PC9 cells treated with miR‐873 inhibitor and GLI‐3’UTR. Suppression of miR‐873 significantly enhanced the viability, angiogenesis, and proliferation of PC9 cells inhibited by gefitinib, followed by upregulation of GLI1.
The Hh signaling pathway is highly activated in NSCLC.32, 33, 34 GLI1 is the transcription factor of the Hh signaling pathway and its downstream target gene. Its expression is highly dependent on the activity of the Hh signal and is often used as a marker for activation of the Hh signaling pathway.35, 36 Furthermore, Gao et al. proved that miR‐873 inhibits the progression of lung adenocarcinoma.37 Zhang et al. confirmed that miR‐873 represses H9C2 cell proliferation by regulating GLI1.31 Thus, we hypothesized that miR‐873 and GLI1 may be linked in NSCLC. As expected, we found that GLI1 is a target gene of miR‐873 in NSCLC. In addition, there is a negative correlation between miR‐873 and GLI1 expression in PC9 cells. MiR‐873 inhibition markedly promoted the expression of GLI1. Luciferase activity was increased in PC9 cells exposed to miR‐873 and GLI‐3’UTR, thus miR‐873 and GLI1 were selected as research objects.
Gefitinib, an EGFR‐TKI, is the first targeted drug to treat advanced‐stage NSCLC.38, 39, 40 Our data showed that gefitinib reduces cell viability and proliferation in PC9 cells. Angiogenesis was also inhibited by gefitinib. However, gefitinib will develop resistance in cancer during its application. Recently, a large number of articles have reported gefitinib resistance in NSCLC cells.40, 41, 42 Herein, we explored the role of miR‐873 in gefitinib resistance in PC9 cells.
It is well known that the development of neovascularization is a necessary condition for tumor growth and metastasis.43, 44 The unrestricted growth, invasion, and metastasis of the tumor are dependent on angiogenesis. Meanwhile, neovascularization provides nutrients for the growth of the tumor and is also an important method to excrete tumor cells. During angiogenesis, tumor growth changes from slow to rapid; angiogenesis is the key step to facilitate this transformation. Inhibition of angiogenesis can obviously control tumor growth and metastasis.45, 46, 47, 48, 49 In this study, we found that miR‐873 suppression conspicuously enhanced the viability and growth of PC9 cells treated with gefitinib. Moreover, the inhibition of miR‐873 increased angiogenesis. These findings suggest that miR‐873 repression heightens gefitinib resistance in NSCLC cells.
Xie et al. reported that a decrease in RKIP increases radioresistance in NSCLC cells by regulating GLI1 signaling.50 Bora‐Singhal et al. reported that silence of GLI1 combined with EGFR inhibitors obviously attenuates the growth of NSCLC cells.51 In our study, we observed that miR‐873 drastically enhanced the level of GLI1 in PC9 cells. Moreover, GLI1 downregulation enhanced apoptosis of PC9 cells, caused by gefitinib. These results imply that the inhibition of miR‐873 exerted in PC9 cells is more likely related to upregulation of GLI1.
GLI1 is a target gene of miR‐873 in NSCLC. Suppression of miR‐873 accelerated the viability, angiogenesis, and proliferation of PC9 cells suppressed by gefitinib. MiR‐873 suppression strengthened gefitinib resistance in NSCLC cells by upregulating GLI1. These results prove that miR‐873‐GLI1 signaling is involved in gefitinib resistance in NSCLC. How to control drug resistance in NSCLC requires further research.
Disclosure
No authors report any conflict of interest.
Acknowledgments
This study was supported by Jiangsu Province Clinical Science and Technology Projects (grant number: BL2012008) and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (grant number: JX10231801).
References
- 1. Castello A, Grizzi F, Toschi L et al Tumor heterogeneity, hypoxia, and immune markers in surgically resected non‐small‐cell lung cancer. Nucl Med Commun 2018; 39: 636–44. [DOI] [PubMed] [Google Scholar]
- 2. Gajra A, Zemla TJ, Jatoi A et al Time‐to‐treatment‐failure and related outcomes among 1000+ advanced non‐small cell lung cancer patients: Comparisons between older versus younger patients (Alliance A151711). J Thorac Oncol 2018; 13: 996–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Niyongere S, Saltos A, Gray JE. Immunotherapy combination strategies (non‐chemotherapy) in non‐small cell lung cancer. J Thorac Dis 2018; 10 (Suppl 3): S433–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takemura Y, Chihara Y, Morimoto Y et al Feasibility study of sequentially alternating EGFR‐TKIs and chemotherapy for patients with non‐small cell lung cancer. Anticancer Res 2018; 38: 2385–90. [DOI] [PubMed] [Google Scholar]
- 5. Tsuboi M, Kondo K, Takizawa H et al A feasibility study of postoperative adjuvant chemotherapy with fluoropyrimidine S‐1 in patients with stage II–IIIA non‐small cell lung cancer. J Med Invest 2018; 65: 90–5. [DOI] [PubMed] [Google Scholar]
- 6. Yano S, Takeuchi S, Nakagawa T, Yamada T. Ligand‐triggered resistance to molecular targeted drugs in lung cancer: Roles of hepatocyte growth factor and epidermal growth factor receptor ligands. Cancer Sci 2012; 103: 1189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Layek B, Sadhukha T, Panyam J, Prabha S. Nano‐engineered mesenchymal stem cells increase therapeutic efficacy of anticancer drug through true active tumor targeting. Mol Cancer Ther 2018; 17: 1196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan S, Wang G. Lung cancer targeted therapy: Folate and transferrin dual targeted, glutathione responsive nanocarriers for the delivery of cisplatin. Biomed Pharmacother 2018; 102: 55–63. [DOI] [PubMed] [Google Scholar]
- 9. Duan X, Shi J. [Advance in microRNAs and EGFR‐TKIs secondary resistance research in non‐small cell lung cancer.]. Zhongguo Fei Ai Za Zhi 2014; 17: 860–4 (In Chinese.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee DH. Treatments for EGFR‐mutant non‐small cell lung cancer (NSCLC): The road to a success, paved with failures. Pharmacol Ther 2017; 174: 1–21. [DOI] [PubMed] [Google Scholar]
- 11. Seike M, Gemma A. [Therapeutic biomarkers of EGFR‐TKI.]. Gan To Kagaku Ryoho 2012; 39: 1613–7 (In Japanese.). [PubMed] [Google Scholar]
- 12. Liu S, Yang Y, Jiang S et al Understanding the role of non‐coding RNA (ncRNA) in stent restenosis. Atherosclerosis 2018; 272: 153–61. [DOI] [PubMed] [Google Scholar]
- 13. Palomer X, Pizarro‐Delgado J, Vázquez‐Carrera M. Emerging actors in diabetic cardiomyopathy: Heartbreaker biomarkers or therapeutic targets? Trends Pharmacol Sci 2018; 39: 452–67. [DOI] [PubMed] [Google Scholar]
- 14. Cooper JB, Ronecker JS, Tobias ME et al Molecular sequence of events and signaling pathways in cerebral metastases. Anticancer Res 2018; 38: 1859–77. [DOI] [PubMed] [Google Scholar]
- 15. Kian R, Moradi S, Ghorbian S. Role of components of microRNA machinery in carcinogenesis. Exp Oncol 2018; 40: 2–9. [PubMed] [Google Scholar]
- 16. Markopoulos GS, Roupakia E, Tokamani M et al Roles of NF‐κB signaling in the regulation of miRNAs impacting on inflammation in cancer. Biomedicines 2018; 6: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schier AF, Giraldez AJ. MicroRNA function and mechanism: insights from zebra fish. Cold Spring Harb Symp Quant Biol 2006; 71: 195–203. [DOI] [PubMed] [Google Scholar]
- 18. Mens MMJ, Ghanbari M. Cell cycle regulation of stem cells by microRNAs. Stem Cell Rev 2018; 14: 309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang D, Si S, Wang Q et al MiR‐27a promotes hemin‐induced erythroid differentiation of K562 cells by targeting CDC25B. Cell Physiol Biochem 2018; 46: 365–74. [DOI] [PubMed] [Google Scholar]
- 20. Chen MJ, Wu DW, Wang GC, Wang YC, Chen CY, Lee H. MicroRNA‐630 may confer favorable cisplatin‐based chemotherapy and clinical outcomes in non‐small cell lung cancer by targeting Bcl‐2. Oncotarget 2018; 9: 13758–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu W, Tan C, He Y, Zhang G, Xu Y, Tang J. Functional miRNAs in breast cancer drug resistance. Onco Targets Ther 2018; 11: 1529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu XG, Xu J, Li F, Li MJ, Hu T. Down‐regulation of miR‐377 contributes to cisplatin resistance by targeting XIAP in osteosarcoma. Eur Rev Med Pharmacol Sci 2018; 22: 1249–57. [DOI] [PubMed] [Google Scholar]
- 23. Sun FD, Wang PC, Luan RL, Zou SH, Du X. MicroRNA‐574 enhances doxorubicin resistance through down‐regulating SMAD4 in breast cancer cells. Eur Rev Med Pharmacol Sci 2018; 22: 1342–50. [DOI] [PubMed] [Google Scholar]
- 24. Xie M, Fu Z, Cao J et al MicroRNA‐132 and microRNA‐212 mediate doxorubicin resistance by down‐regulating the PTEN‐AKT/NF‐κB signaling pathway in breast cancer. Biomed Pharmacother 2018; 102: 286–94. [DOI] [PubMed] [Google Scholar]
- 25. Chen X, Zhang Y, Shi Y et al MiR‐873 acts as a novel sensitizer of glioma cells to cisplatin by targeting Bcl‐2. Int J Oncol 2015; 47: 1603–11. [DOI] [PubMed] [Google Scholar]
- 26. Cui J, Yang Y, Li H et al MiR‐873 regulates ERα transcriptional activity and tamoxifen resistance via targeting CDK3 in breast cancer cells. (Published erratum appears in Oncogene 2015; 34: 4018.). Oncogene 2015; 34: 3895–907. [DOI] [PubMed] [Google Scholar]
- 27. Wu DD, Li XS, Meng XN, Yan J, Zong ZH. MicroRNA‐873 mediates multidrug resistance in ovarian cancer cells by targeting ABCB1. Tumour Biol 2016; 37: 10499–506. [DOI] [PubMed] [Google Scholar]
- 28. Cao D, Yu T, Ou X. MiR‐873‐5P controls gastric cancer progression by targeting hedgehog‐GLI signaling. Pharmazie 2016; 71: 603–6. [DOI] [PubMed] [Google Scholar]
- 29. Skalsky RL, Cullen BR. Reduced expression of brain‐enriched microRNAs in glioblastomas permits targeted regulation of a cell death gene. PLoS One 2011; 6: e24248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang RJ, Li JW, Bao BH et al MicroRNA‐873 (miRNA‐873) inhibits glioblastoma tumorigenesis and metastasis by suppressing the expression of IGF2BP1. J Biol Chem 2015; 290: 8938–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang JS, Zhao Y, Lv Y et al miR‐873 suppresses H9C2 cardiomyocyte proliferation by targeting GLI1. Gene 2017; 626: 426–32. [DOI] [PubMed] [Google Scholar]
- 32. Abe Y, Tanaka N. The hedgehog signaling networks in lung cancer: The mechanisms and roles in tumor progression and implications for cancer therapy. Biomed Res Int 2016; 2016: 7969286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bai XY, Zhang XC, Yang SQ et al Blockade of hedgehog signaling synergistically increases sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in non‐small‐cell lung cancer cell lines. PLoS One 2016; 11: e0149370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gu Y, Pei X, Ren Y et al Oncogenic function of TUSC3 in non‐small cell lung cancer is associated with hedgehog signalling pathway. Biochim Biophys Acta 2017; 1863: 1749–60. [DOI] [PubMed] [Google Scholar]
- 35. Ishiwata T, Iwasawa S, Ebata T et al Inhibition of Gli leads to antitumor growth and enhancement of cisplatin‐induced cytotoxicity in large cell neuroendocrine carcinoma of the lung. Oncol Rep 2018; 39: 1148–54. [DOI] [PubMed] [Google Scholar]
- 36. Pietrobono S, Santini R, Gagliardi S et al Targeted inhibition of hedgehog‐GLI signaling by novel acylguanidine derivatives inhibits melanoma cell growth by inducing replication stress and mitotic catastrophe. Cell Death Dis 2018; 9: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gao Y, Xue Q, Wang D, Du M, Zhang Y, Gao S. miR‐873 induces lung adenocarcinoma cell proliferation and migration by targeting SRCIN1. Am J Transl Res 2015; 7: 2519–26. [PMC free article] [PubMed] [Google Scholar]
- 38. Warthan MM, Jumper CA, Smith JL. Acneiform eruption induced by Iressa (gefitinib) tablets used to treat non‐small cell lung cancer. J Drugs Dermatol 2004; 3: 569–70. [PubMed] [Google Scholar]
- 39. Lin L, Zhao J, Hu J et al Impact of weight loss at presentation on survival in epidermal growth factor receptor tyrosine kinase inhibitors (EGFR‐TKI) sensitive mutant advanced non‐small cell lung cancer (NSCLC) treated with first‐line EGFR‐TKI. J Cancer 2018; 9: 528–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zou B, Lee VHF, Yan H. Prediction of sensitivity to gefitinib/erlotinib for EGFR mutations in NSCLC based on structural interaction fingerprints and multilinear principal component analysis. BMC Bioinformatics 2018; 19: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. de Mello RA, Escriu C, Castelo‐Branco P et al Comparative outcome assessment of epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of advanced non‐small‐cell lung cancer: A network meta‐analysis. Oncotarget 2018; 9: 11805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qi M, Tian Y, Li W et al ERK inhibition represses gefitinib resistance in non‐small cell lung cancer cells. Oncotarget 2018; 9: 12020–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chu YP, Li HC, Ma L, Xia Y. Establishment of a tumor neovascularization animal model with biomaterials in rabbit corneal pouch. Life Sci 2018; 202: 98–102. [DOI] [PubMed] [Google Scholar]
- 44. Wan X, Zhu Y, Zhang L, Hou W. Gefitinib inhibits malignant melanoma cells through the VEGF/AKT signaling pathway. Mol Med Rep 2018; 17: 7351–5. [DOI] [PubMed] [Google Scholar]
- 45. Bullova P, Cougnoux A, Marzouca G, Kopacek J, Pacak K. Bortezomib alone and in combination with salinosporamid A induces apoptosis and promotes pheochromocytoma cell death in vitro and in female nude mice. Endocrinology 2017; 158: 3097–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maishi N, Hida K. Tumor endothelial cells accelerate tumor metastasis. Cancer Sci 2017; 108: 1921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mu J, Zhu D, Shen Z et al The repressive effect of miR‐148a on Wnt/β‐catenin signaling involved in Glabridin‐induced anti‐angiogenesis in human breast cancer cells. BMC Cancer 2017; 17: 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Poggi A, Varesano S, Zocchi MR. How to hit mesenchymal stromal cells and make the tumor microenvironment immunostimulant rather than immunosuppressive. (Published erratum appears in Front Immunol 2018; 9: 1342.). Front Immunol 2018; 9: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sobrinho Santos EM, Guimarães TA, Santos HO et al Leptin acts on neoplastic behavior and expression levels of genes related to hypoxia, angiogenesis, and invasiveness in oral squamous cell carcinoma. Tumour Biol 2017; 39: 1010428317699130. [DOI] [PubMed] [Google Scholar]
- 50. Xie SY, Li G, Han C, Yu YY, Li N. RKIP reduction enhances radioresistance by activating the Shh signaling pathway in non‐small‐cell lung cancer. Onco Targets Ther 2017; 10: 5605–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bora‐Singhal N, Perumal D, Nguyen J, Chellappan S. Gli1‐mediated regulation of Sox2 facilitates self‐renewal of stem‐like cells and confers resistance to EGFR inhibitors in non‐small cell lung cancer. Neoplasia 2015; 17: 538–51. [DOI] [PMC free article] [PubMed] [Google Scholar]