

Abstract
Advances in the genetics, function, and stage-specificity of Plasmodium kinases has driven robust efforts to identify targets for the design of antimalarial therapies. Reverse genomics following phenotypic screening against Plasmodia or related parasites has uncovered vulnerable kinase targets including PI4K, PKG, and GSK-3, an approach bolstered by access to human disease-directed kinase libraries. Alternatively, screening compound libraries against Plasmodium kinases has successfully led to inhibitors with antiplasmodial activity. As with other therapeutic areas, optimizing compound ADMET and PK properties in parallel with target inhibitory potency and whole cell activity becomes paramount toward advancing compounds as clinical candidates. These and other considerations will be discussed in the context of progress achieved toward deriving important, novel mode-of-action kinase-inhibiting antimalarial medicines.
1. Introduction
To this day, the malaria parasite Plasmodium spp. remains a scourge particularly in less developed regions of the world. Of the five Plasmodium species that cause human malaria, Plasmodium falciparum (Pf), which is found predominantly in sub-Saharan Africa, was responsible for 50% of all malaria cases and 91% of the 446,000 deaths worldwide in 2016. The next most widespread is P. vivax (Pv), the dominant species in Latin America and Asia causing a milder form of malaria.1 Other less problematic Plasmodium species are P. ovale (Po), P. malariae (Pm), and P. knowlesi (Pk), the latter much more common in nonhuman primates. Eliminating malaria throughout the world, as has been achieved in many nations, is considered an achievable goal that will incorporate multipronged strategies including the development of new medicines. Currently, the World Health Organization (WHO) recommends that artemisinin-based combination therapy (ACT) and vector control measures are key factors in relieving the burden of malaria.2 However, recent reports of emerging resistance toward the ACT regimen3 and other antimalarial drugs with known mechanisms of action4 emphasize the need to expand the diversity of chemical matter acting against novel targets toward more efficacious drugs with multistage parasite life-cycle activity. In humans, the complex Plasmodium life-cycle encompasses a liver-stage infection wherein motile sporozoites differentiate and proliferate asexually to form merozoites and a blood-stage infection wherein the asexual merozoites replicate within red blood cells (through ring, trophozoite, and schizont substages), egress, and then reinfect red blood cells.5P. vivax and P. ovale sporozoites can also enter a hypnozoite stage that can remain quiescent in the liver for months if not longer before differentiating eventually into merozoites.6 A fraction of the merozoites in red blood cells differentiate and mature to female and male gametocytes that infect the mosquito after transmission from a bite.7 In the mosquito, the gametocytes further differentiate and eventually fuse to form a zygote that further evolves to form sporozoites that get transmitted to people in a mosquito bite.8 Notably, the expression of kinases and their importance to viability vary in the stages and substages of the life-cycle.9
Kinases are key controllers of signal transduction pathways that regulate essential cellular processes such as growth, development, and reproduction in eukaryotic cells.10,11 For this reason, human kinases are pursued as drug targets in a variety of diseases including cancers,12 inflammatory,13 and cardiovascular diseases.14 Since the approval of Gleevec 16 years ago,15 an additional 32 kinase inhibitors targeting the human kinome have been approved by the U.S. Food and Drug Administration (FDA) for clinical use.15 Given the success in developing drugs targeting human kinases, Plasmodium kinases are attractive targets for next generation antimalarials16 as both protein and lipid kinases are involved in key signaling pathways at various stages of the parasite life-cycle.17 The P. falciparum kinome encodes 86 to 99 protein kinase genes16 and a small set of lipid kinase genes. It is highly conserved between Plasmodium species and is much smaller than the human protein kinome of approximately 520 kinases.18Figure 1A shows the phylogenetic tree with a subset of the well-characterized protein kinases of P. falciparum.
Figure 1.

(A) Phylogenetic tree for a set of 38 well-characterized protein kinases in P. falciparum. The bootstrap tree was generated from 500 replicates using the neighbor-joining method on the full protein sequence with evolutionary distances computed using the Poisson correction method in the Mega7 software package.19 Colored labels indicate kinases discussed in this Perspective. Lipid kinases PI3K and PI4K were not included in the analysis. (B) Crystal structure (PDB: 4RZ7) of P. vivax PKG with inhibitor illustrating key interaction in the ATP binding site. (C) Crystal structure (PDB: 4RZ7) of P. vivax PKG inhibitor (cyan) superimposed with dasatinib (purple) from the X-ray structure with activated ABL kinase (PDB: 2GQG). Both inhibitors access the “deep hydrophobic pocket” extending past the threonine gatekeeper residue.
A major challenge when targeting kinases is that inhibitors usually target the highly conserved adenosine triphosphate (ATP)-binding pocket of the enzyme (Figure 1B), and therefore, target selectivity can be difficult to achieve.20 Fortunately, the long independent evolution of the malaria parasite allowed the emergence of distinct features in the malarial kinome. These include kinases that clearly cluster within groups found in the human genome but that can be distinguished from their mammalian homologues (Figure 1C). This would include Plasmodium kinases from given groups that contain characteristics of other families, such as PfPK6 or PfPK7, and composites between mitogen-activated protein kinase (MAPK) and cyclin dependent kinases (CDKs)21 and cyclic adenosine monophosphate (cAMP)-dependent kinase (PKA) and mitogen-activated protein kinase (MEK), respectively.22 This would also include kinases, such as CDPKs, that belong to a specific group but do not have a clear orthologue in mammalians and kinases that do not cluster within any of the established families, for example, Phe (F)–Ile (I)–Lys (K)–Lys (K) (FIKK). These important divergences can be exploited to synthesize compounds that selectively inhibit Plasmodium kinases over mammalian enzymes.23
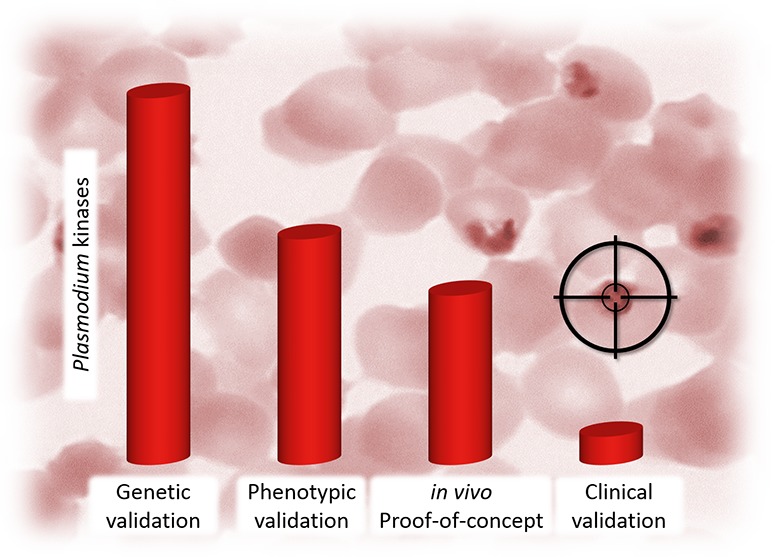
The path to delivering a new antimalarial based on inhibiting a Plasmodium kinase is a multistep process. First, kinase essentiality must be validated by determining the effect of disrupting function or diminishing expression in an organism on proliferation in culture or in the host. This has been achieved for the Plasmodium kinome through kinome-wide reverse genetics studies leading to the identification of 36 protein kinases that are essential (or likely essential) for completion of the erythrocytic cycle in P. falciparum in vitro(20) and of 12 protein kinases that are required for transmission of the rodent malaria parasite P. berghei (Pb) to the mosquito in vivo.(24) Phenotypic validation is a second level of validation defined as a chemical compound inhibiting a kinase target and also demonstrating an effect on the organism (most often cell kill). Target engagement studies need to be carried out to show that the phenotypic effect is due to binding to the intended kinase and not a different mode-of-action. The third level, in vivo validation, oftentimes denoted as in vivo proof-of-concept (POC), refers to the capability of a compound to create the intended pharmacodynamic (PD) effect in an animal model. For an antimalarial drug, this most often is the reduction of parasitaemia in a mouse model of infection. To show efficacy, the compound needs favorable in vivo pharmacokinetic (PK) properties for sufficient exposure in the blood to produce the intended PD response. Finally, as the key goal of drug discovery programs, clinical validation represents the fourth level with a drug molecule working effectively in malaria patients. To achieve clinical validation, the human PK must be favorable, whether it would be exposure in the blood for asexual stages of the malaria parasite or in the liver for liver stage disease. Importantly, sufficient safety for drug must also be demonstrated preclinically to justify human administration in the clinic and progression through Phases 1, 2, and 3. Table 1 represents a summary of the protein and lipid kinases discussed in this Perspective and their respective levels of validation achieved.
Table 1. Level of Validation Achieved for Plasmodium Targets Covered in This Perspectivea.
| P. falciparum protein kinase | genetic validationb | phenotypic validationc | in vivo efficacy | clinical validation |
|---|---|---|---|---|
| CDPK1 | √d | √ | √ | |
| CDPK4 | √ | √ | √e | |
| PKG | √ | √ | √ | |
| PKA | √e | |||
| MRK | √ | √ | ||
| GSK-3 | √ | √ | ||
| NEK-1 | √ | √ | √ | |
| FIKK8 | √ | |||
| PI3Kf | √ | √ | ||
| PI4K | √ | √ | √ | √ |
√ Indicates level of validation was achieved.
Genetic validation of kinases refers to where potential essentiality has been confirmed by knockout, chemical-genetic, or overexpression methods.
The fields are checked if an inhibitor of the kinase target also displayed whole cell activity recognizing that the activity may or may not be due solely to inhibition of the target.
There are conflicting data in the literature as to whether the target has been genetically validated.
Transmission-blocking was shown in an animal model.
DHA, implicated as a PI3K inhibitor, has other modes-of-action that are thought to be primarily responsible for antiplasmodium activity.
In this perspective, the emphasis has been placed on compound series and kinase targets that have a phenotypic level of validation and therefore in vitro antiplasmodial activity due to inhibition of an identified kinase target. Most interesting are those series that show POC in animal models of infection. Beyond animal models, there is a single example of a kinase target, P. falciparum phosphatidylinositol 4-kinase (PI4K), with inhibitor 37 (MMV048) that has progressed to human clinical trials.25
2. Calmodulin-Dependent Kinases (CaMK)
The group of calmodulin-dependent kinases (CaMK) contains the family of calcium-dependent protein kinases (CDPK) that have seven identified members, as well as the sucrose nonfermenting (SNF1)/adenosine monophosphate (AMP)-activated kinase (AMPK) family and other closely related kinases.
2.1. Calcium-Dependent Protein Kinases (CDPKs)
CDPKs, the main transducers of calcium signaling in Plasmodia, are unique to alveolates, plants, and some algae.26 As they do not have human orthologues, it follows that selectivity relative to mammalian kinases would be more easily achieved. Structurally, CDPKs have a catalytic kinase region responsible for phosphorylation, as well as an autoregulatory sequence and so-called EF-hand motifs that are similar to the calcium-binding protein calmodulin.27 Inhibitors of CDPK1 and CDPK4 have been progressed furthest in this family of kinases and will be discussed further. PfCDPK2 has been shown to be nonessential for asexual proliferation but essential for the transition of microgametocytes into male gametes, a process known as exflaggelation, and mosquito infection.28PfCDPK3 is specifically expressed in the sexual erythrocytic stages,29 and its P. berghei homologue has been shown to be required for ookinete gliding motility and invasion of the mosquito’s midgut.30PfCDPK5 is required for parasite egress through microneme discharge.31 However, hyperactivation of P. falciparum cyclic guanosine monophosphate (cGMP)-dependent protein kinase (PfPKG) can overcome the block of egress caused by a PfCDPK5 knock out. To our knowledge, there are no publications of medicinal chemistry programs that have targeted these kinases so far.
For several Plasmodium kinases, chemical genetics approaches have been used involving the mutation of the so-called gatekeeper residue located in the catalytic domain of the kinase. The catalytic domains of kinases have several conserved structural motifs that are common among most kinases. The hinge region connects the N-lobe and the C-lobe and is a frequent binding site for both ATP and kinase inhibitors (Figure 1B).32 The gatekeeper is the first amino acid residue of the hinge region. Depending on its size, a hydrophobic pocket becomes accessible for inhibitors. Mammalian kinases usually possess threonine and larger amino acids in this position. Shokat et al. developed a strategy to achieve selectivity for mammalian kinases that have a small gatekeeper residue by designing kinase inhibitors with a specific substitution termed the “bump”,33 a principle that has been applied to several Plasmodium kinases.34 There are a number of protozoal kinases like PfCDPK4 that possess an atypically small gatekeeper residue and therefore are accessible to inhibition by such bumped kinase inhibitors and offer the opportunity to design selectivity over mammalian kinases.35 Subsequently, mutation of the gatekeeper in these kinases to a larger amino acid offers the opportunity to check whether the antiplasmodial activity is in fact mediated by inhibition of the targeted kinase.
2.1.1. P. falciparum Calcium-Dependent Protein Kinase 1 (PfCDPK1)
PfCDPK1 has been associated with parasite motility, host invasion by P. falciparum, microneme secretion, and gametogenesis.36−38 It is highly expressed in asexual blood and mosquito stages.36 Previous studies hinted at the corresponding cdpk1 gene being essential in P. falciparum in that it is refractory to gene disruption.39 Other studies reported a decrease in parasitaemia due to reduced invasion following conditional knockdown36 or no influence on asexual parasite development using a chemical genetics approach.40 Recently, Bansal et al. were unable to create a knockout (KO) with a wild-type strain but were able to do so with a transgenic line containing a CDPK1 mutant (T145M).38 The phenotype showed slow asexual proliferation compared to the wildtype indicating some degree of ambiguity around the essentiality of PfCDPK1. Interestingly, the CDPK1 KO parasites are defective in the formation of male and female gametes and failed to establish infection in mosquitoes making PfCDPK1 an interesting target for transmission blocking. Increased sensitivity of the CDPK1 KO parasites toward PfPKG inhibitors compared to the wildtype indicated the presence of compensatory mechanisms in the transgenic parasites. Upregulation of kinases such as PfCDPK5 and PfCDPK6 that are downstream from PfPKG has been shown previously for the CDPK1 mutant T145M.41
Considerable work has led to the identification of PfCDPK1 inhibitors with antiplasmodial activity. However, several disconnects between enzyme potency and antiplasmodial activity have been observed and suggest multiple or other targets responsible for the antiplasmodial activity. A high throughput screening (HTS) campaign against PfCDPK1 found a broad array of hits including pyrazolopyrimidines, azabenzimidazoles, isoxazole amides, pyrazinones, and imidazopyridazines with the latter being progressed the farthest.40,42−45 The imidazopyridazine hits with 2-aminoethylpyridines on the left-hand side and phenyl amides on the right-hand side (Figure 2) suffered from high logD (D being the distribution coefficient determined as the ratio of concentrations for molecules partitioned between octanol and water at a given pH) values, low metabolic stability, and poor selectivity relative to a human kinase panel.42 A homology model based on CDPK1 from Toxoplasma gondii (Tg) was created to optimize PfCDPK1 inhibitory potency alongside optimizing absorption, distribution, metabolism, excretion, and toxicology (ADMET) and physicochemical properties. Inhibitors retained potency against PbCDPK1 and PvCDPK1. The pyridine on the left-hand-side contributed little to binding affinity and could be replaced by a basic amine that purportedly formed a salt bridge with a close-by glutamate and accounted for the improved potency against PfCDPK1.43 Compound 2 demonstrated a lower logD, improved metabolic stability in both mouse and human liver microsomes, and better selectivity against a human kinase panel. Ultimately, the work led to compound 4 that demonstrated efficacy in a P. berghei mouse infection model.
Figure 2.

Imidazopyridazine inhibitors of PfCDPK1 and PfPKG.
Toward validating the mode-of-action of the imidazopyridazines and dissecting a disconnect observed between PfCDPK1 inhibitory potency and activity against the Pf3D7 strain, compounds were assessed in a synchronized parasite culture revealing two different classes of compounds (Class 1 and Class 2) with potentially different modes-of-action.40 Compound 3 with a pyrimidine linker (Class 1) acted on a late schizont stage of the parasite life-cycle and showed high inhibitory potency against the alternative PfPKG target. The best imidazopyridazine inhibitor showed an IC50 of 1.6 nM against PfPKG, and the compounds of class 1 showed a very good correlation between PfPKG and Pf3D7 activities. Mutating the gatekeeper residue of PfPKG from threonine to glutamine (T618Q) led to a dramatic loss in inhibitory potency. Replacing wild-type PKG with the T618Q mutant in Pf3D7 correspondingly diminished antiparasitic activity. For class 2 compounds, e.g., 2, PfCDPK1 was excluded as the target due to it not being expressed in earlier stages of the parasite life-cycle where the compound was shown to operate. Pull-down experiments with a biotin modified inhibitor identified P. falciparum heat shock protein 90 (PfHSP90) and in silico methods supported binding to its ATP pocket.40
In another screening campaign against PfCDPK1, Kato et al. identified 2,6,9-trisubstituted purines as inhibitors of PfCDPK1 with IC50s extending down to the low nanomolar range.39 The most potent compound, named purfalcamine 5 (Figure 3), displayed an IC50 value of 17 nM against PfCDPK1 and an EC50 of 230 nM against Pf3D7. As with the imidazopyridazines, a disconnect between enzyme inhibition and antiplasmodial activity was observed. The authors used agarose-immobilized purfalcamine for affinity chromatography toward identifying the target from cell lysates, and PfCDPK1 was the only protein with an ATP binding site that was specifically bound. However, the pulldown experiment could not exclude the possibility that purfalcamine binds to low-abundance proteins to account for the antiplasmodial activity. Nonetheless, in line with the CDPK1 mode-of-action, synchronized parasites treated with purfalcamine progressed through the life-cycle but were arrested at late schizogonic stage where the Pfcdpk1 gene is transcribed.
Figure 3.

2,6,9-Trisubstituted purine inhibitor of PfCDPK1, purfalcamine 5.
Purfalcamine failed to show efficacy in mice infected with P. yoelii (Py) on dosing 10 mg/kg bid. The authors attributed this to the compound being more selective toward PfCDPK1 relative to the orthologue of P. yoelii and to low exposure on oral dosing. This combined with improvement in antiplasmodial activity was deemed necessary for future analogue optimization work.
Lemercier et al. screened a 54,000 compound library against PfCDPK1 to identify 70 compounds with submicromolar Ki values, the most potent being indolizine 6 and imidazopyridazine 7 (Figure 4).46 Testing against a panel of 46 human kinases indicated high selectivity as 6 showed 75 and 54% inhibition at 10 μM only against stress-activated protein kinase-2 alpha (SAPK2α) and fibroblast growth factor receptor 1 (FGFR1), respectively, and 7 showed at least 89% inhibition at 10 μM only against CDK5/p25, Tropomyosin receptor kinase A (TrkA), FGFR1, Lyn, and ribosomal S6 kinase 1 (RSK1). Similar to previous studies, the activity of some compounds against PfCDPK1 correlated well with antiplasmodial activity, while others did not. In the latter case, the low correlation was attributed to variable permeability.
Figure 4.

Indolizine 6 and imidazopyridazine 7 inhibitors of PfCDPK1.
A PfCDPK1 pharmacophore model developed around the imidazopyridazines42−44 for in silico screening identified a number of compounds predicted to have IC50 values below 100 nM.47 Winnowing out compounds predicted to have unfavorable ADMET properties left 12 compounds with predicted IC50s between 15 and 60 nM.
It has been shown in several projects that CDPK1 has not been the efficacious target of the asexual stages despite compounds showing high potency against the enzyme. This relates to the ambiguity of PfCDPK1 essentiality for asexual blood stage proliferation.38 However, it was suggested that PfCDPK1 might be a good target for transmission blocking agents.
2.1.2. P. falciparum Calcium-Dependent Protein Kinase 4 (PfCDPK4)
Knockout experiments in P. berghei have shown the Plasmodium CDPK4 to be essential for exflaggelation48−50 and for sporozoite invasion of hepatocytes.51 The target itself is not vital for the blood stage parasite proliferation, but inhibitors would be of value as part of an extended gametocyte transmission blocking regimen if the drug levels in the blood could be maintained for 3–4 weeks.52 A strong correlation between PfCDPK4 inhibition and suppression of exflaggelation was observed, and bumped kinase inhibitors of PfCDPK4 were developed and shown to block transmission to mosquitoes.53 Inhibitors of PfCDPK4 were shown to be on target by the introduction of a S147M mutation at the gatekeeper residue of the target in P. falciparum, which diminished the suppression of exflaggelation.53
A series of potent 4-amino-pyrazolopyrimidine inhibitors of TgCDPK1 and CpCDPK154,55 were found to also inhibit PfCDPK4,56,57 and more potent inhibitors were developed against PfCDPK4, e.g., 8 and 9. These compounds also blocked exflaggelation in P. falciparum (Figure 5) with EC50s of 35 and 47 nM, respectively.53,56 Notably, 8 did not inhibit Src and Abl kinases at 20 μM, two off-target human kinases that also possess a smaller gatekeeper residue (threonine). Both compounds showed favorable PK in the mouse and were well tolerated in mouse feeding studies at 100 mg/kg bid dosing over 5 days.53 Compound 8 was dosed intraperitoneal (ip) in P. berghei infected mice at 50 mg/kg resulting in suppression of exflaggelation for 0.5 to 14 h after administration.56 When treating P. berghei gametocyte-infected mice with 8 at 10 mg/kg ip and allowing the Anopheles mosquitoes to feed, oocyst formation was completely blocked. Similarly, when Anopheles mosquitoes were fed with human blood infected with the PfNF54 strain of P. falciparum containing 3 μM of either 8 or 9, no sporozoite formation was observed when dissecting the salivary glands.53,56
Figure 5.

Bumped kinase inhibitors 8 and 9.
The initially derived TgCDPK1 inhibitors based on the aminopyrazole-carboxamide (AC) scaffold57 share pharmacophore features with the pyrazolopyrimidine series (Figure 6) and were also tested against PfCDPK4.58 The AC series was about 10-fold less active than the pyrazolopyrimidine series but displayed improved selectivity over human Src kinase, improved margins over the human ether-a-go-go related gene (hERG) channel, and lower cytotoxicity against human liver (HepG2) and human lymphocyte (CRL-8155) cell lines.58,59 Inhibition of exflaggelation was observed at 0.1 μM, concentrations lower than would have been expected from the enzyme IC50 values, suggesting inhibition of other targets. PfCDPK1 and PfPKG also possess small gatekeeper residues and modulate processes that occur prior to the step modulated by PfCDPK4 and were, therefore, suggested as secondary targets.59 One of the most potent compounds in both the enzyme and the exflaggelation assay was 10 (Figure 6) with an IC50 value of 37 nM against PfCDPK4 and 89% inhibition of exflaggelation at 100 nM. Compound 11 showed better exposures on oral dosing in mice but fell short of achieving longer term blood levels deemed necessary for a transmission blocking agent.58,59
Figure 6.

Aminopyrazole-carboxamide inhibitors of PfCDPK4 and Trifluoperazine 12.
The calmodulin (CaM) antagonist trifluoperazine 12 (TFP) was hypothesized to bind to the CaM-like domain of CDPKs rather than the ATP site and displayed a Ki of 150 μM against PfCDPK4.60 It showed an EC50 of 1.9 μM against P. falciparumin vitro(61) suggesting inhibition of other targets that might include other CDPKs.60 To date, no further progress has been reported based on this compound.
3. AGC
The AGC group includes the cyclic adenosine monophosphate (cAMP)-dependent kinase (PKA), protein kinase B (PKB), and cGMP-dependent kinase (PKG). Evidence has been provided for all three of them to be essential for the asexual blood stages of P. falciparum.
3.1. P. falciparum cGMP-Dependent Protein Kinase (PfPKG)
PfPKG is an essential cGMP-dependent protein kinase that targets at least 69 proteins either directly or indirectly through phosphorylation62 and thereby influences multiple cellular activities in Plasmodia at various stages of the life-cycle. The essentiality of PfPKG for blood stage replication in the human host was demonstrated by chemical genetics using an inhibitor-resistant P. falciparum transgenic line with a T618Q gatekeeper mutation.63 Inhibition of PfPKG led to a block of egress with mature schizonts that released noninvasive merozoites when mechanically ruptured.62,64−67 PKG is also involved in gametogenesis68 and ookinete motility69 in the mosquito stages by modulation of calcium signaling,69 as well as in late liver stage development.70PfPKG also regulates calcium levels and can therefore act on calcium-dependent protein kinases like PfCDPK5.69
A series of potent imidazopyridine inhibitors of PfPKG showed correspondingly potent activity against the asexual blood stage of P. falciparum and blocked gametocyte transmission to Anopheles mosquitoes.63 The compounds were derived from PKG inhibitors of the Apicomplexan parasite, Eimeria.(71) The most potent compounds 14 and 13 had IC50s of 160 and 130 pM, respectively, against the wildtype enzyme (Figure 7), and 2 and 102 nM against the wildtype Pf3D7 strain. Binding to the hydrophobic pocket was demonstrated by markedly lower inhibitory potencies against the previously mentioned gatekeeper mutant T618Q enzyme and against the transgenic strain containing the mutant T618Q. A cocrystal structure obtained from PvPKG and 14 (PDB: 5EZR) showed the fluorophenyl group reaching into the hydrophobic pocket that would be blocked by a larger gatekeeper residue of the mutant. Compound 14 showed good margins over cytotoxicity, moderate in vitro metabolic stability, and high selectivity against a panel of 80 human kinases. The killing dynamics of 14 in a parasite reduction ratio (PRR) assay with P. falciparum showed a 24 h lag phase to kill, consistent with PfPKG inhibitors acting at the egress and invasion stage. Twice a day oral administration of 14 (100 mg/kg) over 4 days in P. falciparum infected NOD-scid gamma IL2Rγnull (NSG) mice led to a reduction of parasitaemia below levels of detection. The intense dosing regimen, due in part to relatively rapid compound clearance, was thought to be required to maintain a high enough plasma concentration of the drug to cover the parasite life-cycle through schizont rupture and invasion. The transmission-blocking potential of 14 was demonstrated in a standard membrane feeding assay (SMFA) with mature PfNF54 gametocytes with an IC50 of 41 nM. The SMFA uses a vial with blood covered by an artificial membrane mimicking skin upon which mosquitoes are allowed to feed. Determination of the number of oocysts in the mosquito’s midgut relative to a control is a measure for transmission of the parasite from the host to the vector. It is thus used to predict the effectiveness of transmission blocking agents in vivo.72
Figure 7.

Inhibitors of cGMP-dependent kinase (PKG).
A previously described 2,3-diaryl-pyrrole series showed similarities to the imidazopyridine analogues. It was also derived from a PKG inhibitor program in Eimeria tenella where several core heterocycles and substitution patterns were investigated and in vivo efficacy was shown.73 One of these compounds, compound 15 (Figure 7), was further evaluated against Plasmodia.74 Compound 15 showed an IC50 of 3.5 nM against recombinant PfPKG, similar to the native strain. However, in vitro potency against the chloroquine-sensitive strain PfNF54 and the chloroquine-resistant strain PfDd2 only showed IC50s of 0.49 and 1.3 μM, respectively. Subsequently, 15 did not clear parasites in a P. berghei mouse infection model, when treated ip with 50 mg/kg twice daily for 8 days.
Compound 15 has also been shown to reduce P. berghei sporozoite infection in a HepG2 cell culture in a dose-dependent manner.75 Treatment with 15 at 2 μM decreased the number of liver stage parasites to below the detection limit. It inhibited host cell invasion with an IC50 of less than 1 μM in vitro. However, sporozoites lacking PbPKG remained sensitive to compound 15, indicating that it affected other targets in addition to PbPKG. In a P. yoelii mouse model, which has higher sporozoite infectivity compared to P. berghei, a single dose of 50 mg/kg given ip before infection reduced the parasite burden in the liver by a factor of 1000. With three 50 mg/kg doses, the first dose given 15 min before infection and the second and third doses 6 and 12 h postinfection, all mice were free of blood stage parasites throughout the 3 week time course of the experiment. Compound 15 was also shown to inhibit gametogenesis in a dose-dependent manner68 and to block egress.67
The imidazopyridazine series described above as PfCDPK1 inhibitors were shown to primarily target PfPKG.40 In lieu of what was observed for compound 15, and if the series were assessed to have favorable pharmacokinetic properties, it would be interesting to determine whether the imidazopyridazines would show liver stage activity and transmission blocking potential.
3.2. P. falciparum cAMP-Dependent Protein Kinase (PfPKA)
PfPKA comprises a regulatory and a catalytic subunit, wherein the former blocks the latter to maintain an inactive state. Allosteric binding of cAMP to the regulatory subunit triggers a conformational change that frees and thereby activates the catalytic subunit.76PfPKA is involved in a number of molecular mechanisms including merozoite egress, motility and red blood cell invasion,77,78 schizogony,78 and the progression from schizont to invasive merozoites.79 During merozoite invasion, PfPKA was found to be involved in microneme secretion of erythrocyte host recognition receptors.80 Furthermore, PfPKA was shown to be a key modulator of the cell cycle in the malaria parasite.81 Gene disruption attempts in P. berghei were unsuccessful, suggesting that the Plasmodium PKA might be essential for parasite survival.82 Overexpression of the PfPKA regulatory subunit led to reduced PKA activity and reduced parasite growth.83
An attempt to develop inhibitors against PfPKA was made starting from the commercially available PKA inhibitor 3-methylisoquinoline-4-carbonitrile, previously used for investigations in mammalian systems. A homology model of the PfPKA catalytic subunit was built based on the human crystal structure, and dynamic molecular modeling was used to identify strong binding ligands.84 Synthesis of these ligands and testing against Pf3D7 and multidrug-resistant PfW2 strain showed mid-micromolar activities. In further studies, it was revealed that these compounds have no effect on PfPKA.85 However, the compounds showed inhibition of parasite cytokinesis and erythrocyte invasion at 10 μM, suggesting that other targets were involved. To date, no potent and specific inhibitors against PfPKA have been reported.
4. CMGC
The most prominent group in the P. falciparum kinome, CMGC, includes the following families: the cyclin dependent kinases (CDKs), the mitogen-activated protein kinases (MAPKs), the glycogen synthase kinase 3 (GSK-3), and the CDK-like kinase (CLK) as well as other close relative protein kinases.16−18,23,86 CDKs have been shown to play an essential role in the parasitic growth and development and therefore have received the most attention as targets to treat malaria.
4.1. P. falciparum MO15-Related Kinase (PfMRK)
The PfMRK protein, a member of the CDK family, was isolated and cloned in 1996 and has been suggested to play an essential role in DNA replication and transcriptional control.20,87 Appropriate activation and deactivation of the enzyme at each stage guarantees cell progression in a sequential manner where a fully activated PfMRK requires association with a cyclin subunit PfCYC-1 and an effector protein PfMAT1.88 The structural differentiation within the ATP binding pocket between the parasitic and mammalian (CDK7) enzymes was exploited to identify specific and structurally diverse PfMRK inhibitors as antiplasmodial chemotypes.
Compound 16 (Figure 8) was identified from a series of quinolinones and found to inhibit PfMRK with an IC50 value of 18 μM.89 A search in the Walter Reed Army Institute of Research (WRAIR) internal database identified 17, an oxindole that inhibited PfMRK (IC50 = 1.4 μM) and showed high selectivity relative to the mammalian CDK1 (IC50 = 29 μM). However, the compound displayed only moderate antiplasmodial activity against the sensitive D6 strain of P. falciparum, which was attributed to poor compound permeability.90
Figure 8.

Quinolinone and oxindole-based inhibitors of PfMRK.
In an attempt to identify novel and structurally diverse PfMRK inhibitors from the WRAIR database, Waters and co-workers developed and validated a three-dimensional quantitative structure–activity relationship (3D-QSAR) pharmacophore model.91 The most active compounds clustered around chalcone92 and sulfonamide-based93 molecules (18 and 19, respectively), which formed the basis of medicinal chemistry programs (Figure 9). The most potent PfMRK inhibitors synthesized in these programs also displayed the highest antiplasmodial activity. However, selectivity was low relative to mammalian protein kinases with sulfonamide 19 showing a similar inhibitory potency against CDK7 (IC50 = 0.4 μM), a mammalian homologue of PfMRK.
Figure 9.

Chalcone and sulfonamide-based and flavonoid inhibitors of PfMRK.
Flavonoid natural products are biosynthetically produced from chalcone intermediates prompting Ayuko Akenga and co-workers to evaluate several antiplasmodial flavonoids extracted from the roots and stem bark of Erythrina sp. for inhibitory activity against the PfMRK. As observed in Figure 9, compound 20 was a highly potent inhibitor, approximately 25-fold higher than 18. Despite this higher inhibitory potency, the in vitro antiplasmodial activity was nearly equal to those of the chalcones, indicating either a mixed mode-of-action for the chalcones or diminished cellular permeability for the flavonoids.94 Overall, a direct correlation between PfMRK inhibitory potency and antiplasmodial activity has been difficult to establish, but essentiality of the kinase for the parasite has been previously demonstrated.20
4.2. P. falciparum Glycogen Synthase Kinase 3 (PfGSK-3)
The PfGSK-3 enzyme was identified and cloned in 2004 and is one of three P. falciparum protein kinases related to the mammalian GSK-3.95 The precise biological function of PfGSK-3 has not yet been determined, though it has been demonstrated to be essential for the completion of the parasite asexual blood stage.20,96 As GSK-3 is highly conserved in mammals, sufficient differentiation relative to PfGSK-3 suggested that selectivity could be achieved with inhibitors. A HTS of a collection of structurally diverse compounds against recombinant PfGSK-3 yielded inhibitors that shared two related heterocyclic scaffolds exemplified by compounds 21 and 22 (Figure 10). Screening of an additional nearest neighbor library of 427 compounds afforded 23 with an IC50 of 1.6 μM. From these three hits, analogues were synthesized to explore the SAR and increase inhibitory potency against PfGSK-3 as well as selectivity over the mammalian GSK-3 orthologue. Importantly, antiplasmodial activity was seen for 24 (IC50 = 5.5 μM versus PfNF54), which extended to other Plasmodium strains. When tested against two mammalian kinase panels (77 and 402 kinases from Dundee and DiscoverX, respectively), compound 24 inhibited only a few kinases thus demonstrating broad selectivity. Further optimization of this series is required to demonstrate in vivo activity.96,97
Figure 10.

PfGSK-3 inhibitors.
5. P. falciparum Never-In-Mitosis Gene A (PfNIMA/NEK)
NIMA (never in mitosis, gene A)-related kinases or NEKs are a family of serine/threonine kinases that are generally conserved across eukaryotes.98 There are four representatives of the family in Plasmodia characterized by a shared protein kinase domain normally located at the N-terminus.99 The NEKs play an important role in the life-cycle of the malaria parasite, specifically in mitosis and meiosis where they are associated with centrosomes, spindle poles, and other components of cell machinery involved during division.98,100PfNEK-1 and PfNEK-2–4 are likely essential for the asexual and the sexual cycles, respectively.20 Though the literature covers several programs targeting human NEK kinases, particularly NEK-2, there have not been reports of drug discovery efforts targeting any PfNEK.
Two marine natural product compounds 25 and 26 demonstrated inhibition of PfNEK-1 and activity against P. falciparum (Figure 11); however, the antiplasmodial activity was not confirmed to be due to PfNEK-1 inhibition.101−103 Compound 26 did show activity in a P. berghei NK65 mouse infection model (40% inhibition of parasitaemia at 5 mg/kg over 4 days of dosing), thereby achieving a measure of in vivo validation for the scaffold.103 Higher doses were not tolerated by mice in the model, and better efficacy was not demonstrated. More recently, Mitcheson et al. identified a tool compound that inhibited a genetically modified isozyme of PfNEK-2 (but not the wild-type isozyme) to interrogate enzyme function.104 A recent comprehensive review of chemical starting points for human NEK kinase inhibition may offer an opportunity to cross-screen and identify PfNEK inhibitors.105 As the landscape now stands with little structural information, biological validation, and few chemical starting points, considerable work around Plasmodium NEK kinases is needed to expand upon the biological understanding of PfNEK and to develop drug discovery programs.
Figure 11.

Chemical structures, enzyme, and whole cell activities of PfNEK-1 inhibitors.
6. Phe (F)–Ile (I)–Lys (K)–Lys (K) (FIKK)
The FIKK kinase family, named after the 4-amino acid chain sequence Phe–Ile–Lys–Lys, is specifically found in the Apicomplexan phylum and is mostly expressed in the blood stage of the parasite life-cycle.106 Though most malaria species contain a single FIKK kinase, P. falciparum contains 20 homologues, 18 of them predicted to be functional kinases making them interesting drug targets. Furthermore, their catalytic site contains a small gatekeeper residue, which allows for selectivity over mammalian kinases.107 A few PfFIKK homologues such as FIKK4.2,108 FIKK7.1, and FIKK12109 are implicated in red blood cell remodeling and altering erythrocytic membrane rigidity. Even though none of the three aforementioned kinases was found to be essential for parasite proliferation, little is known about the essentiality and the specific role of the other members of the FIKK family. Compound 27 (Figure 12, emodin) has shown in vitro activity against P. falciparum with an IC50 of 13 μM,110 which was subsequently attributed to inhibition of PfFIKK8 (2 μM), the most similar homologue of Plasmodium species.111 Inhibition of PvFIKK (1.9 μM) has also been demonstrated.112 Otherwise, there has been a drug discovery project targeting Cryptosporidium FIKK, wherein the authors identified a naphthyridine-based low nanomolar inhibitor that, albeit, failed to show antiparasitic activity (Figure 12).113 While several factors make the FIKK family attractive as drug targets in Plasmodia, considerably more work still needs to be conducted to better delineate the roles that FIKKs play within the parasite.
Figure 12.

Chemical structure of emodin and a CpFIKK inhibitor.
7. Phosphoinositide Lipid Kinases (PIKs)
Phosphoinositide lipid kinases (PIKs) generate precise phosphorylated variants of phosphatidylinositols that are crucial for a diverse array of cellular functions including secondary messenger signaling, cellular membrane remodeling, and vesicular trafficking.60,114 The two most widely studied classes of PIKs in Plasmodium species are phosphoinositide 3-kinase (PI3K) and phosphatidylinositol 4-kinase (PI4K), both demonstrated to be essential for the survival of the parasite.115,116 PI3K inhibitors have seen relatively little exploration thus far, while PI4K inhibitors have generated considerable interest within the context of the advancement of antimalarial drug candidates.
7.1. Phosphoinositide 3-Kinases (PI3K)
Though only one PI3K enzyme is encoded in the P. falciparum genome and its essentiality has been demonstrated, there have not been literature reports on medicinal chemistry programs targeting this enzyme. Rather, tool compounds have been described including the mammalian pan-PI3K inhibitors 29 (wortmannin) and 30 (LY294002) (Figure 13). There are no reported IC50 values for wortmannin in standard P. falciparum proliferation assays attributed to its chemical instability, whereas 30 showed an IC50 of 26 μM against the parasite.117
Figure 13.

Chemical structures of compounds known to inhibit PfPI3K.
PI3K has recently been implicated as one of the targets of dihydroartemisinin (DHA), 31, as evidenced by mutations to the regulatory ubiquitination protein PfKelch13118 that were associated with clinical resistance.119 The PfKelch13 mutations led to increased levels of PI3K by abrogating its role in ubiquitination and deactivation of the kinase. PI3K was shown to be essential for parasite growth, and levels of the lipid product phosphatidylinositol-3-phosphate (PI3P) in parasite tissue cultures dropped on treatment with DHA. A quantitative inhibition assay with purified PfPI3K has been derived wherein DHA demonstrated potent (low nanomolar) inhibition. A PfPI3K homology model was also derived from the structure of the class III PI3-kinase from Drosophila (PDB code: 2X6F) and the active site of the human class III PI3K (PDB code 3IHY).119 There is, therefore, considerable interest in developing other scaffolds as antimalarials operating by PI3K inhibition.
7.2. Phosphatidylinositol 4-Kinases (PI4K)
Significant progress in the discovery of compounds inhibiting PI4K in Plasmodium species with two key classes being reported in the literature. A group led by the Genomics Institute of the Novartis Research Foundation (GNF) and the Novartis Institute for Tropical Diseases (NITD) identified antiplasmodial activity for a series of imidazopyridines/pyrazines/pyridazines.115,120 Compound 32 (Figure 14) was identified by phenotypic screening against the P. falciparum blood stage parasite. The compound had moderate P. falciparum activity but was inactive against P. yoelii and P. cynomolgi. Initial SAR around the 6- and the 3-positions identified compound 33 (KAI407), which significantly improved P. falciparum activity and brought in both P. yoelii and P. cynomolgi activity. Compound 33 suffered from poor physicochemical properties and thus modification of the core to an imidazopyrazine reduced lipophilicity leading to compound 34 (KDU691). Compound 34 showed improved physicochemical properties and in vitro activity across Plasmodium species, particularly P. cynomolgi.
Figure 14.

Imidazopyridine/pyrazine compounds as PfPI4K inhibitors.
That the imidazopyridines/imidazopyrazines were active against clinically resistant isolates of all classes of antimalarials suggested a novel mode-of-action. The compounds were active in vitro against liver stage development of the rodent parasite, P. yoelii, and against in vitro cultured liver-resident hypnozoites of P. cynomolgi, indicating potential as a radical cure for P. vivax infection and associated malaria relapse. Activity was also demonstrated against P. vivax field isolates. Compounds showed potential for transmission blocking activity in a gametocyte enriched in vitro assay and in a SMFA. Hence, activity was demonstrated across multiple stages of the parasite life-cycle. PI4K was identified as the target of the series by selection for resistant mutants to 33 and 35 (KAI715) with alterations seen in the single gene, pfpi4k. Full length PvPI4K was expressed, and protein was isolated for assay development wherein 34 exhibited an IC50 of 1.5 nM.
A PfPI4K homology model was generated through which it was suggested that the imidazopyridines are accommodated in the ATP-binding pocket (Figure 15). Compounds 33 and 34 displayed remarkable selectivity over human lipid kinases as well as a selection of human protein kinases. Compound 34 displayed favorable oral pharmacokinetics at 20 mg/kg, prevented colonization of mice with P. berghei with prophylactic dosing, and cured an existing infection in a dose-dependent manner.
Figure 15.
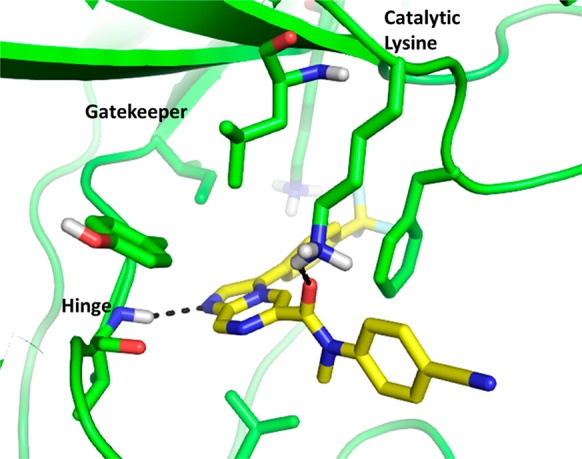
Homology model of PfPI4K with compound 33 (KAI407), illustrating a key interaction in the ATP binding site (Eyermann, unpublished).
A second class of PI4K inhibitor, the aminopyridine/pyrazine class of compounds (Figure 16), was identified at the University of Cape Town (UCT) Drug Discovery and Development Centre (H3D) in collaboration with the Medicines for Malaria Venture (MMV). The initial hits originated from a HTS campaign on a commercial SoftFocus kinase library conducted at the Eskitis Institute, Griffiths University in Brisbane (Australia).121
Figure 16.

Aminopyridine/pyrazine compounds as PfPI4K inhibitors.
The screen identified compound 36, which has a para-sulfonyl phenyl substituent on the aminopyridine 5-position and showed promising activity against PfNF54 (Figure 16). Replacement of the metabolically labile 3-methoxy-4-hydroxy substituent led to 37 (MMV048), a compound with high activity against drug sensitive and drug resistant Plasmodium strains and favorable ADMET and PK properties. Spontaneous resistant mutants to 37 were generated in P. falciparum culture, and whole genome sequencing implicated PI4K as the target. Inhibition of PI4K was also supported via pull-down experiments and confirmed against the PvPI4K with an IC50 of 3.4 nM.25 The compound showed impressive selectivity over human lipid kinases, and the series showed a close correlation between enzyme inhibitory potency and whole cell activity. The speed of kill of the compound was relatively slow as assessed in the in vitro PRR and stage specificity assays but moderate in vivo. Transmission-blocking potential was also evaluated wherein 37 showed antigametocyte activity in vitro and in vivo in a SFMA, supporting potential utility for blocking parasite transmission to the mosquito. The central pyridine core of 37 was replaced with a pyrazine (compound 38) leading to improved in vitro antiplasmodial activity.122 Replacing the sulfone of 38 with a piperazine amide led to 39 (UCT943), a compound with improved solubility as well as higher antiplasmodial activity.123 All four compounds proved highly efficacious in in vivo models of Plasmodium infections, e.g., 37 showing an ED90 of 0.8 mg/kg in a P. berghei mouse model of infection.
Compound 37 was profiled extensively in vitro and in vivo, showing activity across a panel of drug-resistant clinical isolates and therefore a low risk for pre-existing cross-resistance. In vivo, the compound was active in the P. berghei model as described above but was also evaluated in a humanized NSG mouse model that supports infection by P. falciparum, where it similarly exhibited potent activity with an ED90 of 0.57 mg/kg. The compound showed prophylactic activity preventing infection in a P. cynomolgi infected macaque model (2 mg/kg prior to infection). The PK of compound 37 in mice, rats, dogs, and monkeys was characterized by low plasma clearance, moderate to high volume of distribution, and a moderate to long half-life. From this data, a single dose of 80 to 100 mg was estimated to be efficacious in humans. Compound 37 has progressed through preclinical development and Phase 1 clinical trials. During Phase 1, the compound was also evaluated in a volunteer controlled human infection model (CHIM) wherein favorable activity led to its progression to Phase 2a clinical trials.124 Hence, 37 may become the first directed Plasmodium kinase inhibitor to have registered a clinical proof-of-concept.
PI4K inhibitors with antiplasmodial activity have been derived by phenotypic screening and subsequent identification of the target. As the Plasmodium PI4K has emerged as an important target for drug design, it follows that rational inhibitor design would be pursued. Indeed, a PfPI4K homology model and compound 37 were used to create a pharmacophore-based model to perform a computational screen, identifying virtual hits as starting points that were hypothesized to inhibit PfPI4K.125
8. Summary
One of the most persuasive reasons to target Plasmodium kinases for the treatment of malaria is that selectivity relative to problematic human kinases can be achieved and, therefore, less likely to cause kinase-mediated safety issues in humans. Additionally, it can be anticipated that host-directed antimalarial therapies will be developed including those that inhibit human protein kinases.126 For example, inhibition of erythrocyte sik tyrosine kinase by the oncology agent imatinib was perhaps responsible for preventing P. falciparum egress,127 and silencing of Protein Kinase C zeta (PKCζ) led to marked reduction of P. berghei infection in human Huh7 hepatoma cells.128 There would be the added value with such mechanisms-of-action toward circumventing the development of drug resistance.129 Hence, the playbook that has led to upward of 32 small molecule kinase inhibiting therapeutics approved by the FDA can be applied in the parasitology arena.15,130 Knowledge on the essentiality, stage specificity, and mechanism of kinase function in Plasmodia can help triage targets that would be worthwhile pursuing for medicinal chemistry optimization programs. While blood-stage activity is a requirement to treat malaria, transmission-blocking potential is thought to be important toward malaria elimination in the community, and essential targets showing dual-activity can be considered of high value. Comparative gene sequence analyses and homology modeling/structure determination can be used to understand inhibitor binding to targets and mitigate binding to unwanted (most often human kinase) targets. Cross-screening against human kinases has become routine in Plasmodium kinase programs just as cross-screening across human kinase panels are carried out to achieve selectivity for inhibitors directed at a specific human kinase. Conversely, collections of human kinase inhibitors are valuable to screen against Plasmodium kinases toward obtaining hits that can eventually be optimized for higher Plasmodium and lower human kinase potencies. Alternatively, considerable success has been achieved by screening libraries of human kinase inhibitors phenotypically against Plasmodium followed by subsequent determination of their mode-of-action.
In fact, it is this latter approach that led to 37 (MMV048), the only Plasmodium kinase inhibiting compound to have achieved a measure of clinical validation. However, expectations are high that other Plasmodium targets already known will yield new classes of antimalarial drugs both in terms of chemical scaffold and mode-of-action. At least eight kinase targets delineated herein have led to phenotypic levels of validation, albeit sometimes it might not be clear that the Plasmodium kill is due to the originally targeted kinase. Notwithstanding the inability to always directly link kinase inhibition to the mode-of-action responsible for parasite kill, the work around protein and lipid kinases to date point to an impressive synergy created between fundamental research into the Plasmodium kinome and applied drug discovery research. A number of kinases that are likely to be essential have not been targeted in medicinal chemistry programs yet and certainly offer potential for the development of future antimalarials.20
Acknowledgments
The Novartis Research Foundation, Bill and Melinda Gates Foundation (grant # OPP1066878), Medicines for Malaria Venture (MMV), Technology Innovation Agency (TIA), University of Cape Town, South African Medical Research Council, and South African Department of Science and Technology/National Research Foundation Research Chairs Initiative are gratefully acknowledged for support.
Glossary
ABBREVIATIONS USED
- Pf
P. falciparum, Plasmodium falciparum
- WHO
World Health Organization
- ACT
artemisinin-based combination therapy
- FDA
Food and Drug Administration
- ATP
adenosine triphosphate
- POC
proof-of-concept
- PD
pharmacodynamic
- PK
pharmacokinetic
- PI4K
phosphatidylinositol 4-kinase
- CamK
calmodulin-dependent kinases
- CDPK
calcium-dependent protein kinases
- SNF1
sucrose nonfermenting
- AMPK
adenosine monophosphate-activated kinase
- PKG
cyclic guanosine monophosphate (cGMP)-dependent protein kinase
- HTS
high throughput screening
- Tg
Toxoplasma gondii
- ADMET
absorption, distribution, metabolism, excretion, and toxicology
- PfHSP90
P. falciparum heat shock protein 90
- Bid
bis in die
- SAPK2α
stress-activated protein kinase-2 alpha
- FGFR1
fibroblast growth factor receptor 1
- TrkA
tropomyosin receptor kinase A
- RSK1
ribosomal S6 kinase 1
- Cp
Cryptosporidium parvum
- Ip
intraperitoneal
- AC
aminopyrazole-carboxamide
- hERG
human Ether-a-go-go related gene
- CaM
calmodulin
- TPF
trifluoperazine
- cAMP
cyclic adenosine monophosphate
- PKA
protein kinase A
- PKB
protein kinase B
- PRR
parasite reduction ratio
- NSG
NOD-scid gamma
- SMFA
standard membrane feeding assay
- CDK
cyclin dependent kinase
- MAPKs
mitogen-activated protein kinase
- GSK-3
glycogen-synthase kinase 3
- CLK
CDK-like kinase
- NIMA
never in mitosis, gene A
- FIKK
Phe (F)–Ile (I)–Lys (K)–Lys (K)
- PIK
phosphoinositide lipid kinase
- PI3K
phosphoinositide 3-kinase
- PI4K
phosphatidylinositol 4-kinase
- DHA
dihydroartemisinin
- GNF
Genomics Institute of the Novartis Research Foundation
- NITD
Novartis Institute for Tropical Diseases
- UCT
University of Cape Town
- H3D
Drug Discovery and Development Centre
Biographies
Diego Gonzalez Cabrera obtained his M.Sc. (2004) and Ph.D. (2008) at The University of Edinburgh under the supervision of Prof. David A. Leigh, working on the preparation and the development of novel hydrogen-bond assembled [2]Rotaxanes. He took up a postdoctoral position with Prof. Kelly Chibale focusing on the development of new affordable therapeutic agents for the treatment of uncomplicated Plasmodium falciparum, before taking up his current position as a Senior Investigator at H3D in 2015.
André Horatscheck received his Dr. rer. nat. in Chemistry from the Freie Universität Berlin (Germany) in 2011 under the supervision of Prof Jörg Rademann. After his postdoctoral work in the field of Medicinal Chemistry at the Leibniz-Institut für Molekulare Pharmakologie Berlin, he joined H3D in 2016 where he is a Senior Investigator working on malaria related drug discovery projects.
Colin Wilson received his Ph.D. from the University of KwaZulu-Natal in South Africa under the supervision of Prof. Orde Munro where he worked on the discovery of novel topoisomerase inhibitors. He then held a National Research Foundation Scare Skills postdoctoral fellowship at the University of Cape Town under the supervision of Prof. Kelly Chibale where he worked on a number projects focused on tuberculosis drug discovery. He is now a Senior Investigator at H3D where he leads malaria focused discovery projects.
Greg Basarab has an appointment as Principal Research Officer and Associate Director at H3D. He leads research directed toward the eradication of resistant infectious diseases including malaria and tuberculosis. Previously, he was an Associate Director at AstraZeneca where he led multidisciplinary teams for the design of novel mode-of-action antibacterials and delivered three drug candidates that progressed into human clinical trials including one currently in Phase 3. Previous to that, he worked at DuPont leading projects in three departments: Central Research & Development, Biochemicals and Agricultural Products in the antifungal arena and in automated chemical synthesis. He received a B.S. in Chemistry from Penn State University and a Ph.D. in Chemistry from MIT.
Joe Eyermann is Head of Computer-Aided Drug Design at H3D. He has been especially involved in drug discovery programs against infectious diseases including HIV, Gram-negative bacteria, and, most recently, tuberculosis and malaria. Previous to joining H3D, he worked at DuPont, DuPont Merck, ARIAD, and AstraZeneca. He received a B.S. in Chemistry from Marietta College and a Ph.D. in Chemistry from Miami University of Ohio.
Kelly Chibale obtained his Ph.D. in Synthetic Organic Chemistry from the University of Cambridge with Stuart Warren (1989–1992). This was followed by postdoctoral stints at the University of Liverpool as a British Ramsay Fellow with Nick Greeves (1992–1994) and at the Scripps Research Institute as a Wellcome Trust International Prize Research Fellow with K. C. Nicolaou (1994–1996). His research is in the field of global health drug discovery. He is the Director of the University of Cape Town (UCT) Drug Discovery and Development Centre (H3D) and the South African Medical Research Council Drug Discovery and Development Research Unit at UCT.
Author Contributions
∥ These authors contributed equally.
The authors declare no competing financial interest.
References
- World Health Organization (WHO) . World Malaria Report. http://www.who.int/malaria/publications/world-malaria-report-2017/report/en/ (accessed Feb 19, 2018).
- Blasco B.; Leroy D.; Fidock D. A. Antimalarial Drug Resistance: Linking Plasmodium Falciparum Parasite Biology to the Clinic. Nat. Med. 2017, 23 (8), 917–928. 10.1038/nm.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P. J.; Lindegardh N.; Socheat D.; White N. J. Artemisinin Resistance in Plasmodium Falciparum Malaria. N. Engl. J. Med. 2009, 361 (5), 455–467. 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F. J. Antimalarial Drug Resistance: New Treatments Options for Plasmodium. Drug Discovery Today: Technol. 2014, 11 (1), 81–88. 10.1016/j.ddtec.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Phillips M. A.; Burrows J. N.; Manyando C.; van Huijsduijnen R. H.; Van Voorhis W. C.; Wells T. N. C. Malaria. Nat. Rev. Dis. Prim. 2017, 3, 17050. 10.1038/nrdp.2017.50. [DOI] [PubMed] [Google Scholar]
- Campo B.; Vandal O.; Wesche D. L.; Burrows J. N. Killing the Hypnozoite – Drug Discovery Approaches to Prevent Relapse in Plasmodium vivax. Pathog. Global Health 2015, 109 (3), 107–122. 10.1179/2047773215Y.0000000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reader J.; Botha M.; Theron A.; Lauterbach S. B.; Rossouw C.; Engelbrecht D.; Wepener M.; Smit A.; Leroy D.; Mancama D.; Coetzer T. L.; Birkholtz L. M. Nowhere to Hide: Interrogating Different Metabolic Parameters of Plasmodium Falciparum Gametocytes in a Transmission Blocking Drug Discovery Pipeline towards Malaria Elimination. Malar. J. 2015, 14 (1), 1–17. 10.1186/s12936-015-0718-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aly A. S. I.; Vaughan A. M.; Kappe S. H. I. Malaria Parasite Development in the Mosquito and Infection of the Mammalian Host. Annu. Rev. Microbiol. 2009, 63 (1), 195–221. 10.1146/annurev.micro.091208.073403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho T. G.; Morahan B.; John von Freyend S.; Boeuf P.; Grau G.; Garcia-Bustos J.; Doerig C. The Ins and Outs of Phosphosignalling in Plasmodium: Parasite Regulation and Host Cell Manipulation. Mol. Biochem. Parasitol. 2016, 208 (1), 2–15. 10.1016/j.molbiopara.2016.05.006. [DOI] [PubMed] [Google Scholar]
- Hubbard S. R.; Till J. H. Protein Tyrosine Kinase Structure and Function. Annu. Rev. Biochem. 2000, 69 (1), 373–398. 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- Rauch J.; Volinsky N.; Romano D.; Kolch W. The Secret Life of Kinases: Functions beyond Catalysis. Cell Commun. Signaling 2011, 9, 23. 10.1186/1478-811X-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.; Shen A.; Ding J.; Geng M. Molecularly Targeted Cancer Therapy: Some Lessons from the Past Decade. Trends Pharmacol. Sci. 2014, 35 (1), 41–50. 10.1016/j.tips.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Clark J. D.; Flanagan M. E.; Telliez J. B. Discovery and Development of Janus Kinase (JAK) Inhibitors for Inflammatory Diseases. J. Med. Chem. 2014, 57 (12), 5023–5038. 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]
- Kikuchi R.; Nakamura K.; Maclauchlan S.; Ngo D. T.; Shimizu I.; Fuster J. J.; Katanasaka Y.; Yoshida S.; Qiu Y.; Yamaguchi T. P.; Matsushita T.; Murohara T.; Gokce N.; Bates D. O.; Hamburg N. M.; Walsh K. An Anti-Angiogenic Isoform of VEGF-A Contributes to Impaired Vascularization in Peripheral Artery Disease. Nat. Med. 2014, 20 (12), 1464–1471. 10.1038/nm.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P.; Nielsen T. E.; Clausen M. H. FDA-Approved Small-Molecule Kinase Inhibitors. Trends Pharmacol. Sci. 2015, 36 (7), 422–439. 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Doerig C.; Billker O.; Haystead T.; Sharma P.; Tobin A. B.; Waters N. C. Protein Kinases of Malaria Parasites: An Update. Trends Parasitol. 2008, 24 (12), 570–577. 10.1016/j.pt.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Kappes B.; Doerig C.; Graeser R. An Overview of Plasmodium Protein Kinases. Parasitol. Today 1999, 15 (11), 449–454. 10.1016/S0169-4758(99)01527-6. [DOI] [PubMed] [Google Scholar]
- Talevich E.; Tobin A. B.; Kannan N.; Doerig C. An Evolutionary Perspective on the Kinome of Malaria Parasites. Philos. Trans. R. Soc., B 2012, 367 (1602), 2607–2618. 10.1098/rstb.2012.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Stecher G.; Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33 (7), 1870–1874. 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solyakov L.; Halbert J.; Alam M. M.; Semblat J.-P.; Dorin-Semblat D.; Reininger L.; Bottrill A. R.; Mistry S.; Abdi A.; Fennell C.; Holland Z.; Demarta C.; Bouza Y.; Sicard A.; Nivez M.-P.; Eschenlauer S.; Lama T.; Thomas D. C.; Sharma P.; Agarwal S.; Kern S.; Pradel G.; Graciotti M.; Tobin A. B.; Doerig C. Global Kinomic and Phospho-Proteomic Analyses of the Human Malaria Parasite Plasmodium Falciparum. Nat. Commun. 2011, 2, 565. 10.1038/ncomms1558. [DOI] [PubMed] [Google Scholar]
- Bracchi-Ricard V.; Barik S.; Delvecchio C.; Doerig C.; Chakrabarti R.; Chakrabarti D. PfPK6, a Novel Cyclin-Dependent Kinase/Mitogen-Activated Protein Kinase-Related Protein Kinase from Plasmodium Falciparum. Biochem. J. 2000, 347 (1), 255–263. 10.1042/0264-6021:3470255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorin D.; Semblat J. P.; Poullet P.; Alano P.; Goldring J. P. D.; Whittle C.; Patterson S.; Chakrabarti D.; Doerig C. PfPK7, an Atypical MEK-Related Protein Kinase, Reflects the Absence of Classical Three-Component MAPK Pathways in the Human Malaria Parasite Plasmodium Falciparum. Mol. Microbiol. 2005, 55 (1), 184–196. 10.1111/j.1365-2958.2004.04393.x. [DOI] [PubMed] [Google Scholar]
- Ward P.; Equinet L.; Packer J.; Doerig C. Protein Kinases of the Human Malaria Parasite Plasmodium Falciparum: The Kinome of a Divergent Eukaryote. BMC Genomics 2004, 5, 79. 10.1186/1471-2164-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari R.; Straschil U.; Bateman A.; Böhme U.; Cherevach I.; Gong P.; Pain A.; Billker O. The Systematic Functional Analysis of Plasmodium Protein Kinases Identifies Essential Regulators of Mosquito Transmission. Cell Host Microbe 2010, 8 (4), 377–387. 10.1016/j.chom.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet T.; Le Manach C.; Cabrera D. G.; Younis Y.; Henrich P. P.; Abraham T. S.; Lee M. C. S.; Basak R.; Ghidelli-Disse S.; Lafuente-Monasterio M. J.; Bantscheff M.; Ruecker A.; Blagborough A. M.; Zakutansky S. E.; Zeeman A.-M.; White K. L.; Shackleford D. M.; Mannila J.; Morizzi J.; Scheurer C.; Angulo-Barturen I.; Martínez M. S.; Ferrer S.; Sanz L. M.; Gamo F. J.; Reader J.; Botha M.; Dechering K. J.; Sauerwein R. W.; Tungtaeng A.; Vanachayangkul P.; Lim C. S.; Burrows J.; Witty M. J.; Marsh K. C.; Bodenreider C.; Rochford R.; Solapure S. M.; Jiménez-Díaz M. B.; Wittlin S.; Charman S. A.; Donini C.; Campo B.; Birkholtz L.-M.; Hanson K. K.; Drewes G.; Kocken C. H. M.; Delves M. J.; Leroy D.; Fidock D. A.; Waterson D.; Street L. J.; Chibale K. Antimalarial Efficacy of MMV390048, an Inhibitor of Plasmodium Phosphatidylinositol 4-Kinase. Sci. Transl. Med. 2017, 9 (387), eaad9735. 10.1126/scitranslmed.aad9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J. F.; Harmon A. Plants, Symbiosis and Parasites: A Calcium Signalling Connection. Nat. Rev. Mol. Cell Biol. 2005, 6 (7), 555–566. 10.1038/nrm1679. [DOI] [PubMed] [Google Scholar]
- Wernimont A. K.; Amani M.; Qiu W.; Pizarro J. C.; Artz J. D.; Lin Y. H.; Lew J.; Hutchinson A.; Hui R. Structures of Parasitic CDPK Domains Point to a Common Mechanism of Activation. Proteins: Struct., Funct., Genet. 2011, 79 (3), 803–820. 10.1002/prot.22919. [DOI] [PubMed] [Google Scholar]
- Bansal A.; Molina-Cruz A.; Brzostowski J.; Mu J. Plasmodium Falciparum Calcium- Dependent Protein Kinase 2 Is Critical for Male Gametocyte Exflagellation But. mBio 2017, 8 (5), e01656-17. 10.1128/mBio.01656-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. L.; Baker D. A.; Cox L. S. Sexual Stage-Specific Expression of a Third Calcium-Dependent Protein Kinase from Plasmodium Falciparum. Biochim. Biophys. Acta, Gene Struct. Expression 2000, 1491 (1–3), 341–349. 10.1016/S0167-4781(00)00032-4. [DOI] [PubMed] [Google Scholar]
- Siden-Kiamos I.; Ecker A.; Nybäck S.; Louis C.; Sinden R. E.; Billker O. Plasmodium Berghei Calcium-Dependent Protein Kinase 3 Is Required for Ookinete Gliding Motility and Mosquito Midgut Invasion. Mol. Microbiol. 2006, 60 (6), 1355–1363. 10.1111/j.1365-2958.2006.05189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Absalon S.; Blomqvist K.; Rudlaff R. M.; DeLano T. J.; Pollastri M. P.; Dvorin J. D. Calcium-Dependent Protein Kinase 5 Is Required for Release of Egress-Specific Organelles in Plasmodium Falciparum. mBio 2018, 9 (1), e00130-18. 10.1128/mBio.00130-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccotto F.; Ardini E.; Casale E.; Angiolini M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Conformation. J. Med. Chem. 2010, 53 (7), 2681–2694. 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
- Bishop A. C.; Ubersax J. A.; Petsch D. T.; Matheos D. P.; Grayk N. S.; Blethrow J.; Shimizu E.; Tsien J. Z.; Schultzk P. G.; Rose M. D.; Wood J. L.; Morgan D. O.; Shokat K. M. A Chemical Switch for Inhibitor- Sensitive Alleles of Any Protein Kinase. Nature 2000, 407 (6802), 395–401. 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Doerig C.; Meijer L.; Mottram J. C. Protein Kinases as Drug Targets in Parasitic Protozoa. Trends Parasitol. 2002, 18 (8), 366–371. 10.1016/S1471-4922(02)02321-8. [DOI] [PubMed] [Google Scholar]
- Van Voorhis W. C.; Doggett J. S.; Parsons M.; Hulverson M. A.; Choi R.; Arnold S. L. M.; Riggs M. W.; Hemphill A.; Howe D. K.; Mealey R. H.; Lau A. O. T.; Merritt E. A.; Maly D. J.; Fan E.; Ojo K. K. Extended-Spectrum Antiprotozoal Bumped Kinase Inhibitors: A Review. Exp. Parasitol. 2017, 180, 71–83. 10.1016/j.exppara.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Kumar M.; Ekka R.; Dvorin J. D.; Paul A. S.; Madugundu A. K.; Gilberger T.; Gowda H.; Duraisingh M. T.; Keshava Prasad T. S.; Sharma P. PfCDPK1 Mediated Signaling in Erythrocytic Stages of Plasmodium Falciparum. Nat. Commun. 2017, 8 (1), 63. 10.1038/s41467-017-00053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal A.; Singh S.; More K. R.; Hans D.; Nangalia K.; Yogavel M.; Sharma A.; Chitnis C. E. Characterization of Plasmodium Falciparum Calcium-Dependent Protein Kinase 1 (PfCDPK1) and Its Role in Microneme Secretion during Erythrocyte Invasion. J. Biol. Chem. 2013, 288 (3), 1590–1602. 10.1074/jbc.M112.411934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal A.; Molina-Cruz A.; Brzostowski J.; Liu P.; Luo Y.; Gunalan K.; Li Y.; Ribeiro J. M. C.; Miller L. H. PfCDPK1 Is Critical for Malaria Parasite Gametogenesis and Mosquito Infection. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (4), 774–779. 10.1073/pnas.1715443115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N.; Sakata T.; Breton G.; Le Roch K. G.; Nagle A.; Andersen C.; Bursulaya B.; Henson K.; Johnson J.; Kumar K. A.; Felix M.; Mason D.; McNamara C.; Plouffe D.; Vandana Ramachandran M. S.; Tuntland T.; Zhou Y.; Peters E. C.; Chatterjee A.; Schultz P. G.; Ward G. E.; Gray N.; Harper J.; Winzeler E. A. Gene Expression Signatures and Small-Molecule Compounds Link a Protein Kinase to Plasmodium Falciparum Motility. Nat. Chem. Biol. 2008, 4 (6), 347–356. 10.1038/nchembio.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J. L.; Moon R. W.; Whalley D.; Bowyer P. W.; Wallace C.; Rochani A.; Nageshan R. K.; Howell S. A.; Grainger M.; Jones H. M.; Ansell K. H.; Chapman T. M.; Taylor D. L.; Osborne S. A.; Baker D. A.; Tatu U.; Holder A. A. Imidazopyridazine Inhibitors of Plasmodium falciparum Calcium-Dependent Protein Kinase 1 Also Target Cyclic GMP-Dependent Protein Kinase and Heat Shock Protein 90 To Kill the Parasite at Different Stages of Intracellular Development. Antimicrob. Agents Chemother. 2016, 60 (3), 1464–1475. 10.1128/AAC.01748-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal A.; Ojo K. K.; Mu J.; Maly D. J.; Van Voorhis W. C.; Miller L. H. Reduced Activity of Mutant Calcium-Dependent Protein Kinase 1 Is Compensated in Plasmodium Falciparum through the Action of Protein Kinase G. mBio 2016, 7 (6), e02011-16. 10.1128/mBio.02011-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman T. M.; Osborne S. A.; Bouloc N.; Large J. M.; Wallace C.; Birchall K.; Ansell K. H.; Jones H. M.; Taylor D.; Clough B.; Green J. L.; Holder A. A. Substituted Imidazopyridazines Are Potent and Selective Inhibitors of Plasmodium Falciparum Calcium-Dependent Protein Kinase 1 (PfCDPK1). Bioorg. Med. Chem. Lett. 2013, 23 (10), 3064–3069. 10.1016/j.bmcl.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman T. M.; Osborne S. A.; Wallace C.; Birchall K.; Bouloc N.; Jones H. M.; Ansell K. H.; Taylor D. L.; Clough B.; Green J. L.; Holder A. A. Optimisation of an Imidazopyridazine Series of Inhibitors of Plasmodium Falciparum Calcium-Dependent Protein Kinase 1 (PfCDPK1). J. Med. Chem. 2014, 1, 3570–3587. 10.1021/jm500342d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Large J. M.; Osborne S. A.; Smiljanic-Hurley E.; Ansell K. H.; Jones H. M.; Taylor D. L.; Clough B.; Green J. L.; Holder A. A. Imidazopyridazines as Potent Inhibitors of Plasmodium Falciparum Calcium-Dependent Protein Kinase 1 (PfCDPK1): Preparation and Evaluation of Pyrazole Linked Analogues. Bioorg. Med. Chem. Lett. 2013, 23 (21), 6019–6024. 10.1016/j.bmcl.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell K. H.; Jones H. M.; Whalley D.; Hearn A.; Taylor D. L.; Patin E. C.; Chapman T. M.; Osborne S. A.; Wallace C.; Birchall K.; Large J.; Bouloc N.; Smiljanic-Hurley E.; Clough B.; Moon R. W.; Green J. L.; Holder A. A.; Anseil K. H.; Jones H. M.; Whalley D.; Hearn A.; Taylor D. L.; Patin E. C.; Chapman T. M.; Osborne S. A.; Wallace C.; Birchall K.; Large J.; Bouloc N.; Smiljanic-Hurley E.; Clough B.; Moon R. W.; Green J. L.; Holder A. A. Biochemical and Antiparasitic Properties of Inhibitors of the Plasmodium Falciparum Calcium-Dependent Protein Kinase PfCDPK1. Antimicrob. Agents Chemother. 2014, 58 (10), 6032–6043. 10.1128/AAC.02959-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemercier G.; Fernandez-Montalvan A.; Shaw J. P.; Kugelstadt D.; Bomke J.; Domostoj M.; Schwarz M. K.; Scheer A.; Kappes B.; Leroy D. Identification and Characterization of Novel Small Molecules as Potent Inhibitors of the Plasmodial Calcium-Dependent Protein Kinase 1. Biochemistry 2009, 48 (27), 6379–6389. 10.1021/bi9005122. [DOI] [PubMed] [Google Scholar]
- Aher R. B.; Roy K. Exploring the Structural Requirements in Multiple Chemical Scaffolds for the Selective Inhibition of Plasmodium Falciparum Calcium-Dependent Protein Kinase-1 (PfCDPK-1) by 3D-Pharmacophore Modelling, and Docking Studies. SAR QSAR Environ. Res. 2017, 28 (5), 390–414. 10.1080/1062936X.2017.1326401. [DOI] [PubMed] [Google Scholar]
- Fang H.; Klages N.; Baechler B.; Hillner E.; Yu L.; Pardo M.; Choudhary J.; Brochet M. Multiple Short Windows of Calcium-Dependent Protein Kinase 4 Activity Coordinate Distinct Cell Cycle Events during Plasmodium Gametogenesis. eLife 2017, 6, 26524. 10.7554/eLife.26524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billker O.; Dechamps S.; Tewari R.; Wenig G.; Franke-Fayard B.; Brinkmann V. Calcium and a Calcium-Dependent Protein Kinase Regulate Gamete Formation and Mosquito Transmission in a Malaria Parasite. Cell 2004, 117 (4), 503–514. 10.1016/S0092-8674(04)00449-0. [DOI] [PubMed] [Google Scholar]
- Kato K.; Sudo A.; Kobayashi K.; Sugi T.; Tohya Y.; Akashi H. Characterization of Plasmodium Falciparum Calcium-Dependent Protein Kinase 4. Parasitol. Int. 2009, 58 (4), 394–400. 10.1016/j.parint.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Govindasamy K.; Jebiwott S.; Jaijyan D. K.; Davidow A.; Ojo K. K.; Van Voorhis W. C.; Brochet M.; Billker O.; Bhanot P. Invasion of Hepatocytes by Plasmodium Sporozoites Requires cGMP-Dependent Protein Kinase and Calcium Dependent Protein Kinase 4. Mol. Microbiol. 2016, 102 (2), 349–363. 10.1111/mmi.13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidadala R. S. R.; Ojo K. K.; Johnson S. M.; Zhang Z.; Leonard S. E.; Mitra A.; Choi R.; Reid M. C.; Keyloun K. R.; Fox A. M. W.; Kennedy M.; Silver-Brace T.; Hume J. C. C.; Kappe S.; Verlinde C. L. M. J.; Fan E.; Merritt E. A.; Van Voorhis W. C.; Maly D. J. Development of Potent and Selective Plasmodium Falciparum Calcium-Dependent Protein Kinase 4 (PfCDPK4) Inhibitors That Block the Transmission of Malaria to Mosquitoes. Eur. J. Med. Chem. 2014, 74, 562–573. 10.1016/j.ejmech.2013.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo K. K.; Eastman R. T.; Vidadala R.; Zhang Z.; Rivas K. L.; Choi R.; Lutz J. D.; Reid M. C.; Fox A. M. W.; Hulverson M. A.; Kennedy M.; Isoherranen N.; Kim L. M.; Comess K. M.; Kempf D. J.; Verlinde C. L. M. J.; Su X. Z.; Kappe S. H. I.; Maly D. J.; Fan E.; Van Voorhis W. C. A Specific Inhibitor of PfCDPK4 Blocks Malaria Transmission: Chemical-Genetic Validation. J. Infect. Dis. 2014, 209 (2), 275–284. 10.1093/infdis/jit522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo K. K.; Larson E. T.; Keyloun K. R.; Castaneda L. J.; Derocher A. E.; Inampudi K. K.; Kim J. E.; Arakaki T. L.; Murphy R. C.; Zhang L.; Napuli A. J.; Maly D. J.; Verlinde C. L. M. J.; Buckner F. S.; Parsons M.; Hol W. G. J.; Merritt E. A.; Van Voorhis W. C. Toxoplasma Gondii Calcium-Dependent Protein Kinase 1 Is a Target for Selective Kinase Inhibitors. Nat. Struct. Mol. Biol. 2010, 17 (5), 602–607. 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R. C.; Ojo K. K.; Larson E. T.; Castellanos-Gonzalez A.; Perera B. G. K.; Keyloun K. R.; Kim J. E.; Bhandari J. G.; Muller N. R.; Verlinde C. L. M. J.; White A. C.; Merritt E. A.; Van Voorhis W. C.; Maly D. J. Discovery of Potent and Selective Inhibitors of CDPK1 from C. Parvum and T. Gondii. ACS Med. Chem. Lett. 2010, 1 (7), 331–335. 10.1021/ml100096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo K. K.; Pfander C.; Mueller N. R.; Burstroem C.; Larson E. T.; Bryan C. M.; Fox A. M. W.; Reid M. C.; Johnson S. M.; Murphy R. C.; Kennedy M.; Mann H.; Leibly D. J.; Hewitt S. N.; Verlinde C. L. M. J.; Kappe S.; Merritt E. A.; Maly D. J.; Billker O.; Van Voorhis W. C. Transmission of Malaria to Mosquitoes Blocked by Bumped Kinase Inhibitors. J. Clin. Invest. 2012, 122 (6), 2301–2305. 10.1172/JCI61822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Ojo K. K.; Vidadala R.; Huang W.; Geiger J. A.; Scheele S.; Choi R.; Reid M. C.; Keyloun K. R.; Rivas K.; Kallur Siddaramaiah L.; Comess K. M.; Robinson K. P.; Merta P. J.; Kifle L.; Hol W. G. J.; Parsons M.; Merritt E. A.; Maly D. J.; Verlinde C. L. M. J.; Van Voorhis W. C.; Fan E. Potent and Selective Inhibitors of CDPK1 from T. Gondii and C. Parvum Based on a 5-Aminopyrazole-4-Carboxamide Scaffold. ACS Med. Chem. Lett. 2014, 5 (1), 40–44. 10.1021/ml400315s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Ojo K. K.; Zhang Z.; Rivas K.; Vidadala R. S. R.; Scheele S.; DeRocher A. E.; Choi R.; Hulverson M. A.; Barrett L. K.; Bruzual I.; Siddaramaiah L. K.; Kerchner K. M.; Kurnick M. D.; Freiberg G. M.; Kempf D.; Hol W. G. J.; Merritt E. A.; Neckermann G.; de Hostos E. L.; Isoherranen N.; Maly D. J.; Parsons M.; Doggett J. S.; Van Voorhis W. C.; Fan E. SAR Studies of 5-Aminopyrazole-4-Carboxamide Analogues as Potent and Selective Inhibitors of Toxoplasma Gondii CDPK1. ACS Med. Chem. Lett. 2015, 6 (12), 1184–1189. 10.1021/acsmedchemlett.5b00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Hulverson M. A.; Zhang Z.; Choi R.; Hart K. J.; Kennedy M.; Vidadala R. S. R.; Maly D. J.; Van Voorhis W. C.; Lindner S. E.; Fan E.; Ojo K. K. 5-Aminopyrazole-4-Carboxamide Analogues Are Selective Inhibitors of Plasmodium Falciparum Microgametocyte Exflagellation and Potential Malaria Transmission Blocking Agents. Bioorg. Med. Chem. Lett. 2016, 26 (22), 5487–5491. 10.1016/j.bmcl.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavagnino A.; Rossi F.; Rizzi M. The Potent Antiplasmodial Calmodulin-Antagonist Trifluoperazine Inhibits Plasmodium Falciparum Calcium-Dependent Protein Kinase 4. Protein Pept. Lett. 2011, 18 (12), 1273–1279. 10.2174/092986611797642742. [DOI] [PubMed] [Google Scholar]
- Scheibel L. W.; Colombani P. M.; Hess A. D.; Aikawa M.; Atkinson C. T.; Milhous W. K. Calcium and Calmodulin Antagonists Inhibit Human Malaria Parasites (Plasmodium Falciparum): Implications for Drug Design. Proc. Natl. Acad. Sci. U. S. A. 1987, 84 (20), 7310–7314. 10.1073/pnas.84.20.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M. M.; Solyakov L.; Bottrill A. R.; Flueck C.; Siddiqui F. A.; Singh S.; Mistry S.; Viskaduraki M.; Lee K.; Hopp C. S.; Chitnis C. E.; Doerig C.; Moon R. W.; Green J. L.; Holder A. A.; Baker D. A.; Tobin A. B. Phosphoproteomics Reveals Malaria Parasite Protein Kinase G as a Signalling Hub Regulating Egress and Invasion. Nat. Commun. 2015, 6, 7285. 10.1038/ncomms8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D. A.; Stewart L. B.; Large J. M.; Bowyer P. W.; Ansell K. H.; Jiménez-Díaz M. B.; El Bakkouri M.; Birchall K.; Dechering K. J.; Bouloc N. S.; Coombs P. J.; Whalley D.; Harding D. J.; Smiljanic-Hurley E.; Wheldon M. C.; Walker E. M.; Dessens J. T.; Lafuente M. J.; Sanz L. M.; Gamo F.-J.; Ferrer S. B.; Hui R.; Bousema T.; Angulo-Barturén I.; Merritt A. T.; Croft S. L.; Gutteridge W. E.; Kettleborough C. A.; Osborne S. A. A Potent Series Targeting the Malarial CGMP-Dependent Protein Kinase Clears Infection and Blocks Transmission. Nat. Commun. 2017, 8 (1), 430. 10.1038/s41467-017-00572-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale V. L.; Watermeyer J. M.; Hackett F.; Vizcay-Barrena G.; van Ooij C.; Thomas J. A.; Spink M. C.; Harkiolaki M.; Duke E.; Fleck R. A.; Blackman M. J.; Saibil H. R. Parasitophorous Vacuole Poration Precedes Its Rupture and Rapid Host Erythrocyte Cytoskeleton Collapse in Plasmodium Falciparum Egress. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (13), 3439–3444. 10.1073/pnas.1619441114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C. R.; Hackett F.; Strath M.; Penzo M.; Withers-Martinez C.; Baker D. A.; Blackman M. J. Malaria Parasite cGMP-Dependent Protein Kinase Regulates Blood Stage Merozoite Secretory Organelle Discharge and Egress. PLoS Pathog. 2013, 9 (5), e1003344. 10.1371/journal.ppat.1003344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorin J. D.; Martyn D. C.; Patel S. D.; Grimley J. S.; Collins C. R.; Hopp C. S.; Bright A. T.; Westenberger S.; Winzeler E.; Blackman M. J.; Baker D. A.; Wandless T. J.; Duraisingh M. T. A Plant-Like Kinase in Plasmodium Falciparum Regulates Parasite Egress from Erythrocytes. Science 2010, 328 (5980), 910–912. 10.1126/science.1188191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor H. M.; McRobert L.; Grainger M.; Sicard A.; Dluzewski A. R.; Hopp C. S.; Holder A. A.; Baker D. A. The Malaria Parasite Cyclic GMP-Dependent Protein Kinase Plays a Central Role in Blood-Stage Schizogony. Eukaryotic Cell 2010, 9 (1), 37–45. 10.1128/EC.00186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRobert L.; Taylor C. J.; Deng W.; Fivelman Q. L.; Cummings R. M.; Polley S. D.; Billker O.; Baker D. A. Gametogenesis in Malaria Parasites Is Mediated by the cGMP-Dependent Protein Kinase. PLoS Biol. 2008, 6 (6), e139. 10.1371/journal.pbio.0060139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochet M.; Collins M. O.; Smith T. K.; Thompson E.; Sebastian S.; Volkmann K.; Schwach F.; Chappell L.; Gomes A. R.; Berriman M.; Rayner J. C.; Baker D. A.; Choudhary J.; Billker O. Phosphoinositide Metabolism Links cGMP-Dependent Protein Kinase G to Essential Ca2+ Signals at Key Decision Points in the Life Cycle of Malaria Parasites. PLoS Biol. 2014, 12 (3), e1001806. 10.1371/journal.pbio.1001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falae A.; Combe A.; Amaladoss A.; Carvalho T.; Menard R.; Bhanot P. Role of Plasmodium Berghei CGMP-Dependent Protein Kinase in Late Liver Stage Development. J. Biol. Chem. 2010, 285 (5), 3282–3288. 10.1074/jbc.M109.070367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biftu T.; Feng D.; Fisher M.; Liang G. B.; Qian X.; Scribner A.; Dennis R.; Lee S.; Liberator P. A.; Brown C.; Gurnett A.; Leavitt P. S.; Thompson D.; Mathew J.; Misura A.; Samaras S.; Tamas T.; Sina J. F.; McNulty K. A.; McKnight C. G.; Schmatz D. M.; Wyvratt M. Synthesis and SAR Studies of Very Potent Imidazopyridine Antiprotozoal Agents. Bioorg. Med. Chem. Lett. 2006, 16 (9), 2479–2483. 10.1016/j.bmcl.2006.01.092. [DOI] [PubMed] [Google Scholar]
- Churcher T. S.; Blagborough A. M.; Delves M.; Ramakrishnan C.; Kapulu M. C.; Williams A. R.; Biswas S.; Da D. F.; Cohuet A.; Sinden R. E. Measuring the Blockade of Malaria Transmission - An Analysis of the Standard Membrane Feeding Assay. Int. J. Parasitol. 2012, 42 (11), 1037–1044. 10.1016/j.ijpara.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Biftu T.; Feng D.; Ponpipom M.; Girotra N.; Liang G. B.; Qian X.; Bugianesi R.; Simeone J.; Chang L.; Gurnett A.; Liberator P.; Dulski P.; Leavitt P. S.; Crumley T.; Misura A.; Murphy T.; Rattray S.; Samaras S.; Tamas T.; Mathew J.; Brown C.; Thompson D.; Schmatz D.; Fisher M.; Wyvratt M. Synthesis and SAR of 2,3-Diarylpyrrole Inhibitors of Parasite cGMP-Dependent Protein Kinase as Novel Anticoccidial Agents. Bioorg. Med. Chem. Lett. 2005, 15 (13), 3296–3301. 10.1016/j.bmcl.2005.04.060. [DOI] [PubMed] [Google Scholar]
- Diaz C. A.; Allocco J.; Powles M. A.; Yeung L.; Donald R. G. K.; Anderson J. W.; Liberator P. A. Characterization of Plasmodium Falciparum CGMP-Dependent Protein Kinase (PfPKG): Antiparasitic Activity of a PKG Inhibitor. Mol. Biochem. Parasitol. 2006, 146 (1), 78–88. 10.1016/j.molbiopara.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Panchal D.; Bhanot P. Activity of a Trisubstituted Pyrrole in Inhibiting Sporozoite Invasion and Blocking Malaria Infection. Antimicrob. Agents Chemother. 2010, 54 (10), 4269–4274. 10.1128/AAC.00420-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler D. R.; Bullen H. E.; Harvey K. L.; Beddoe T.; Crabb B. S.; Rossjohn J.; Gilson P. R. Disrupting the Allosteric Interaction between the Plasmodium Falciparum cAMP-Dependent Kinase and Its Regulatory Subunit. J. Biol. Chem. 2016, 291 (49), 25375–25386. 10.1074/jbc.M116.750174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leykauf K.; Treeck M.; Gilson P. R.; Nebl T.; Braulke T.; Cowman A. F.; Gilberger T. W.; Crabb B. S. Protein Kinase a Dependent Phosphorylation of Apical Membrane Antigen 1 Plays an Important Role in Erythrocyte Invasion by the Malaria Parasite. PLoS Pathog. 2010, 6 (6), e1000941. 10.1371/journal.ppat.1000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasonder E.; Green J. L.; Camarda G.; Talabani H.; Holder A. A.; Langsley G.; Alano P. The Plasmodium Falciparum Schizont Phosphoproteome Reveals Extensive Phosphatidylinositol and cAMP-Protein Kinase A Signaling. J. Proteome Res. 2012, 11 (11), 5323–5337. 10.1021/pr300557m. [DOI] [PubMed] [Google Scholar]
- Lasonder E.; Green J. L.; Grainger M.; Langsley G.; Holder A. A. Extensive Differential Protein Phosphorylation as Intraerythrocytic Plasmodium Falciparum Schizonts Develop into Extracellular Invasive Merozoites. Proteomics 2015, 15 (15), 2716–2729. 10.1002/pmic.201400508. [DOI] [PubMed] [Google Scholar]
- Dawn A.; Singh S.; More K. R.; Siddiqui F. A.; Pachikara N.; Ramdani G.; Langsley G.; Chitnis C. E. The Central Role of cAMP in Regulating Plasmodium Falciparum Merozoite Invasion of Human Erythrocytes. PLoS Pathog. 2014, 10 (12), e1004520. 10.1371/journal.ppat.1004520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraldo F. H.; Almeida F. M.; Da Silva A. M.; Garcia C. R. S. Cyclic AMP and Calcium Interplay as Second Messengers in Melatonin-Dependent Regulation of Plasmodium Falciparum Cell Cycle. J. Cell Biol. 2005, 170 (4), 551–557. 10.1083/jcb.200505117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo A.; Kato K.; Kobayashi K.; Tohya Y.; Akashi H. Susceptibility of Plasmodium Falciparum Cyclic AMP-Dependent Protein Kinase and Its Mammalian Homologue to the Inhibitors. Mol. Biochem. Parasitol. 2008, 160 (2), 138–142. 10.1016/j.molbiopara.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Merckx A.; Nivez M. P.; Bouyer G.; Alano P.; Langsley G.; Deitsch K.; Thomas S.; Doerig C.; Egée S. Plasmodium Falciparum Regulatory Subunit of CAMP-Dependent PKA and Anion Channel Conductance. PLoS Pathog. 2008, 4 (2), e19. 10.1371/journal.ppat.0040019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buskes M. J.; Harvey K. L.; Prinz B.; Crabb B. S.; Gilson P. R.; Wilson D. J. D.; Abbott B. M. Exploration of 3-Methylisoquinoline-4-Carbonitriles as Protein Kinase A Inhibitors of Plasmodium Falciparum. Bioorg. Med. Chem. 2016, 24 (11), 2389–2396. 10.1016/j.bmc.2016.03.048. [DOI] [PubMed] [Google Scholar]
- Buskes M. J.; Harvey K. L.; Richards B. J.; Kalhor R.; Christoff R. M.; Gardhi C. K.; Littler D. R.; Cope E. D.; Prinz B.; Weiss G. E.; O’Brien N. J.; Crabb B. S.; Deady L. W.; Gilson P. R.; Abbott B. M. Antimalarial Activity of Novel 4-Cyano-3-Methylisoquinoline Inhibitors against Plasmodium Falciparum: Design, Synthesis and Biological Evaluation. Org. Biomol. Chem. 2016, 14 (20), 4617–4639. 10.1039/C5OB02517F. [DOI] [PubMed] [Google Scholar]
- Doerig C.; Billker O.; Pratt D.; Endicott J. Protein Kinases as Targets for Antimalarial Intervention: Kinomics, Structure-Based Design, Transmission-Blockade, and Targeting Host Cell Enzymes. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1754 (1–2), 132–150. 10.1016/j.bbapap.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Li J. L.; Robson K. J.; Chen J. L.; Targett G. A.; Baker D. A. Pfmrk, a MO15-Related Protein Kinase from Plasmodium Falciparum. Gene Cloning, Sequence, Stage-Specific Expression and Chromosome Localization. Eur. J. Biochem. 1996, 241 (3), 805–813. 10.1111/j.1432-1033.1996.00805.x. [DOI] [PubMed] [Google Scholar]
- Jirage D.; Chen Y.; Caridha D.; O’Neil M. T.; Eyase F.; Witola W. H.; Ben Mamoun C.; Waters N. C. The Malarial CDK Pfmrk and Its Effector PfMAT1 Phosphorylate DNA Replication Proteins and Co-Localize in the Nucleus. Mol. Biochem. Parasitol. 2010, 172 (1), 9–18. 10.1016/j.molbiopara.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Waters N. C.; Woodard C. L.; Li Z.; Li P. K. Design and Synthesis of Pfmrk Inhibitors as Potential Antimalarial Agents. Bioorg. Med. Chem. Lett. 2001, 11 (21), 2875–2878. 10.1016/S0960-894X(01)00578-9. [DOI] [PubMed] [Google Scholar]
- Woodard C. L.; Li Z.; Kathcart A. K.; Terrell J.; Gerena L.; Lopez-Sanchez M.; Kyle D. E.; Bhattacharjee A. K.; Nichols D. A.; Ellis W.; Prigge S. T.; Geyer J. A.; Waters N. C. Oxindole-Based Compounds Are Selective Inhibitors of Plasmodium Falciparum Cyclin Dependent Protein Kinases. J. Med. Chem. 2003, 46 (18), 3877–3882. 10.1021/jm0300983. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A. K.; Geyer J. A.; Woodard C. L.; Kathcart A. K.; Nichols D. A.; Prigge S. T.; Li Z.; Mott B. T.; Waters N. C. A Three-Dimensional in Silico Pharmacophore Model for Inhibition of Plasmodium Falciparum Cyclin-Dependent Kinases and Discovery of Different Classes of Novel Pfmrk Specific Inhibitors. J. Med. Chem. 2004, 47 (22), 5418–5426. 10.1021/jm040108f. [DOI] [PubMed] [Google Scholar]
- Geyer J. A.; Keenan S. M.; Woodard C. L.; Thompson P. A.; Gerena L.; Nichols D. A.; Gutteridge C. E.; Waters N. C. Selective Inhibition of Pfmrk, a Plasmodium Falciparum CDK, by Antimalarial 1,3-Diaryl-2-Propenones. Bioorg. Med. Chem. Lett. 2009, 19 (7), 1982–1985. 10.1016/j.bmcl.2009.02.042. [DOI] [PubMed] [Google Scholar]
- Caridha D.; Kathcart A. K.; Jirage D.; Waters N. C. Activity of Substituted Thiophene Sulfonamides against Malarial and Mammalian Cyclin Dependent Protein Kinases. Bioorg. Med. Chem. Lett. 2010, 20 (13), 3863–3867. 10.1016/j.bmcl.2010.05.039. [DOI] [PubMed] [Google Scholar]
- Akala H. M.; Waters C. N.; Yenesew A.; Wanjala C.; Akenga T. A. In Vitro Antiplasmodial and Cyclin- Dependent Protein Kinase (Pfmrk) Inhibitory Activities of Sele c Ted Flavonoids in Combination with Chloroquine (CQ) and Artemisinin. Res. Pharm. Biotechnol. 2010, 2 (4), 40–50. [Google Scholar]
- Droucheau E.; Primot A.; Thomas V.; Mattei D.; Knockaert M.; Richardson C.; Sallicandro P.; Alano P.; Jafarshad A.; Baratte B.; Kunick C.; Parzy D.; Pearl L. H.; Doerig C.; Meijer L. Plasmodium Falciparum Glycogen Synthase Kinase-3: Molecular Model, Expression, Intracellular Localisation and Selective Inhibitors. Biochim. Biophys. Acta, Proteins Proteomics 2004, 1697 (1–2), 181–196. 10.1016/j.bbapap.2003.11.023. [DOI] [PubMed] [Google Scholar]
- Masch A.; Kunick C. Selective Inhibitors of Plasmodium Falciparum Glycogen Synthase-3 (PfGSK-3): New Antimalarial Agents?. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854 (10), 1644–1649. 10.1016/j.bbapap.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Fugel W.; Oberholzer A. E.; Gschloessl B.; Dzikowski R.; Pressburger N.; Preu L.; Pearl L. H.; Baratte B.; Ratin M.; Okun I.; Doerig C.; Kruggel S.; Lemcke T.; Meijer L.; Kunick C. 3,6-Diamino-4-(2-Halophenyl)-2-Benzoylthieno[2,3- b ]Pyridine-5- Carbonitriles Are Selective Inhibitors of Plasmodium Falciparum Glycogen Synthase Kinase-3. J. Med. Chem. 2013, 56 (1), 264–275. 10.1021/jm301575n. [DOI] [PubMed] [Google Scholar]
- Reininger L.; Garcia M.; Tomlins A.; Müller S.; Doerig C. The Plasmodium Falciparum, Nima-Related Kinase Pfnek-4: A Marker for Asexual Parasites Committed to Sexual Differentiation. Malar. J. 2012, 11, 250. 10.1186/1475-2875-11-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A. M.; Bayliss R.; Roig J. Mitotic Regulation by NEK Kinase Networks. Front. Cell Dev. Biol. 2017, 5, 102. 10.3389/fcell.2017.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorin-Semblat D.; Schmitt S.; Semblat J. P.; Sicard A.; Reininger L.; Goldring D.; Patterson S.; Quashie N.; Chakrabarti D.; Meijer L.; Doerig C. Plasmodium Falciparum NIMA-Related Kinase Pfnek-1: Sex Specificity and Assessment of Essentiality for the Erythrocytic Asexual Cycle. Microbiology 2011, 157 (10), 2785–2794. 10.1099/mic.0.049023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desoubzdanne D.; Marcourt L.; Raux R.; Chevalley S.; Dorin D.; Doerig C.; Valentin A.; Ausseil F.; Debitus C. Alisiaquinones and Alisiaquinol, Dual Inhibitors of Plasmodium Falciparum Enzyme Targets from a New Caledonian Deep Water Sponge Alisiaquinones and Alisiaquinol, Dual Inhibitors of Plasmodium Falciparum Enzyme Targets from a New Caledonian Deep Water Sp. J. Nat. Prod. 2008, 71 (7), 1189–1192. 10.1021/np8000909. [DOI] [PubMed] [Google Scholar]
- Lebouvier N.; Jullian V.; Desvignes I.; Maurel S.; Parenty A.; Dorin-Semblat D.; Doerig C.; Sauvain M.; Laurent D. Antiplasmodial Activities of Homogentisic Acid Derivative Protein Kinase Inhibitors Isolated from a Vanuatu Marine Sponge Pseudoceratina Sp.. Mar. Drugs 2009, 7 (4), 640–653. 10.3390/md7040640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent D.; Jullian V.; Parenty A.; Knibiehler M.; Dorin D.; Schmitt S.; Lozach O.; Lebouvier N.; Frostin M.; Alby F.; Maurel S.; Doerig C.; Meijer L.; Sauvain M. Antimalarial Potential of Xestoquinone, a Protein Kinase Inhibitor Isolated from a Vanuatu Marine Sponge Xestospongia Sp. Bioorg. Med. Chem. 2006, 14 (13), 4477–4482. 10.1016/j.bmc.2006.02.026. [DOI] [PubMed] [Google Scholar]
- Mitcheson D. F.; Bottrill A. R.; Carr K.; Coxon C. R.; Cano C.; Golding B. T.; Griffin R. J.; Fry A. M.; Doerig C.; Bayliss R.; Tobin A. B. A New Tool for the Chemical Genetic Investigation of the Plasmodium Falciparum Pfnek-2 NIMA-Related Kinase. Malar. J. 2016, 15 (1), 535. 10.1186/s12936-016-1580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells C. I.; Kapadia N. R.; Counago R. M.; Drewry D. H. In Depth Analysis of Kinase Cross Screening Data to Identify Chemical Starting Points for Inhibition of the Nek Family of Kinases. MedChemComm 2018, 9, 44–66. 10.1039/C7MD00510E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes M. C.; Goldring J. P. D.; Doerig C.; Scherf A. A Novel Protein Kinase Family in Plasmodium Falciparum Is Differentially Transcribed and Secreted to Various Cellular Compartments of the Host Cell.. Mol. Microbiol. 2007, 63 (2), 391–403. 10.1111/j.1365-2958.2006.05521.x. [DOI] [PubMed] [Google Scholar]
- Jaijyan D. K.; Verma P. K.; Singh A. P. A Novel FIKK Kinase Regulates the Development of Mosquito and Liver Stages of the Malaria. Sci. Rep. 2016, 6, 39285. 10.1038/srep39285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kats L. M.; Fernandez K. M.; Glenister F. K.; Herrmann S.; Buckingham D. W.; Siddiqui G.; Sharma L.; Bamert R.; Lucet I.; Guillotte M.; Mercereau-Puijalon O.; Cooke B. M. An Exported Kinase (FIKK4.2) That Mediates Virulence-Associated Changes in Plasmodium Falciparum-Infected Red Blood Cells. Int. J. Parasitol. 2014, 44 (5), 319–328. 10.1016/j.ijpara.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Nunes M. C.; Okada M.; Scheidig-Benatar C.; Cooke B. M.; Scherf A. Plasmodium Falciparum Fikk Kinase Members Target Distinct Components of the Erythrocyte Membrane. PLoS One 2010, 5 (7), e11747. 10.1371/journal.pone.0011747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J.; Johnson R. L.; Huang R.; Wichterman J.; Jiang H.; Hayton K.; Fidock D. A.; Wellems T. E.; Inglese J.; Austin C. P.; Su X. Z. Genetic Mapping of Targets Mediating Differential Chemical Phenotypes in Plasmodium Falciparum. Nat. Chem. Biol. 2009, 5 (10), 765–771. 10.1038/nchembio.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B. C.; Harris D. R.; Kirkman L. M. D.; Perez A. M.; Qian Y.; Schermerhorn J. T.; Hong M. Y.; Winston D. S.; Xu L.; Lieber A. M.; Hamilton M.; Brandt G. S. The Anthraquinone Emodin Inhibits the Non-Exported FIKK Kinase from Plasmodium Falciparum. Bioorg. Chem. 2017, 75, 217–223. 10.1016/j.bioorg.2017.09.011. [DOI] [PubMed] [Google Scholar]
- Lin B. C.; Harris D. R.; Kirkman L. M. D.; Perez A. M.; Qian Y.; Schermerhorn J. T.; Hong M. Y.; Winston D. S.; Xu L.; Brandt G. S. FIKK Kinase, a Ser/Thr Kinase Important to Malaria Parasites, Is Inhibited by Tyrosine Kinase Inhibitors. ACS Omega 2017, 2 (10), 6605–6612. 10.1021/acsomega.7b00997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman K. T.; Ye J.; Shi Z.; Toker C.; Lovato D.; Jumani R. S.; Zuercher W.; Huston C. D.; Edwards A. M.; Lautens M.; Santhakumar V.; Hui R. Discovery and Structure Activity Relationship of the First Potent Cryptosporidium FIKK Kinase Inhibitor. Bioorg. Med. Chem. 2017, 25 (5), 1672–1680. 10.1016/j.bmc.2017.01.036. [DOI] [PubMed] [Google Scholar]
- Brown J. R.; Auger K. R. Phylogenomics of Phosphoinositide Lipid Kinases: Perspectives on the Evolution of Second Messenger Signaling and Drug Discovery. BMC Evol. Biol. 2011, 11 (1), 4. 10.1186/1471-2148-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara C. W.; Lee M. C. S.; Lim C. S.; Lim S. H.; Roland J.; Nagle A.; Simon O.; Yeung B. K. S.; Chatterjee A. K.; McCormack S. L.; Manary M. J.; Zeeman A. M.; Dechering K. J.; Kumar T. R. S.; Henrich P. P.; Gagaring K.; Ibanez M.; Kato N.; Kuhen K. L.; Fischli C.; Rottmann M.; Plouffe D. M.; Bursulaya B.; Meister S.; Rameh L.; Trappe J.; Haasen D.; Timmerman M.; Sauerwein R. W.; Suwanarusk R.; Russell B.; Renia L.; Nosten F.; Tully D. C.; Kocken C. H. M.; Glynne R. J.; Bodenreider C.; Fidock D. A.; Diagana T. T.; Winzeler E. A. Targeting Plasmodium PI(4)K to Eliminate Malaria. Nature 2013, 504 (7479), 248–253. 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassett M. R.; Sternberg A. R.; Riegel B. E.; Thomas C. J.; Roepe P. D. Heterologous Expression, Purification, and Functional Analysis of Plasmodium Falciparum Phosphatidylinositol 3′-Kinase. Biochemistry 2017, 56, 4335–4345. 10.1021/acs.biochem.7b00416. [DOI] [PubMed] [Google Scholar]
- Vaid A.; Ranjan R.; Smythe W. A.; Hoppe H. C.; Sharma P. PfPI3K, a Phosphatidylinositol-3 Kinase from Plasmodium Falciparum, Is Exported to the Host Erythrocyte and Is Involved in Hemoglobin Trafficking. Blood 2010, 115 (12), 2500–2507. 10.1182/blood-2009-08-238972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdech Z.; Ferreira P. E.; Mok S. A Crucial Piece in the Puzzle of the Artemisinin Resistance Mechanism in Plasmodium Falciparum. Trends Parasitol. 2015, 31 (8), 345–346. 10.1016/j.pt.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Mbengue A.; Bhattacharjee S.; Pandharkar T.; Liu H.; Estiu G.; Stahelin R. V.; Rizk S. S.; Njimoh D. L.; Ryan Y.; Chotivanich K.; Nguon C.; Ghorbal M.; Lopez-Rubio J.-J.; Pfrender M.; Emrich S.; Mohandas N.; Dondorp A. M.; Wiest O.; Haldar K. A Molecular Mechanism of Artemisinin Resistance in Plasmodium Falciparum Malaria. Nature 2015, 520 (7549), 683–687. 10.1038/nature14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou B.; Nagle A.; Chatterjee A. K.; Leong S. Y.; Tan L. J.; Sim W. L. S.; Mishra P.; Guntapalli P.; Tully D. C.; Lakshminarayana S. B.; Lim C. S.; Tan Y. C.; Abas S. N.; Bodenreider C.; Kuhen K. L.; Gagaring K.; Borboa R.; Chang J.; Li C.; Hollenbeck T.; Tuntland T.; Zeeman A. M.; Kocken C. H. M.; McNamara C.; Kato N.; Winzeler E. A.; Yeung B. K. S.; Diagana T. T.; Smith P. W.; Roland J. Lead Optimization of Imidazopyrazines: A New Class of Antimalarial with Activity on Plasmodium Liver Stages. ACS Med. Chem. Lett. 2014, 5 (8), 947–950. 10.1021/ml500244m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younis Y.; Douelle F.; Feng T. S.; Cabrera D. G.; Le Manach C.; Nchinda A. T.; Duffy S.; White K. L.; Shackleford D. M.; Morizzi J.; Mannila J.; Katneni K.; Bhamidipati R.; Zabiulla K. M.; Joseph J. T.; Bashyam S.; Waterson D.; Witty M. J.; Hardick D.; Wittlin S.; Avery V.; Charman S. A. Chibale, K. 3,5-Diaryl-2-Aminopyridines As a Novel Class of Orally Active Antimalarials Demonstrating Single Dose Cure in Mice and Clinical Candidate Potential. J. Med. Chem. 2012, 55 (7), 3479–3487. 10.1021/jm3001373. [DOI] [PubMed] [Google Scholar]
- Younis Y.; Douelle F.; González Cabrera D.; Le Manach C.; Nchinda A. T.; Paquet T.; Street L. J.; White K. L.; Zabiulla K. M.; Joseph J. T.; Bashyam S.; Waterson D.; Witty M. J.; Wittlin S.; Charman S. A.; Chibale K. Structure-Activity-Relationship Studies around the 2-Amino Group and Pyridine Core of Antimalarial 3,5-Diarylaminopyridines Lead to a Novel Series of Pyrazine Analogues with Oral in Vivo Activity. J. Med. Chem. 2013, 56 (21), 8860–8871. 10.1021/jm401278d. [DOI] [PubMed] [Google Scholar]
- Le Manach C.; Nchinda A. T.; Paquet T.; Gonzàlez Cabrera D.; Younis Y.; Han Z.; Bashyam S.; Zabiulla M.; Taylor D.; Lawrence N.; White K. L.; Charman S. A.; Waterson D.; Witty M. J.; Wittlin S.; Botha M. E.; Nondaba S. H.; Reader J.; Birkholtz L. M.; Jiménez-Díaz M. B.; Martínez M. S.; Ferrer S.; Angulo-Barturen I.; Meister S.; Antonova-Koch Y.; Winzeler E. A.; Street L. J.; Chibale K. Identification of a Potential Antimalarial Drug Candidate from a Series of 2-Aminopyrazines by Optimization of Aqueous Solubility and Potency across the Parasite Life Cycle. J. Med. Chem. 2016, 59 (21), 9890–9905. 10.1021/acs.jmedchem.6b01265. [DOI] [PubMed] [Google Scholar]
- Medicines for Malaria Venture (MMV) . MMV048. https://www.mmv.org/research-development/project-portfolio/mmv048 (accessed Feb 27, 2018).
- Ren J. X.; Gao N. N.; Cao X. S.; Hu Q. A.; Xie Y. Homology Modeling and Virtual Screening for Inhibitors of Lipid Kinase PI(4)K from Plasmodium. Biomed. Pharmacother. 2016, 83, 798–808. 10.1016/j.biopha.2016.07.048. [DOI] [PubMed] [Google Scholar]
- Lelliott P. M.; McMorran B. J.; Foote S. J.; Burgio G. The Influence of Host Genetics on Erythrocytes and Malaria Infection: Is There Therapeutic Potential?. Malar. J. 2015, 14 (1), 289. 10.1186/s12936-015-0809-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesely K. R.; Pantaleo A.; Turrini F. M.; Olupot-Olupot P.; Low P. S. Inhibition of an Erythrocyte Tyrosine Kinase with Imatinib Prevents Plasmodium Falciparum Egress and Terminates Parasitemia. PLoS One 2016, 11 (10), e0164895. 10.1371/journal.pone.0164895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudêncio M.; Rodrigues C. D.; Hannus M.; Martin C.; Real E.; Gonçalves L. A.; Carret C.; Dorkin R.; Röhl I.; Jahn-Hoffmann K.; Luty A. J. F.; Sauerwein R.; Echeverri C. J.; Mota M. M. Kinome-Wide RNAi Screen Implicates at Least 5 Host Hepatocyte Kinases in Plasmodium Sporozoite Infection. PLoS Pathog. 2008, 4 (11), e1000201. 10.1371/journal.ppat.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudêncio M.; Mota M. M. Targeting Host Factors to Circumvent Anti-Malarial Drug Resistance. Curr. Pharm. Des. 2012, 19 (2), 290–299. 10.2174/1381612811306020290. [DOI] [PubMed] [Google Scholar]
- Wu P.; Nielsen T. E.; Clausen M. H. Small-Molecule Kinase Inhibitors: An Analysis of FDA-Approved Drugs. Drug Discov. Drug Discovery Today 2016, 21 (1), 5–10. 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]