

Abstract
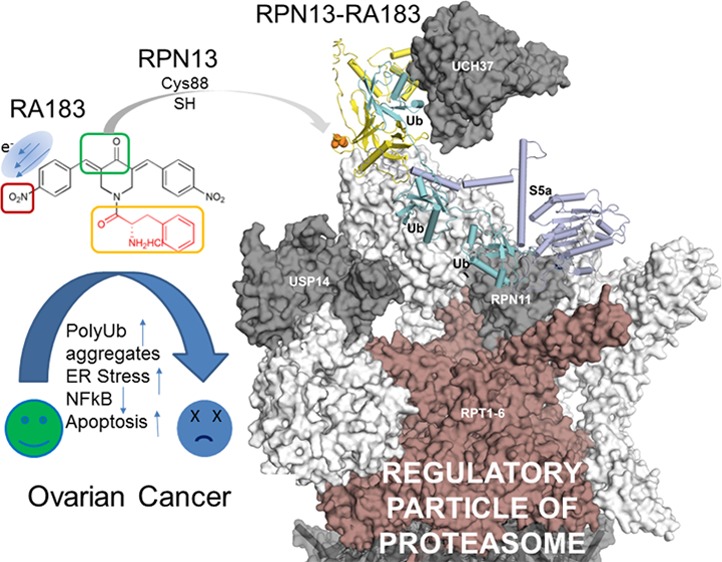
Substitution of the m,p-chloro groups of bis-benzylidinepiperidone RA190 for p-nitro, generating RA183, enhanced covalent drug binding to Cys88 of RPN13. Treatment of cancer cell lines with RA183 inhibited ubiquitin-mediated protein degradation, resulting in rapid accumulation of high-molecular-weight polyubiquitinated proteins, blockade of NFκB signaling, endoplasmic reticulum stress, an unfolded protein response, production of reactive oxygen species, and apoptotic cell death. High-grade ovarian cancer, triple-negative breast cancer, and multiple myeloma cell lines were particularly vulnerable to RA183. RA183 stabilized a tetraubiquitin-linked firefly luciferase reporter protein in cancer cell lines and mice, demonstrating in vitro and in vivo proteasomal inhibition, respectively. However, RA183 was rapidly cleared from plasma, likely reflecting its rapid degradation to the active compound RA9, as seen in human liver microsomes. Intraperitoneal administration of RA183 inhibited proteasome function and orthotopic tumor growth in mice bearing human ovarian cancer model ES2-luc ascites or syngeneic ID8-luc tumor.
Introduction
Ovarian cancer exhibits an increased accumulation of misfolded and polyubiquitinated proteins that is associated with a stressed ubiquitin–proteasome system (UPS), and this occurs despite the coordinate upregulation of proteasome components.1 The resulting unfolded protein response (UPR) is associated with an increased sensitivity to proteasomal inhibition.2,3ADRM1 is amplified and overexpressed in a significant fraction of several cancers, most notably ovarian carcinoma.4−6ADRM1 encodes human RPN13 that is associated with the 19S regulatory particle (RP) of the proteasome by binding to RPN2.7−10 Knockdown of RPN13 expression in ovarian and colorectal cancer cells by ADRM1 small interfering RNA (siRNA) produces a toxic accumulation of polyubiquitinated proteins and apoptosis, whereas overexpression is associated with proliferation, migration, and growth in soft agar.11−15 In one study, RPN13-deficient mice not only survive but reach normal body weight.16 A later study17 found that loss of RPN13 causes neonatal lethality in mice, but that RPN10 can compensate for RPN13 in the liver. Thus, RPN13 inhibitors could potentially be tolerable and efficacious against ovarian and other cancers.
The proteasome inhibitors bortezomib (Bz), carfilzomib, and ixazomib all target the same β5 subunit of the 20S catalytic subunit, and iβ5 of the immunoproteasome,18 and are licensed for treating multiple myeloma (MM).19 However, they are associated with thrombocytopenia, neuropathy, and the eventual emergence of resistance to the proteasome inhibitor.20 Despite activity against MM and promising in vitro activity against ovarian cancer cell models, intravenous (i.v.) bortezomib had no appreciable clinical activity against ovarian cancer in early phase trials, either alone or in combination with carboplatin.21 New drugs targeting the 19S regulatory particle (RP) of the proteasome may be able to improve activity against ovarian cancer, overcome resistance in MM, and reduce side effects, including immunosuppression.
Recently, we have described a bis-benzylidinepiperidone, RA190, which covalently binds to cysteine residue 88 (Cys88) of RPN13 via Michael addition and is active against models of MM and ovarian cancer.15,22−25 In seeking to improve its potency, we identified RA183. We also noted structural similarities between RA183 and the previously described cancer drug candidates b-AP1526 and RA93,27 that act as 19S RP deubiquitatinase inhibitors. Here, we compare their properties and examine whether RA183 inhibits proteasomal degradation and its potential for therapeutic activity against ovarian and other cancer models.
Results and Discussion
RA183 Binds to RPN13 More Strongly Than RA190
RA190 is structurally distinct from typical Michael acceptors in that the carbonyl moiety shares two alkenes and is attached at their β-carbon to two substituted aromatic rings.22 We hypothesized that substitution of the m,p-chloro groups of RA190 with p-nitro (RA183, see Figure 1A), given its greater electron-withdrawing effect, might enhance the nucleophilic attack of Cys88 from the 42 kDa proteasome RP protein RPN13. To evaluate whether this substitution improves covalent binding to RPN13, we also conjugated biotin to RA183 (RA183B) as a tag. By comparison to the biotinylated version of RA190 (RA190B),22 a significantly stronger labeling of a 42 kDa band (the apparent molecular weight of RPN13) was observed in the RA183B-treated 19S RP (Figure 1B). Labeling of RPN13 was lost in 19S RP pretreated with RA183 and then incubated with RA190B or when the 19S RP sample was pretreated with RA190 and then labeled with RA183B (Figure 1B). These results indicate that RA183 reacts with RPN13 more strongly than RA190.
Figure 1.

RA183 binding to RPN13 is stronger than RA190. (A) Chemical structures of RA9, b-AP15, RA190, RA183, and RA183B, in which a biotin moiety (blue) is linked to RA183, and likewise for RA190B. (B) Purified 19S RP (200 ng) was labeled with RA183, RA190, RA183B, or RA190B (20 μM) alone or in the presence of competitor RA183 and RA190 (100 or 200 μM) for 1 h at 37 °C. After labeling, equal aliquots were boiled in sodium dodecyl sulfate (SDS) sample buffer, separated by SDS-polyacrylamide gel electrophoresis (PAGE), transferred to a poly(vinylidene difluoride) (PVDF) membrane, probed with streptavidin peroxidase, and developed with chemiluminescence for the recognition of biotinylated proteins. (C, D) Cell lines from cancers of different tissue origins were lysed in Mammalian Protein Extraction Reagent (M-PER). In addition, Escherichia coli transduced with a plasmid expressing 6His and T7 tag-labeled RPN13 or irrelevant antigen and induced with isopropyl β-d-1-thiogalactopyranoside (0.1 mM, 2 h) were lysed in B-PER. Cell lysates (40 μg of protein) were treated with 5 μM RA183B (L) or not (C) for 45 min at 4 °C and subjected to SDS-PAGE, transferred to PVDF membrane, and probed with horseradish peroxidase (HRP)–streptavidin. The asterisk corresponds to ∼42 kDa RPN13 band.
RA183 Binding Pattern in Cell Lines
We analyzed the binding pattern of RA183B in extracts of cell lines of ovarian, breast, colon, and cervical cancers and MM origin. A band of 42 kDa was detected in each, suggesting RPN13 as the common primary target for RA183 in cells of diverse origin (Figure 1C,D). Several, but not all cell lines showed weaker labeling at ∼50 kDa, also seen upon labeling of 19S RP (Figure 1B). Notably, extracts of bacteria induced to express tagged full-length human RPN13 labeled at the expected size (Figure 1D).
RA183B Binds RPN13 Directly in Cell Lysate
We sought to further confirm direct binding to RPN13 in cell lysates using a precipitation assay. Briefly, RA183B-pretreated or untreated SKOV3 cell lysate was denatured with 1% (w/v) SDS at 70 °C, then cooled, and finally diluted 10-fold. The biotinylated proteins were collected on streptavidin-coated magnetic beads, separated with a magnet and washed exhaustively. Bead eluate was subjected to Western blot analysis with antibody to RPN13 (Figure S1A). We observed pulldown of RPN13 from the RA183B-treated lysate. This band was also labeled with HRP–streptavidin (Figure S1B), which indicates that RA183B directly binds to RPN13. Diminished labeling of the 42 kDa target of RA183B was observed in ADRM1 knockdown HeLa cells compared to control, consistent with lower levels of RPN13 available for reaction (Figure S1C–E).
RA183 Binds to Cys88 of RPN13 Pru Domain, from Where It Interacts with a Hydrophobic Cavity
RA190 adducts Cys88 of the RPN13 Pru domain.22 Comparison by 1H–15N heteronuclear single quantum coherence (HSQC) NMR spectroscopy of the chemical shifts of 15N-labeled human RPN13 Pru domain alone or preincubated with RA183 revealed shifted signals for several residues (Figure 2A), including Cys88. A similar subset of RPN13 amino acids was affected by RA183 addition as for RA190;22 however, RA183-bound RPN13 Pru yielded strong signals in contrast to the signal loss observed for RA190-bound RPN13 Pru (Figure 2B). A peak consistent with adduct formation between RA183 and the RPN13 Pru domain was observed by liquid chromatography–mass spectrometry (LC–MS) (Figure 2C), indicating that RA183, like RA190, binds covalently.
Figure 2.

RA183 adducts to RPN13 Pru domain C88, making hydrophobic interactions in a neighboring pocket. (A) (1H, 15N) HSQC spectra of 15N-labeled RPN13 Pru domain alone (black) and after incubation with 10-fold molar excess of RA183, followed by dialysis (orange). Signals affected by RA183 are labeled. (B) Comparison of RPN13 Pru domain with RA190 (left panels) and RA183 (right panels) for G91 and C88 as indicated. In contrast to RA190, which causes severe attenuation, most signals shift to an observable new location after RA183 addition, as exemplified by G91 and C88. (C) LC–MS experiment for RA183-exposed RPN13 Pru domain. Unmodified Rpn13 is present (expected molecular weight of 17 017 Da) as well as an additional species at a molecular weight shifted by 512.3 Da. The expected molecular weight shift caused by RA183 attachment is 513 Da. The sample used for this experiment is identical to (A). (D) Normalized peak intensity attenuation (Δ) of RPN13 Pru domain backbone (top) and side-chain (bottom) amide groups upon binding RA183. The dashed line indicates 1 standard deviation (SD) above average. Unassigned, overlapping, or proline groups are excluded from this analysis and indicated by *. (E) Lowest-energy structure of RPN13 Pru with RA183 adducted. The data from (D) are mapped onto an RPN13 Pru domain ribbon diagram with a red gradient, as indicated, and RA183 carbon, nitrogen, and oxygen atoms are displayed in blue, indigo, and red, respectively. (F) Expanded view of the RPN13 Pru–RA183 complex, as shown in (E) to illustrate interactions at the contact surface with key amino acids displayed and labeled. (G) Selected regions of (1H, 13C) HSQC spectra acquired on 13C-labeled RPN13 Pru domain alone (top) and after incubation with 10-fold molar excess unlabeled RA183 (bottom). The M31 methyl group splits into multiple states following RA183 addition.
In the presence of RA183, signal shifting was not observed for 15N RPN13 Pru C88A (in which cysteine 88 is replaced with alanine) by 1H–15N HSQC experiments (Figure S2A). In contrast, RA183 addition induced changes to the 1H–15N HSQC spectrum of 15N RPN13 Pru C60/80/121A (in which cysteine 60 and 80 and 121 of the Pru domain are all replaced with alanine) (Figure S2B). Thus, RA183, like RA190, reacts with the thiol of Cys88 of RPN13 Pru.
We quantified the loss of signal from the RPN13 Pru unbound state caused by RA183 incubation (Figure 2D). As previously measured for RA190,22 we found the most effected amino acids to localize to a surface opposite to the ubiquitin-binding loops, which as expected contains C88. We used the data plotted in Figure 2D to generate model structures for RPN13 Pru–RA183 by using High Ambiguity Driven protein–protein DOCKing (HADDOCK) and crystallography and NMR system (CNS). The structure calculations yielded three clusters (Figure S2C), similar to RA190, that all place RA183 in a cavity rich with hydrophobic amino acids and with C88 at the periphery (Figures 2E and S2D).
In all cases, interactions are formed between either RA183 or RA190 and RPN13 Pru M31, V85, P89, V93, and F106; W108 also interacts with the most populated cluster from RA190 and all structures calculated for RA183. The most populated (56%) and lowest-energy configuration for RPN13 Pru–RA183 directs the RA183 phenylalanine group toward the side-chain atoms of RPN13 M31, T37, T39, and P40 (cluster 1); the lowest-energy structure from this cluster is presented (Figure 2E). By contrast, the RA190 phenylalanine group in the lowest-energy RPN13–RA190 structures is closer to F106 (Figure S2E).22 The placement for RA183 enables its longer p-nitro group on the benzylidene ring to be directed toward RPN13 R104 and F106 (Figure 2E). A p-nitro benzylidene group adopts a similar configuration in the RPN13–RA183 structures from cluster 2 (Figure S2D, left panel), although these structures direct the RA183 phenylalanine group toward V85 and P89 (Figure S2F). Cluster 3, which contains 8% of the RPN13–RA183 structures, is similar to cluster 1, but with the p-nitro benzylidene farther from R104 (Figure S2G, green vs blue). Compared to the structures in clusters 1 and 2, the electrostatic energy of structures in cluster 3 is higher because of lost interaction with R104 (Figure S2C). This configuration, however, features additional interactions for the other p-nitro benzylidene group in RA183 with M31, P40, V93, and W108 of RPN13 Pru (Figure S2G, green vs blue). All RPN13 Pru–RA183 structures from the three clusters have an exposed amine group (Figures 2E and S2D), which is consistent with the ability of biotinylated RA183 to bind to RPN13 Pru.
We propose that RA183 likely undergoes chemical exchange between the various configurations represented in our three clusters. In particular, our experimental data indicate the presence of multiple bound-state configurations for RPN13 Pru–RA183, as illustrated by multiple bound-state signals for the G91 amide group (Figure 2B) or M31 methyl group (Figure 2G).
RA183 Impacts RPN13 Interdomain Interactions but Not Binding to Ubiquitin
While RPN13 Pru domain binds to ubiquitin, its other C-terminal DEUBAD domain binds to the deubiquitinating enzyme UCH37.8−10,28,29 An 1H–15N HSQC study of full-length RPN13 revealed signal shifting in the presence of RA183 for amino acids of both domains (Figure 3A,B). As expected, amino acids from the RPN13 Pru domain were affected by RA183, as demonstrated for T39 and G91 (Figure 3B), and notably, so were residues L314 and M342 from the DEUBAD domain and T273 of the interdomain region (Figure 3A,B). When not interacting with binding partners, the two domains of RPN13 interact with each other, which causes signal shifting for the RPN13 full-length protein in 1H–15N HSQC NMR spectra compared to RPN13 fragments that contain only the Pru or DEUBAD domain.30 Therefore, we compared spectra recorded on full-length RPN13 and RPN13 DEUBAD domain to those recorded on full-length RPN13 mixed with RA183. The comparison revealed RA183 to cause signal shifting that reflects the truncated protein and thus loss of interaction with RPN13 Pru domain, as exemplified by T273, L314, and M342 (Figure 3B).
Figure 3.

RA183 impacts RPN13 interdomain interactions, but not binding to ubiquitin. (A) Full-length RPN13 structure with amino acids highlighted in (B) displayed and labeled. The RPN13 Pru and DEUBAD domains are colored wheat and gray, respectively, with T39 and G91 in purple; T273, L314, and M342 in green; and C88 in yellow; the side-chain nitrogen, oxygen, and sulfur atoms of these amino acids are in blue, red, and yellow, respectively. (B, top) Expanded regions highlighting T39 and G91 of the RPN13 Pru domain from (1H, 15N) HSQC spectra recorded on 15N-labeled RPN13 (black, left) and RPN13 Pru domain (black, right) and after incubation with 10-fold molar excess RA183, followed by dialysis for removal of excess and labile RA183 (orange). (B, bottom) Expanded regions highlighting T273, L314, and M342 from (1H, 15N) HSQC spectra recorded on 15N-labeled RPN13 (black) and after incubation with 10-fold molar excess RA183, followed by dialysis for removal of excess and labile RA183 (orange), and RPN13 DEUBAD domain (third row). The top two panels are derived from the same spectra as used to generate those for T39 and G91. A merger of the top three rows is displayed in the bottom row with RPN13, RPN13 with RA183, and RPN13 DEUBAD in black, orange, and blue, respectively. (C–E) LC–MS analysis of 2 μM RPN13 DEUBAD (C, left), Pru without (D, black) or with equimolar RPN2 (940–953) (D, orange), or full-length RPN13 (E) following 2 h of incubation with 20 μM RA183. (C, right) Expanded regions highlighting RPN13 Pru domain C88 (boxed inset) and RPN13 DEUBAD domain L311, C357, Q358, and F359 from (1H, 15N) HSQC spectra recorded on 15N-labeled RPN13 (black) and after incubation with 10-fold molar excess RA183, followed by buffer exchange to remove excess and labile RA183 (orange). (F) (1H, 15N) HSQC spectra of 15N-labeled RPN13 Pru domain after incubating with 10-fold molar excess of RA183, followed by dialysis to remove excess and labile RA183 (black) and with equimolar ubiquitin (orange). Resonances effected significantly are labeled, and a comparison to unligated RPN13 (black) with ubiquitin (orange) is provided for I75, which is at the ubiquitin-binding surface.
We previously found that RA190 adducts to C357 in the RPN13 DEUBAD domain; this interaction, however, is more labile than RA190 covalent modification of C88,22,31 most likely because this site is surface exposed and does not offer a favorable binding pocket. We tested for RA183 interaction with the DEUBAD domain cysteine by LC–MS. Indeed, we observed RPN13 DEUBAD domain to be adducted by RA183 (Figure 3C, left), but in contrast to the Pru domain, this interaction was lost following exchange to a buffer with no RA183 (Figure 3C, right), thus supporting a more labile interaction at this location.
RPN13 C88 is proximal to the proteasome-binding site, which is contributed by the C-terminal 14 amino acids in RPN2.32 Therefore, we tested whether RPN2 (940–953) interferes with RA183 adduction to RPN13 Pru by using LC–MS. We added RA183 to RPN13 Pru domain preincubated with RPN2 (940–953). RA183-conjugated RPN13 Pru domain was detected by LC–MS (Figure 3D, black); however, this species was not detected in the presence of RPN2 (940–953) (Figure 3D, orange). Moreover, only one RA183 molecule conjugated to full-length RPN13 preincubated with RPN2 (940–953) (Figure 3E). Collectively, these findings suggest that RA183 targets the RPN13 Pru domain when RPN13 is free from the proteasome, and the DEUBAD domain when it is proteasome-bound.
We tested whether RA183 interaction affects RPN13 Pru binding to ubiquitin. RPN13 Pru was preincubated with RA183 and interaction was confirmed by a 1H–15N HSQC experiment (Figure 3F, black). Next, ubiquitin was added to equimolarity. Signal shifting reflective of interaction with ubiquitin was observed, indicating that RA183 does not inhibit RPN13 Pru interaction with ubiquitin (Figure 3F, orange). For example, I75 shifts in an identical manner whether RA183 is present or not (Figure 3F, inset).
Promiscuity in RA183 Binding in Vitro
We noted the structural similarity of RA183 to b-AP1526 and RA9,3,27 previously identified as DUB inhibitors and candidate cancer therapeutics (Figure 1A), and in a previous study, found RA190 to interact with UCH37 in vitro.31 Indeed, b-AP15 binds promiscuously in vitro, not only to its known targets UCH37 and USP14, but also to RPN13 (Figure S3A–C) and adducts RPN13 Pru C88 (Figure S4). VLX1570, an analogue of b-AP15 being tested in MM patients (NCT02372240), competed with binding of RA183B to RPN13 only at high concentrations, well above its IC50 for cancer cell cytotoxicity (Figure S3D).
At 5 μM, RA183 exhibited 19S RP DUB inhibition in vitro (Figure S5A) and competed with HA-UbVS labeling of USP14. However, only limited inhibition of HA-UbVS labeling of UCH37 was apparent at even 25 μM RA183 (Figure S5B), although it could bind directly to purified UCH37 (Figure S5C). RA183 did not impact 20S CP proteolytic activities (Figure S5D–F).
RA183 Inhibits NF-κB Activation
We hypothesized that RA183 treatment inhibits the activation of NF-κB by preventing the proteasomal degradation of IκB-α. IκB-α degradation at 20 min after the addition of tumor necrosis factor α (TNFα) was inhibited by RA183 (Figure 4A,B), and this coincides with an accumulation of total polyubiquitinated proteins (Figure 4C). Further, in HEK 293 cells carrying an NF-κB-Luc reporter, RA183 produced a dose-dependent decrease in NF-κB-associated promoter activity (Figure 4D). Similar results were also obtained with RA190 and bortezomib (Figure 4D).
Figure 4.

RA183 blocks NFκB signaling and irreversibly induces cell death. (A) HeLa cells were simultaneously treated with 10 ng/mL TNFα and 1 μM RA183 (right) or 10 ng/mL TNFα only as a control (left). Whole cell lysates were prepared at the indicated times and analyzed by Western blot for IκB and β-tubulin levels. (B) HeLa cells were treated with 10 ng/mL TNFα (left) or not (right) and 1 μM RA183 or bortezomib (Bz) or dimethyl sulfoxide (DMSO) only as a control. Whole cell lysates were harvested 10 or 20 min later and analyzed by Western blot for IκB and β-tubulin levels. (C) Same as in (A), but probed with antibody to Lys48-coupled ubiquitin. (D) Two hundred ninety-three cells transiently transfected 1 day earlier with a luciferase reporter construct driven by either an NFκB-dependent promotor (NFκB-Luc) or a constitutive promoter (Luc) were treated with the compounds indicated and TNFα (10 ng/mL) for 7 h. Upon the addition of luciferin, bioluminescence was measured in cell lysates using a luminometer. The % activation of the NFκB-dependent promoter (normalized by the constitutive reporter construct) as compared to TNFα stimulation in the absence of compound is presented. (E) SKOV3 cells were treated with compounds (0.5 μM) for 4 h, lysed, and subjected to Western blot analysis with Ub antibody. (F) SKOV3 cells were treated with bortezomib, RA190, cisplatin, or RA183 (1 μM) for 12 h. Cell lysates were analyzed by Western blot analysis with antibody to ubiquitin. (G, H) The viability of HeLa cells treated with bortezomib (G) or RA183 (H) or at the indicated concentrations for 24 h (+, +) was compared to 1 h treatment with the same compounds at the same concentrations, after which the cells were washed and the medium was replaced without compounds and incubated for a further 23 h (+, −). Cell viability was measured by the XTT method.
Activity of RA183 against Cancer Cells
An initial survey of cytotoxicity for a panel of epithelial cancer cell lines suggested that ovarian, triple-negative breast cancer (TNBC), and colon cancer cells were sensitive to RA183 (Table S1). Bortezomib-treated patients often develop resistance.33 We tested RA183 against two MM cell lines that were selected for resistance by extended culture in bortezomib and it was similarly efficacious against both the bortezomib-resistant derivative lines and their parental lines, consistent with a distinct mode of action from bortezomib (Table S1).
RA183 Triggers Rapid Accumulation of High-Molecular-Weight Polyubiquitinated Proteins
Treatment with RA183 caused a rapid accumulation of Lys48-linked polyubiquitinated proteins (Figure 4E,F), but b-AP15 was less potent. Importantly, bortezomib produced a buildup of lower-molecular-weight Lys48-linked polyubiquitinated proteins compared to RA183, consistent with its targeting of the 20S subunit and suggesting that RA183 prevents deubiquitination of proteasome substrates by 19S RP.
Irreversible Action of RA183
The cytotoxicity of RA183 was not reversed by washout, consistent with an irreversible inhibitor, whereas the cells recovered significantly after washout of bortezomib, as expected (Figure 4G,H).
RA183 Induces Unresolved Unfolded Protein Response (UPR), Reactive Oxygen Species (ROS), and Apoptosis
Accumulation of unfolded proteins upon the inhibition of proteasome function by RA183 rapidly induces an unresolved unfolded protein response (UPR) and apoptosis, a mechanism associated with bortezomib-induced cytotoxicity,34 as demonstrated by rapid upregulation of CHOP-10 (Figure 5A), BIP (Figure 5B), and spliced XBP1 mRNA (Figure 5C) and ATF4, all important effectors of UPR (Figure 5D). Cleavage of PARP, elevated levels of the apoptotic protein BAX (Figure 5E), and production of reactive oxygen species (Figure 5F) and apoptotic cells were detected 12 h after incubation with RA183 (Figure 5G,H).
Figure 5.

RA183 triggers unresolved UPS stress and apoptosis. (A) After treatment of ES2 cells with bortezomib or RA183 (1 μM) for the indicated times, mRNA was extracted and CHOP-10 mRNA levels were assessed by quantitative real-time polymerase chain reaction (PCR) and normalized to GAPDH expression. Results are expressed as fold change over control-treated cells and represent the average and SD of three independent experiments. (B) Same as in (A), except BIP mRNA levels were assessed. (C) Same as in (A), except XBP1 spliced mRNA levels were assessed. (D) Western blot analysis for ATF-4 and actin in OVCAR3 (left) and ES2 (right) cells either untreated (control) or treated with 1 μM RA183 for 6 h. (E) After treatment of ES2 cells with 1 μM RA183 for the indicated times, ES2 cell lysate was subjected to Western blot analysis and probed with antibody to PARP (top) and Bax (middle) or actin (bottom). (F) OAW42 cells were treated with RA183 (empty) or vehicle control (filled) for 12 h, respectively. After treatment, the cells were harvested and incubated with the cell-permeable dye H2DCFDA. Release of dichlorofluorescein was analyzed by flow cytometry as a measure of reactive oxygen species (ROS). H2O2 was used as a positive control (not shown). (G, H) SKOV3 and TOV21G cells were treated with 1 μM b-AP15 or RA183 at 0.5 or 0.25 μM for 12 h and analyzed by flow cytometry after staining with fluorescein-labeled annexin-V.
Inhibition of Proteasome Function in Mice by RA183
Treatment of HEK 293 cells expressing tetraubiquitin firefly luciferase (4UbFL) with RA183 induced a dramatic increase in luciferase activity (Figure 6A) consistent with inhibition of proteasomal 4UbFL degradation (Figure 6B).
Figure 6.

RA183 inhibits proteasomal activity and tumor growth in mice. (A, B) 293TT cells were stably transfected with 4UbFL expressing plasmid35 and treated with titrations of the indicated compounds for 4 h. Lysates were prepared and luciferase activity was measured (A). Data are presented as fold change in luciferase activity compared to untreated cells. (B) Levels of Lys48-linked ubiquitin in lysates were analyzed by Western blot. (C) Mice were administered 4UbFL DNA to the skin via gene gun (i.d.). After 24 h, the site of DNA administration was treated topically by application of 4% (w/v) RA183 (40 μL) in emu oil or oil alone (10 mice/group) respectively. After the indicated time points post-treatment, bioluminescence was measured by intraperitoneal (i.p.) injection of luciferin and imaging with an IVIS 200 at the site of DNA delivery (* p <0.05, ** p <0.01). (D) Mice were administered 4UbFL DNA to the leg muscle (intramuscularly (i.m.)) via in vivo electroporation. RA183 was given i.p. at 20 mg/kg. After the indicated time points post-treatment, bioluminescence was measured in control and RA183-treated animals (10 mice/group) by intraperitoneal injection of luciferin and imaging with an IVIS 200 at the site of DNA delivery. (E, F) 3 × 105 ID8-Luc cells (a syngeneic mouse ovarian cell line expressing luciferase) were injected into the peritoneal cavity of C57BL6 mice (five per group) on day 0. The mice were initially treated daily i.p. with either vehicle or 5 mg/kg RA183 dissolved in 2% DMSO and 25% β-hydroxypropyl cyclodextrin on days 3–17 and then RA183 dose was escalated to 10 mg/kg on days 17–24. The tumor growth was detected by imaging the mice through IVIS system and measuring the luciferase activity (relative light units (RLUs)) before, at the interval, and after the treatment. (G) Nude mice were administered ES2-luc cells i.p., and 2 days later, a basal bioluminescent level was determined for each mouse. Nude mice bearing ES2-luciferase cells i.p. were treated with either 6.6 mg/kg RA183 (dissolved in 2% DMSO and 25% β-hydroxypropyl cyclodextrin) (i.p.) or vehicle alone (n = 9 per group) 6 days on and 1 day off. Prior to and both 7 and 14 days after initiation of treatment, the mice were imaged for their luciferase activity (RLU) and fold change over baseline was determined.
To test for proteasome inhibition by RA183 in vivo, we first employed biolistic delivery of the 4UbFL reporter plasmid via gene gun to transduce the skin of mice. After i.p. injection of luciferin, the enzymatic activity of luciferase in the skin was visualized as bioluminescence using an IVIS imager. At 2 h post gene gun delivery of the 4UbFL DNA, the mice were imaged and baseline luminescence was recorded. The control group (n = 10) of mice was treated topically at the site of 4UbFL delivery with vehicle alone (emu oil) and another group (n = 10) was treated topically with 40 μL of RA183 (4% w/v in 5% emu oil) (Figure 6C). At 4, 18, and 42 h post-treatment, the mice were imaged and luminescence was quantified; RA183 treatment was associated with 4UbFL stabilization at 4 and 18 h, but returned to near baseline by 42 h (Figure 6C). In a similar experiment, the 4UbFL reporter DNA construct was delivered i.m. by in vivo electroporation, and we chose to administer RA183 i.p. because recent clinical data36 suggest that this is more effective than i.v. administration of proteasome inhibitor for the treatment of ovarian cancer (Figure 6D). One day after electroporation i.m. with the 4UbFL reporter, the mice were imaged to obtain baseline bioluminescence. One dose of RA183 (20 mg/kg in 25% β-hydroxypropyl cyclodextrin in water) was administered i.p., and the mice were imaged again at 4 and 24 h to determine the luciferase activity. RA183 caused a rapid increase in bioluminescence, which was sustained for >24 h.
In Vitro Microsomal Stability and Metabolite Identification of RA183
In our previous study,22 RA190 was detected in the plasma of mice 1 h after i.p. injection, and it exhibited an initial half-life of ∼4 h. In contrast, upon i.p. administration, no RA183 was detected in plasma at 1 h, suggesting that it is degraded rapidly in vivo, possibly by the liver. Indeed, reduced nicotinamide adenine dinucleotide phosphate (NADPH)-dependent metabolism was observed when RA183 and RA190 were incubated with human liver microsomes. Most RA183 (∼93%) was converted to metabolites after 1 h incubation with human microsomes, whereas RA190 was more stable such that only ∼41% was converted to metabolites in 1 h (Figure S6). One major metabolite of RA183 was observed with loss of mass that is consistent with the RA183 structure (Figure S7). The proposed metabolite structure was confirmed by chemical synthesis as the active compound RA9.3,27 Since ovarian cancer is routinely treated with i.p. chemotherapy, and this potentially enhances the tumor dosing while limiting systemic toxicity,36 we elected to proceed with i.p. administration for studies to assess the antitumor activity of RA183 in mouse models of ovarian cancer.
Tumor Therapy with RA183
We first examined in immunocompetent mice the therapeutic effect of RA183 against the syngeneic murine ovarian tumor model ID8-luc expressing luciferase.24,37 C57BL6 mice were challenged i.p. with ID8-luc tumor as this is the site through which ovarian cancer primarily develops and spreads. Treatment was initiated 3 days later. The mice were administered i.p. either 6.6 mg/kg RA183 in 25% β-hydroxypropyl cyclodextrin in water or vehicle alone for 12 days daily and tumor growth was measured using bioluminescent imaging. On day 17, the RA183-treated mice showed a significantly lower tumor burden (Figure 6E,F). The dosing was increased to 10 mg/(kg day) RA183 for 5 more days and on day 24, the tumor burden had not increased further, whereas the control mice demonstrated continued tumor growth.
Nude mice were inoculated i.p. with human ES2-luc ovarian clear cell carcinoma cells. After 4 days, treatment i.p. with vehicle alone or 6.6 mg/kg RA183 daily was initiated for 12 days. Bioluminescent imaging on days 7, 10, and 13 indicated that treatment with RA183 significantly reduced ES2 xenograft tumor burden compared to vehicle-treated mice (Figure 6G).
Studies indicating an important biological role for RPN13 in ovarian and other cancers and resistance to cisplatin treatment,38,39 as well as the preclinical antitumor activity of RA183 and RA190 indicate the promise for this approach to treat ovarian and TNBC in addition to MM.25 Our data provide information about the pharmacophore of the α,β-unsaturated carbonyls of this series of bischalcones and the influence of the side chains upon metabolism and specificity for RPN13, with which to further improve their druglike properties.
Methods
Cell Lines and Viability Assays
All cell lines were obtained from American Type Culture Collection (Manassas, VA) and cultured in specified medium supplemented with 10% fetal bovine serum, 100 IU/mL penicillin, and 100 μg/mL streptomycin at 5% CO2. OV2008, HeLa, SiHa, ME180, HT3, and C33A were grown in Dulbecco’s modified Eagle’s medium-, OVCAR3-, CaSki-, ES2-, TOV21G-, NCI-H929-, MM.1S-, RPMI-8226-, U266-, ANBL6-, and Bortezomib-resistant lines in RPMI-1640, and SKOV3 and HCT116 cell lines in McCoy’s 5A medium. Cell viability was assayed using CellTiter 96 AQ ueous One Solution Reagent (Promega, Madison, WI). Cells seeded at a concentration of 1000 cells/well for HeLa, 5000 cells/well for ES2 and HCT116, 3000 cells/well for ovarian cancer lines, and 10 000–20 000 cells/well for other cell lines in 100 μL medium in 96-well plate were treated with compounds at specified concentrations. After the indicated periods, the cells were incubated according to the manufacturer’s protocol with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide labeling mixture (Sigma) for 1–2 h and absorbance at 570 nm was measured using a Benchmark Plus microplate spectrophotometer (Bio-Rad).
Antibodies and Western Blot Analysis
Total cellular protein (10–20 μg) from each sample was subjected to SDS-PAGE, transferred to PVDF membranes, and analyzed by Western blot. Antibodies for Western Blot analysis were obtained by the following commercial sources: anti-p21(WAF1), Bax (Cell Signaling Technology, Danvers, MA), antiubiquitin (SC8017), anti-IkB-α (C-15), anti-ATF-4 (SC-200) (Santa Cruz Biotechnology, Santa Cruz, CA), anti-PARP (BD Pharmingen, San Diego, California), (Stressgen Corp., Victoria, BC, Canada), antitubulin (Sigma, St. Louis, MO), anti-actin, anti-ADRM1 (anti-RPN13) (Sigma-Aldrich), antiubiquitin, Lys48 (05-1307) (Millipore), streptavidin Dynabeads (Invitrogen), HRP–streptavidin (Invitrogen), peroxidase-linked antimouse immunoglobulin G (IgG), and antirabbit IgG (GE Healthcare U.K. Ltd., U.K.) and utilized at the concentration recommended by the manufacturer.
Flow Cytometry
Induction of apoptosis was determined using annexin V-PE Apoptosis Detection Kit I (BD Pharmingen, San Diego, CA) according to the manufacturer’s protocol. Briefly, 1 × 105 cells were resuspended in binding buffer and 5 μL of annexin V-PE and 5 μL of 7-AAD were then added into the cells, which were then incubated at room temperature for 15 min and analyzed by flow cytometry on a FACSCalibur using CellQuest software (Becton Dickinson, Mountain View, CA). Active caspase-3 was measured using PE-Active Caspase-3 antibody (550821) and 1× BD Cytofix/Cytoperm fixation buffer (51-6896 kc) (BD Pharmingen, San Diego, CA) according to the manufacturer’s protocol and flow cytometry. To assay ROS, 2 × 105 cells were added to a six-well plate the day before use. After treatment and incubation, the plates were washed once with phosphate-buffered saline (PBS) and the cells were harvested with trypsin–ethylenediaminetetraacetic acid. The cells were subsequently washed again with PBS and then suspended in 1 mL of PBS. The cells were incubated with 2,7-dichlorofluorescein diacetate (H2DCFDA; 1 μM) in a CO2 incubator at 37 °C for 60 min. The cells were then washed twice with PBS and analyzed by flow cytometry.
Biotin Labeling Assay
19S purified proteasome (R&D Systems, Cat. No. E-366) (500 ng) in 19S buffer (20 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid, 20 mM NaCl, 1 mM dithiothreitol (DTT), 15% glycerol) was incubated with compounds (20 μM) for a period of 30 min at room temperature, mixed with an equal volume of Laemmli sample buffer (20 μL) (Bio-Rad), and boiled for 5 min. The total sample was mixed with an equal volume of Laemmli sample buffer (20 μL) (Bio-Rad) and boiled for 5 min. The proteins were separated using a 4–15% Bio-Rad Mini-PROTEAN SDS-PAGE gel (1 h at 100 V) and transferred to PVDF membrane overnight at 4 °C (24 V). The membrane was blocked with 5% bovine serum albumin in PBST for 1 h at room temperature and washed for 20 min (3× with PBST). Then, the membrane was probed with HRP–streptavidin (1:10 000 in PBST) for a period of 1 h at room temperature and washed for 30 min (3× with PBST) and developed using HyGLO chemiluminescent detection reagent (Denville) for biotin recognition.
Recombinant RPN13 Preparation
Human RPN13 full-length, Pru domain, DEUBAD domain, RPN2 (940–953), and His-tagged UCH37 were prepared as described previously.30,31,40
NMR Spectroscopy
All NMR experiments were performed at 25 °C on Bruker AVANCE 700, 800, or 850 MHz spectrometers equipped with cryogenically cooled probes. Processing was performed in NMRPipe,41 and the resulting spectra were visualized with XEASY.42 Protein concentrations were calculated by extinction coefficients based on amino acid composition and absorbance at 280 nm for protein dissolved in 6 M guanidine–HCl. Buffer A (20 mM NaPO4, 50 mM NaCl, 2 mM DTT, 0.1% NaN3, 5% D2O pH 6.5) was used for all NMR samples except those containing RA183 or b-AP15, which were performed in buffer B (buffer A with no DTT present). For all NMR samples, 10-fold molar excess RA183 or b-AP15 (5 mM stock in DMSO) was preincubated with RPN13 protein at 4 °C overnight and unreacted or labile RA183 or b-AP15 was removed by extensive dialysis against buffer C (20 mM NaPO4, 50 mM NaCl, pH 6.5).
LC–MS Analysis of RA183 Adducts
Human RPN13 full-length, Pru domain, DEUBAD domain, RPN2 (940–953), or His-UCH37 was dialyzed extensively against buffer C to remove DTT. Complexes of RPN13 full-length:RPN2 (940–953) or RPN13 Pru:RPN2 (940–953) were formed by incubating sample mixtures on ice for >1 h. Next, 20 or 50 μM RA183 (5 mM stock in DMSO) was added to 100 μL of 2 μM target sample and incubated at 4 °C for 2 h while rotating. After 2 h, the samples were immediately analyzed by LC–MS without any dialysis. For LC–MS analysis, acetonitrile was added to RA183-treated proteins to a final concentration of 10%. LC–MS was performed on either an Agilent (Agilent Technologies, Inc., Santa Clara, CA) 6100 Series Quadrupole LC/MS System or 6520 Accurate-Mass Q-TOF LC/MS System, each equipped with an electrospray source, operated in the positive-ion mode. Data acquisition and analysis were performed by OpenLAB CDS ChemStation Edition C.01.05 or MassHunter Workstation (version B.06.01). For data analysis and deconvolution of accurate mass spectra, MassHunter Qualitative Analysis software (version B.07.00) with BioConfirm Workflow was used.
RPN13 Pru–RA183 Complex Calculation
RPN13 Pru–RA183 complexes were generated by using High Ambiguity Driven protein–protein DOCKing (HADDOCK) 2.243 in combination with CNS44 via a method described previously.22 The atomic coordinates for human RPN13 Pru domain were obtained from PDB 5IRS.45 Four distance restraints were defined between C88 Sγ of RPN13 and four atoms of RA183 to recapitulate the sulfur–carbon bond; HADDOCK requires two distinct molecules for docking. 1H,15N HSQC experiments were recorded on 15N-labeled RPN13 Pru without/with RA183. The ratio of peak intensity values (Δ) was plotted for each RPN13 backbone and side-chain (Figure 2D) amide group according to eq 1, in which I represents peak intensity and 0.65 is a scaling factor derived by setting the randomly coiled N- and C-terminal ends of RPN13 Pru domain as unaffected by RA183.
| 1 |
RPN13 Pru residues with Δ values greater than 1 standard deviation (SD) above average (Figure 2D), and >40% accessibility were defined as “active” and their neighbors as “passive” with the exception of M31, V93, V95, F106, and W108. The accessibility of these amino acids is <40%; however, their side chains are located in the binding surface and these residues were therefore also defined as active. Ambiguous interaction restraints (AIRs) were imposed to restrict hRPN13 Pru active residues to be within 2.0 Å of any RA183 atom.43 A total of 1000 structures were subjected to rigid-body energy minimization, and 250 lowest-energy structures were chosen for semiflexible simulated annealing in torsion angle space, followed by refinement in explicit water. During semiflexible simulated annealing, atoms at the interface were allowed to move, but constrained by the AIRs and unambiguous distance constraints defining the sulfur–carbon bond. After water refinement, 250 RPN13 Pru–RA183 structures were sorted into three clusters by using a 1.5 Å cutoff criterion on the root-mean-square deviation of RA183. The lowest-energy structure of each cluster was energy-minimized by Schrödinger after explicit introduction of a covalent bond between RPN13 C88 Sγ and the RA183 reacted carbon.
Mapping the RPN13 Pru Domain Surface That Interacts with b-AP15
1H,15N HSQC experiments were recorded on 15N-labeled RPN13 Pru without/with b-AP15. The ratio of peak intensity values (Δ) were calculated for each RPN13 Pru domain backbone amide group, according to eq 2, in which I represents peak intensity and 0.259 is a scaling factor to adjust for reduced protein concentration in the RPN13 Pru–b-AP15 sample; this value was derived by setting the randomly coiled N- and C-terminal ends of RPN13 Pru domain as unaffected by b-AP15.
| 2 |
RPN13 Pru residues with Δ values greater than 1 SD value above average were mapped onto an RPN13 Pru domain ribbon diagram with a red gradient by using PyMOL software.
Immunoprecipitation Assay
SKOV3 cells (5 × 107 cells) were lysed in 2 mL of M-PER buffer, and lysate was cleared using a tabletop centrifuge (2 min, 13 000 rpm). The collected supernatant was precleared with Dynabeads MyOne Streptavidin T1 beads (100 μL) (Invitrogen, Cat. No. 65601) for 1 h at 4 °C. Beads were removed by magnet, and the supernatant was either treated with RA183B (1 μM) or untreated for 45 min at 4 °C. Lysate was denatured using SDS (1% final concentration) at 70 °C for 5 min and diluted 10-fold. Dynabeads MyOne Streptavidin T1 beads (200 μL) were added to both samples and incubated at 4 °C overnight with continuous gentle rotation. The beads were separated using a magnet and washed with M-PER (200 μL, 3×). The supernatant was saved for Western blotting. The collected beads were eluted by heating at 70 °C for 10 min with 25 μL of M-PER and 25 μL of Laemmli buffer. The beads were removed using magnet, and the sample was separated by SDS-PAGE and transferred to PVDF membrane. The membrane was blocked with 5% milk and treated with RPN13 antibody (SAB1404889, Sigma). The membrane was treated with HRP-linked antimouse IgG secondary antibody and developed using chemiluminesence reagent.
siRNA Knockdown Studies
HeLa cells were transfected with Dharmacon siRNA for RPN13 (L-012340-01-0005) (50 nM) or nonspecific control (D-001810-10-05) for 48 h. The cells were lysed and subjected to the labeling experiment similar to the previous experiments.
Luciferase Assay
Subconfluent cultures of cells were transfected with 4UbFL or FL plasmids using Lipofectamine 2000 reagent (Life Technologies, Carlsbad, CA). The cells were seeded at 10 000 cells/well in a 96-well plate 48 h post-transfection and incubated with compounds or vehicle (DMSO) at the doses or time indicated for individual experiments. Luciferase activity in cell lysate was determined with a Luciferase Assay Kit (Promega, Madison, WI) according to the manufacturer’s instructions. Bioluminescence was measured by using a GloMax-Multi Detection System (Promega, Madison, WI).
Microsomal Stability and Metabolite Assays
Human and mouse liver microsomes and the NADPH regenerating systems were used to characterize the stability and metabolism of RA183 that were purchased from Corning Life Sciences (Tewksbury, MA) and BD Gentest Products and Services (Woburn, MA). RA183 and its proposed metabolite were synthesized at Johns Hopkins University and verified by NMR and mass spectrometry analysis.
Quantitative PCR To Measure mRNA Levels
Total RNA was isolated from cells using the RNeasy Mini Kit (Qiagen). Extracted RNA was normalized for concentration and reverse-transcribed using iScript cDNA synthesis kit (Bio-Rad). CHOP10 expression levels were measured by TaqMan Gene Expression Assays with TaqMan Gene Expression Master Mix (Applied Biosystems) and run with the standard thermal cycling protocol. Spliced XBP1 mRNA was assayed with SsoFast EvaGreen Supermix (Bio-Rad) following the protocol for the iCycler System. Forward and reverse primers were 5′-TGCTGAGTCCGCAGCAGGTG and 5′-TGGGTCCAAGTTGTCCAGAATGCC, respectively. Calculations were done according to the Livak method46 and normalized to reference gene GAPDH. Each condition was replicated three times, and each sample was run in triplicate.
In Vivo DNA Delivery, Imaging, and Treatment
All animal procedures were performed according to approved protocols and in accordance with the recommendations for the proper use and care of laboratory animals. Nude, C57BL/6, Balb/c female mice (4–6 weeks old) were purchased from the NCI-Frederick Animal Production Area (Frederick, MD). Gene gun particle-mediated DNA delivery was performed using a helium-driven gene gun (Bio-Rad, Hercules, CA), as described previously.47 For electroporation, a patch of the mouse leg was shaved and 10 μg of 4UbFL or FL DNA plasmid in 20 μL of PBS was injected into the quadriceps femoralis muscle, followed immediately by injection of the 2 Needle Array to 5 mm depth encompassing the injection site and square-wave electroporation (ElectroSquarePorator 833, BTX-2 Needle Array 5 mm gap, Harvard Apparatus) delivered as eight pulses at 106 V for 20 ms with 200 ms intervals. Two hours after gene gun delivery of the plasmid or 1 day post-electroporation, the mice were injected with luciferin (0.3 mg in 100 μL of water i.p.) and anesthetized with isoflurane, and optical imaging was performed for basal level luciferase expression. Images were acquired for 10 min with a Xenogen IVIS 200 (Caliper, Hopkinton, MA). Equally sized areas were analyzed using Living Image 2.20 software. The mice were again imaged at 4 and 24 h post-treatment.
Tumor Treatment Studies
Nude mice (eight per group) were inoculated with 1 × 106 ES2-Luc cells i.p. in 100 μL of PBS. On day 3, the mice were imaged for basal level luminescence expression. The mice were randomized into two groups, treated daily i.p. with RA183 (6.6 mg/kg) or vehicle, and imaged again on days 7 and 14. Results are reported as mean ± standard deviation (SD). ID8-Luc cells (3 × 105) were injected into the peritoneal cavity of C57BL6 mice (n = 5 per group) on day 0. The tumor growth was detected by monitoring luciferase activity after injection of luciferin. The mice were treated daily with either vehicle alone or RA183 (5 mg/kg) on days 3–17. The daily RA183 dose was increased to 10 mg/kg on days 18–24. The mice were imaged on days 3, 10, 17, and 24 for their luciferase activity.
Statistical Analysis
Statistical significance of differences was assessed by two-tailed Student’s t using Prism (v.5 GraphPad, San Diego, CA) and Excel. The level of significance was set at p ≤ 0.05.
Acknowledgments
The authors thank D. Piwnica-Worms (Washington University, St. Louis, MO) for the 4UbFL plasmid. This work was supported by the Intramural Research Program of the NCI, National Institutes of Health Center for Cancer Research (to K.J.W.), and grant support was provided by National Institutes of Health grant P50 CA098252, the Alleghany Health Network-Johns Hopkins Cancer Research Fund, and the Ovarian Cancer Research Fund Alliance #458972 (to R.K.A., C.-F.H., and R.B.S.R.). The project was supported in part by the Sidney Kimmel Comprehensive Cancer Center Analytical Pharmacology Core at Johns Hopkins (P30 CA006973 and UL1 RR 025005) and the Dr. Richard W. TeLinde endowment. This project was funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01479.
RA183’s cellular target (Figure S1); RPN13 adduct structure (Figure S2); binding and competition by RA183-related compounds (Figures S3 and S4); proteasomal binding by RA183 (Figure S5); in vitro microsomal stability of RA183 and its metabolites (Figures S6 and S7); and cell killing by RA183 (Table S1) (PDF)
The authors declare the following competing financial interest(s): Under a licensing agreement between Pontifax/PI Therapeutics and Johns Hopkins University, Drs. Anchoori and Roden are entitled to royalties on an invention described in this article. This arrangement has been reviewed and approved by Johns Hopkins University in accordance with its conflict of interest policies.
Supplementary Material
References
- Bazzaro M.; Lee M. K.; Zoso A.; Stirling W. L.; Santillan A.; Shih I.-M.; Roden R. B. Ubiquitin-proteasome system stress sensitizes ovarian cancer to proteasome inhibitor-induced apoptosis. Cancer Res. 2006, 66, 3754–3763. 10.1158/0008-5472.CAN-05-2321. [DOI] [PubMed] [Google Scholar]
- Bazzaro M.; Lin Z.; Santillan A.; Lee M. K.; Wang M. C.; Chan K. C.; Bristow R. E.; Mazitschek R.; Bradner J.; Roden R. B. Ubiquitin proteasome system stress underlies synergistic killing of ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor. Clin. Cancer Res. 2008, 14, 7340–7347. 10.1158/1078-0432.CCR-08-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin K.; Anchoori R.; Iizuka Y.; Meints J.; MacNeill L.; Vogel R. I.; Orlowski R. Z.; Lee M. K.; Roden R. B.; Bazzaro M. Small-molecule RA-9 inhibits proteasome-associated DUBs and ovarian cancer in vitro and in vivo via exacerbating unfolded protein responses. Clin. Cancer Res. 2014, 20, 3174–3186. 10.1158/1078-0432.CCR-13-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilarsky C.; Wenzig M.; Specht T.; Saeger H. D.; Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia 2004, 6, 744–750. 10.1593/neo.04277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejzo M. S.; Dering J.; Ginther C.; Anderson L.; Ramos L.; Walsh C.; Karlan B.; Slamon D. J. Comprehensive analysis of 20q13 genes in ovarian cancer identifies ADRM1 as amplification target. Genes, Chromosomes Cancer 2008, 47, 873–883. 10.1002/gcc.20592. [DOI] [PubMed] [Google Scholar]
- Carvalho B.; Postma C.; Mongera S.; Hopmans E.; Diskin S.; van de Wiel M. A.; van Criekinge W.; Thas O.; Matthai A.; Cuesta M. A.; Terhaar Sive Droste J. S.; Craanen M.; Schrock E.; Ylstra B.; Meijer G. A. Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression. Gut 2009, 58, 79–89. 10.1136/gut.2007.143065. [DOI] [PubMed] [Google Scholar]
- Jørgensen J. P.; Lauridsen A. M.; Kristensen P.; Dissing K.; Johnsen A. H.; Hendil K. B.; Hartmann-Petersen R. Adrm1, a putative cell adhesion regulating protein, is a novel proteasome-associated factor. J. Mol. Biol. 2006, 360, 1043–1052. 10.1016/j.jmb.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Hamazaki J.; Iemura S.; Natsume T.; Yashiroda H.; Tanaka K.; Murata S. A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO J. 2006, 25, 4524–4536. 10.1038/sj.emboj.7601338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T.; Song L.; Xu W.; DeMartino G. N.; Florens L.; Swanson S. K.; Washburn M. P.; Conaway R. C.; Conaway J. W.; Cohen R. E. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat. Cell Biol. 2006, 8, 994–1002. 10.1038/ncb1460. [DOI] [PubMed] [Google Scholar]
- Gandhi T. K.; Zhong J.; Mathivanan S.; Karthick L.; Chandrika K. N.; Mohan S. S.; Sharma S.; Pinkert S.; Nagaraju S.; Periaswamy B.; Mishra G.; Nandakumar K.; Shen B.; Deshpande N.; Nayak R.; Sarker M.; Boeke J. D.; Parmigiani G.; Schultz J.; Bader J. S.; Pandey A. Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat. Genet. 2006, 38, 285–293. 10.1038/ng1747. [DOI] [PubMed] [Google Scholar]
- Chen W.; Hu X.-T.; Shi Q.-L.; Zhang F.-B.; He C. Knockdown of the novel proteasome subunit Adrm1 located on the 20q13 amplicon inhibits colorectal cancer cell migration, survival and tumorigenicity. Oncol. Rep. 2009, 21, 531–537. 10.3892/or_00000254. [DOI] [PubMed] [Google Scholar]
- Mazumdar T.; Gorgun F. M.; Sha Y.; Tyryshkin A.; Zeng S.; Hartmann-Petersen R.; Jorgensen J. P.; Hendil K. B.; Eissa N. T. Regulation of NF-kappaB activity and inducible nitric oxide synthase by regulatory particle non-ATPase subunit 13 (Rpn13). Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 13854–13859. 10.1073/pnas.0913495107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejzo M. S.; Ginther C.; Dering J.; Anderson L.; Venkatesan N.; Konecny G.; Karlan B.; Slamon D. J. Knockdown of ovarian cancer amplification target ADRM1 leads to downregulation of GIPC1 and upregulation of RECK. Genes, Chromosomes Cancer 2011, 50, 434–441. 10.1002/gcc.20868. [DOI] [PubMed] [Google Scholar]
- Fejzo M. S.; Anderson L.; von Euw E. M.; Kalous O.; Avliyakulov N. K.; Haykinson M. J.; Konecny G. E.; Finn R. S.; Slamon D. J. Amplification Target ADRM1: Role as an Oncogene and Therapeutic Target for Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 3094–3109. 10.3390/ijms14023094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejzo M. S.; Anderson L.; Chen H. W.; Anghel A.; Zhuo J.; Anchoori R.; Roden R.; Slamon D. J. ADRM1-amplified metastasis gene in gastric cancer. Genes, Chromosomes Cancer 2015, 54, 506–515. 10.1002/gcc.22262. [DOI] [PubMed] [Google Scholar]
- Al-Shami A.; Jhaver K. G.; Vogel P.; Wilkins C.; Humphries J.; Davis J. J.; Xu N.; Potter D. G.; Gerhardt B.; Mullinax R.; Shirley C. R.; Anderson S. J.; Oravecz T. Regulators of the proteasome pathway, Uch37 and Rpn13, play distinct roles in mouse development. PLoS One 2010, 5, e13654 10.1371/journal.pone.0013654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki J.; Hirayama S.; Murata S. Redundant Roles of Rpn10 and Rpn13 in Recognition of Ubiquitinated Proteins and Cellular Homeostasis. PLoS Genet. 2015, 11, e1005401 10.1371/journal.pgen.1005401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromm P. M.; Crews C. M. The Proteasome in Modern Drug Discovery: Second Life of a Highly Valuable Drug Target. ACS Cent. Sci. 2017, 3, 830–838. 10.1021/acscentsci.7b00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Desai A.; Zeng D.; Gong T.; Lu P.; Wang M. Magic year for multiple myeloma therapeutics: Key takeaways from the ASH 2015 annual meeting. Oncotarget 2017, 8, 10748–10759. 10.18632/oncotarget.13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlafer D.; Shah K. S.; Panjic E. H.; Lonial S. Safety of proteasome inhibitors for treatment of multiple myeloma. Expert Opin. Drug Saf. 2017, 16, 167–183. 10.1080/14740338.2017.1259310. [DOI] [PubMed] [Google Scholar]
- Aghajanian C.; Blessing J. A.; Darcy K. M.; Reid G.; DeGeest K.; Rubin S. C.; Mannel R. S.; Rotmensch J.; Schilder R. J.; Riordan W. A phase II evaluation of bortezomib in the treatment of recurrent platinum-sensitive ovarian or primary peritoneal cancer: a Gynecologic Oncology Group study. Gynecol. Oncol. 2009, 115, 215–220. 10.1016/j.ygyno.2009.07.023. [DOI] [PubMed] [Google Scholar]
- Anchoori R. K.; Karanam B.; Peng S.; Wang J. W.; Jiang R.; Tanno T.; Orlowski R. Z.; Matsui W.; Zhao M.; Rudek M. A.; Hung C. F.; Chen X.; Walters K. J.; Roden R. B. A bis-Benzylidine Piperidone Targeting Proteasome Ubiquitin Receptor RPN13/ADRM1 as a Therapy for Cancer. Cancer Cell 2013, 24, 791–805. 10.1016/j.ccr.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randles L.; Anchoori R. K.; Roden R. B.; Walters K. J. Proteasome Ubiquitin Receptor hRpn13 and its Interacting Deubiquitinating Enzyme Uch37 are Required for Proper Cell Cycle Progression. J. Biol. Chem. 2016, 291, 8773–8783. 10.1074/jbc.M115.694588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong R. S.; Anchoori R. K.; Yang B.; Yang A.; Tseng S. H.; He L.; Tsai Y. C.; Roden R. B.; Hung C. F. RPN13/ADRM1 inhibitor reverses immunosuppression by myeloid-derived suppressor cells. Oncotarget 2016, 7, 68489–68502. 10.18632/oncotarget.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Ray A.; Li S.; Das D. S.; Tai Y. T.; Carrasco R. D.; Chauhan D.; Anderson K. C. Targeting proteasome ubiquitin receptor Rpn13 in multiple myeloma. Leukemia 2016, 30, 1877–1886. 10.1038/leu.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arcy P.; Brnjic S.; Olofsson M. H.; Fryknas M.; Lindsten K.; De Cesare M.; Perego P.; Sadeghi B.; Hassan M.; Larsson R.; Linder S. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011, 17, 1636–1640. 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- Vogel R. I.; Coughlin K.; Scotti A.; Iizuka Y.; Anchoori R.; Roden R. B.; Marastoni M.; Bazzaro M. Simultaneous inhibition of deubiquitinating enzymes (DUBs) and autophagy synergistically kills breast cancer cells. Oncotarget 2015, 6, 4159–4170. 10.18632/oncotarget.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X. B.; Ouyang S. Y.; Li C. J.; Miao S.; Wang L.; Goldberg A. L. hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO J. 2006, 25, 5742–5753. 10.1038/sj.emboj.7601450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnjak K.; Elsasser S.; Zhang N.; Chen X.; Randles L.; Shi Y.; Hofmann K.; Walters K. J.; Finley D.; Dikic I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008, 453, 481–488. 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Lee B. H.; Finley D.; Walters K. J. Structure of proteasome ubiquitin receptor hRpn13 and its activation by the scaffolding protein hRpn2. Mol. Cell 2010, 38, 404–415. 10.1016/j.molcel.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Nowicka U.; Sridharan V.; Liu F.; Randles L.; Hymel D.; Dyba M.; Tarasov S. G.; Tarasova N. I.; Zhao X. Z.; Hamazaki J.; Murata S.; Burke T. R. Jr.; Walters K. J. Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets. Nat. Commun. 2017, 8, 15540 10.1038/ncomms15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Liu F.; Durham S. E.; Tarasov S. G.; Walters K. J. A High Affinity hRpn2-Derived Peptide That Displaces Human Rpn13 from Proteasome in 293T Cells. PLoS One 2015, 10, e0140518 10.1371/journal.pone.0140518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ri M.; Iida S.; Nakashima T.; Miyazaki H.; Mori F.; Ito A.; Inagaki A.; Kusumoto S.; Ishida T.; Komatsu H.; Shiotsu Y.; Ueda R. Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010, 24, 1506–1512. 10.1038/leu.2010.137. [DOI] [PubMed] [Google Scholar]
- McConkey D. J. The integrated stress response and proteotoxicity in cancer therapy. Biochem. Biophys. Res. Commun. 2017, 482, 450–453. 10.1016/j.bbrc.2016.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker G. D.; Pica C. M.; Song J.; Luker K. E.; Piwnica-Worms D. Imaging 26S proteasome activity and inhibition in living mice. Nat. Med. 2003, 9, 969–973. 10.1038/nm894. [DOI] [PubMed] [Google Scholar]
- Jandial D. A.; Brady W. E.; Howell S. B.; Lankes H. A.; Schilder R. J.; Beumer J. H.; Christner S. M.; Strychor S.; Powell M. A.; Hagemann A. R.; Moore K. N.; Walker J. L.; DiSilvestro P. A.; Duska L. R.; Fracasso P. M.; Dizon D. S. A phase I pharmacokinetic study of intraperitoneal bortezomib and carboplatin in patients with persistent or recurrent ovarian cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 145, 236–242. 10.1016/j.ygyno.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roby K. F.; Taylor C. C.; Sweetwood J. P.; Cheng Y.; Pace J. L.; Tawfik O.; Persons D. L.; Smith P. G.; Terranova P. F. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000, 21, 585–591. 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Ratovitski E. A. Phosphorylated TP63 induces transcription of RPN13, leading to NOS2 protein degradation. J. Biol. Chem. 2010, 285, 41422–41431. 10.1074/jbc.M110.158642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Ratovitski E. A. Phospho-ΔNp63α/Rpn13-dependent regulation of LKB1 degradation modulates autophagy in cancer cells. Aging 2010, 2, 959–968. 10.18632/aging.100249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner P.; Chen X.; Husnjak K.; Randles L.; Zhang N.; Elsasser S.; Finley D.; Dikic I.; Walters K. J.; Groll M. Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature 2008, 453, 548–552. 10.1038/nature06924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Bartels C.; Xia T. H.; Billeter M.; Guntert P.; Wuthrich K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 1995, 6, 1–10. 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- Dominguez C.; Boelens R.; Bonvin A. M. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Brünger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998, 54, 905–921. 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- Chen X.; Randles L.; Shi K.; Tarasov S. G.; Aihara H.; Walters K. J. Structures of Rpn1 T1:Rad23 and hRpn13:hPLIC2 Reveal Distinct Binding Mechanisms between Substrate Receptors and Shuttle Factors of the Proteasome. Structure 2016, 24, 1257–1270. 10.1016/j.str.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen T. D.; Livak K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Trimble C.; Lin C. T.; Hung C. F.; Pai S.; Juang J.; He L.; Gillison M.; Pardoll D.; Wu L.; Wu T. C. Comparison of the CD8+ T cell responses and antitumor effects generated by DNA vaccine administered through gene gun, biojector, and syringe. Vaccine 2003, 21, 4036–4042. 10.1016/S0264-410X(03)00275-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.