

SUMMARY
Transcription factors bind to their binding sites over a wide range of affinities, yet how differences in affinity are encoded in DNA sequences is not well understood. Here, we report X-ray crystal structures of four heterodimers of the Hox protein AbdominalB bound with its cofactor Extradenticle to four target DNA molecules that differ in affinity by up to ~20-fold. Remarkably, despite large differences in affinity, the overall structures are very similar in all four complexes. In contrast, the predicted shapes of the DNA binding sites (i.e., the intrinsic DNA shape) in the absence of bound protein are strikingly different from each other and correlate with affinity: binding sites that must change conformations upon protein binding have lower affinities than binding sites that have more optimal conformations prior to binding. Together, these observations suggest that intrinsic differences in DNA shape provide a robust mechanism for modulating affinity without affecting other protein-DNA interactions.
In Brief
By solving the structures of four ternary Hox-Exd-DNA complexes, Zeiske et al. show that lower-affinity binding sites have intrinsic DNA shapes that must change conformation upon protein binding, and that the paths of Hox N-terminal arms determine the extent to which DNA shape can be read out.
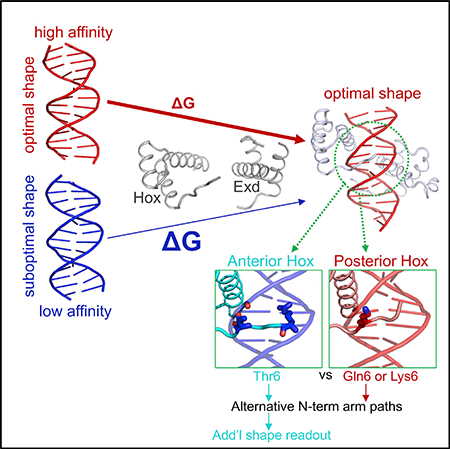
Graphical Abstract
INTRODUCTION
To execute appropriate gene regulatory functions, transcription factors (TFs) must select the correct subset of DNA binding sites from a very large number of potential sites that are typically present in eukaryotic genomes. In many cases, TFs are limited to binding sites that lie within DNA regions that are more accessible in the genome and that can be mapped in a cell-type-specific manner using several powerful techniques (Guertin and Lis, 2010; Mahony and Pugh, 2015). These more accessible regions presumably are a consequence of earlier acting pioneer TFs, which have the ability to bind to nucleosome-coated DNA and alter chromatin structure, thereby allowing other TFs access to their binding sites (Farley et al., 2015a; Guertin and Lis, 2010). Yet, even within more accessible regions, TFs only choose a subset of potential binding sites, raising the fundamental question of how TFs identify correct binding sites in vivo.
Affinity is another likely parameter for influencing TF binding site selection, which for many TFs can vary more than three orders of magnitude for different DNA sequences. In principle, the TF binding site selection problem could be solved in part by TFs choosing only the highest affinity binding sites. Consistent with this idea, good correlation is often observed between occupancy in vivo and affinity of the underlying binding site for TFs that have large DNA footprints, such as p53 tetramers, as approximated by how closely the binding site matches the optimal consensus site defined in vitro (Weinberg et al., 2004). In contrast, for many other TFs and TF complexes, binding in vivo often depends upon the recognition of sub-optimal or low-affinity binding sites that have poor matches to optimal consensus sites (Crocker et al., 2015, 2016; Farley et al., 2015b). In addition, low-affinity interactions can also be a consequence of suboptimal spacing of binding sites for interacting TFs. For some TF families, such as the Hox family of homeodomain TFs, the use of low-affinity binding sites is essential for specificity, i.e., the ability of closely related TF family members to selectively bind specific DNA sequences (Crocker et al., 2015). This problem is particularly striking for the Hox TFs, which all bind closely related TAAT-containing DNA motifs as monomers. However, upon heterodimerization with the Hox cofactor Extradenticle (Exd in Drosophila; Pbx in vertebrates) distinct DNA binding preferences between different Hox-Exd heterodimers emerge (Merabet and Mann, 2016; Slattery et al., 2011). This phenomenon has been termed “latent specificity,” and depends on cofactor-mediated conformational stabilization of the N-terminal arms (NTAs) of Hox homeodomains. Both phenomena—the use of low-affinity binding sites and latent specificity—compounds the challenge for identifying bona fide TF binding sites in vivo (Crocker et al., 2016; Slattery et al., 2014).
Despite direct relevance to binding site selection in vivo, a structural understanding of how differential TF affinity and specificity are encoded in DNA sequences has been achieved in very few instances. One example is the Drosophila Hox protein Sex combs reduced (Scr), which binds cooperatively with the Hox cofactor Exd to two different DNA binding sites, fkh250 and fkh250con, with a similar affinity (Kd ≈10 nM) (Joshi et al., 2007). In contrast, other Hox-Exd complexes bind with very different affinities to these two DNA sequences. For example, although Ultrabithorax (Ubx)-Exd is able to bind fkh250con with a Kd≈20 nM, the binding of Ubx-Exd to fkh250 is weaker than what can be reliably measured in standard in vitro DNA binding assays. Structural studies suggest that Scr-Exd’s affinity for fkh250 depends on Scr’s ability to recognize a novel DNA shape: fkh250, but not fkh250con, has an additional local minimum in minor groove width that is read by the insertion of two basic side chains present in Scr (Joshi et al., 2007; Rohs et al., 2010). Due to differences in the sequences of their homeodomain N-terminal arms, these residues are either absent or in a different conformation in Ubx, thus reducing the affinity of this Hox protein to fkh250. Removing these basic side chains in Scr, by mutation to alanine, reduced Scr-Exd’s affinity to fkh250 by ~6-fold and eliminated Scr’s ability to regulate Scr-specific target genes in vivo, but had <2-fold effect on affinity to fkh250con. Moreover, changing the context of these basic side chains, which presumably altered their conformations, also eliminated the ability to read a specific DNA shape and, consequently, specificity (Abe et al., 2015). Thus, the recognition of DNA shape by TFs, specifically minor groove width, can have profound consequences on both binding affinity and specificity (Rohs et al., 2010).
In the work presented here, we expand understanding of how DNA sequences encode differences in TF affinity, focusing on the most posteriorly expressed Drosophila Hox protein, Abdominal-B (AbdB). We determined X-ray crystal structures of four AbdB-Exd-DNA ternary complexes that subtly differ in the sequences of the binding site and, as a consequence, have different affinities for AbdB-Exd. Strikingly, although the overall ternary structures are very similar, affinity correlates with the predicted shape of the DNA binding site prior to protein binding: binding sites that must structurally adapt upon protein binding generally have a lower affinity than binding sites that are optimally pre-formed for protein binding. Furthermore, comparison of all four AbdB-Exd-DNA structures to previously solved Scr-Exd-DNA complexes (Joshi et al., 2007) reveals consistent differences between the N-terminal arms of anterior (e.g., Scr) versus posterior (e.g., AbdB) Hox proteins that contribute to their ability to read differences in DNA shape. Together, these observations support a general model in which TF-DNA affinity is sensitive to differences in intrinsic DNA shape, thus providing a mechanism for varying affinity that is independent of direct contacts between protein side chains and DNA base pairs.
RESULTS
Overview of AbdB-Exd Bound to Four Different Binding Sites
Using SELEX-seq experiments, Slattery et al. (2011) described a set of 10 Hox-Exd binding sites that differ in relative affinity for each of the eight Drosophila Hox-Exd heterodimers. These 10 motifs, named after different colors, were defined by their central 8 base pairs. To gain insight into the structural basis for differences in Hox-Exd relative affinity, we used X-ray crystallography to solve the structures of AbdB-Exd bound to four of these binding sites (core 8-mer is in caps), red (gcaTGATTTATgac), magenta (gcaTGATTACgac), blue (gcaTGATTAATgac), and black (gcaTGATAAATgac) (Figures 1A–1D). For the crystallography studies, AbdB included residues 146–229 (the homeodomain is 164–223) and Exd included residues 237–310 (the homeodomain is 238–300). We also measured the relative affinities of AbdB-Exd to each of these DNA sequences using competition electrophoretic mobility shift assays (compEMSAs, see Experimental Procedures for details) (Figure 1I). These measurements agreed well with the relative affinities obtained from previous SELEX-seq experiments (Figure 1J) (Abe et al., 2015; Slattery et al., 2011) and ranged more than 20-fold, providing a powerful dataset for understanding how differences in affinity are encoded in DNA binding sites.
Figure 1. Overview of AbdB-Exd-DNA Ternary Complexes.

(A–D) Similar head-to-tail arrangement of four AbdB-Exd heterodimers (A, red; B, magenta; C, blue; D, black), bound to different DNA sequences. The structures are viewable from the PDB with the following identifiers: red, 5ZJQ; magenta, 5ZJR; blue, 5ZJS; and black, 5ZJT.
(E–H) The W motif binds to the TALE motif of Exd in all four complexes (A, red; B, magenta; C, blue; D, black). In the blue structure (G), the Trp was more difficult to model due to poorer density, suggesting a flexible and dynamic binding mode. The black structure had the greatest number of well-resolved residues, perhaps due in part to stabilizing crystal contacts.
(I)Results from competition EMSAs showing the relative affinities of each of the four binding sites to AbdB-Exd.
(J)Comparison of the relative affinities obtained from the competitive EMSAs with measurements from previous SELEX-seq experiments (Slattery et al., 2011).
The structures of the red, blue, magenta, and black crystals were refined to final resolutions of 2.44, 2.90, 3.0, and 2.4 Å, respectively. Despite these moderate resolutions, composite omit maps confirmed key features of these structures (Figure S1). The red, blue, and magenta crystals were in the C2 space group, all with similar unit cell dimensions (Tables S1 and S2). The black complex was in the P1 space group. The asymmetric units for the first three samples crystallize as ternary complexes with a single DNA duplex bound to one homeodomain of AbdB and one homeodomain of Exd in the asymmetric unit. In contrast, the asymmetric unit for the black DNA sample contains an additional DNA duplex bound by a single AbdB homeodomain, in addition to the ternary complex. Below, we first discuss the four ternary complexes and then discuss the additional AbdB-DNA binary complex.
AbdB, like all Hox proteins, has a homeodomain that consists of three α helices and an N-terminal arm (NTA, residues 1–9 of the homeodomain) and a linker region N-terminal to the homeodomain. The linker region includes a W motif that directly contacts the three amino acid loop extension (TALE) motif of the Exd (or Pbx) homeodomain in all previously characterized Hoxcofactor-DNA ternary structures (Foos et al., 2015; Joshi et al., 2007; LaRonde-LeBlanc and Wolberger, 2003; Passner et al., 1999; Piper et al., 1999). All four ternary complexes solved here show the typical binding mode observed in previous ternary structures. AbdB and Exd bind in head-to-tail fashion to opposite faces of the DNA, using overlapping binding sites, with their respective recognition helices (helix 3 of the homeodomain) lying in the major groove of the DNA and their side chains making direct contacts with DNA bases (Figures 1A–1D and S2). The protein backbones of all four complexes superpose very well, with a Cα root-mean-square deviation (RMSD) of <1 Å for any pair of homeodomains, when aligned using the DNA as a template.
In most Hox proteins, the W motif is related to the sequence YPWM (Merabet and Mann, 2016). In contrast, AbdB and its vertebrate orthologs rely on a distinct W-containing motif, which in AbdB is HEWT. As with YPWM, the conserved tryptophan in the HEWT sequence is responsible for interaction with the Exd homeodomain by inserting into a hydrophobic pocket formed by Exd’s TALE motif. AbdB also has the shortest linker region of all the Drosophila Hox paralogs, consisting of only 3 residues between the homeodomain and W motif, compared to 8–109 residues for the other Drosophila Hox paralogs. This feature is conserved in vertebrate orthologs of AbdB (LaRonde-LeBlanc and Wolberger, 2003). While density for the W motif is present in the TALE binding pocket of Exd for all four ternary complexes, occupancies vary between the structures (Figures 1E–1H). In particular, well-defined densities for the W motif were observed in the red and black structures, with poorer densities and higher B factors for the magenta and blue structures. Further, the partial density of the W motif in the blue structure leads to a model in which the tryptophan is not as buried as deeply inside the TALE hydrophobic pocket as in the other structures (Figures 1E–1H). Taken together, these observations suggest that, depending on the complex, the stability of the W motif-TALE interaction may differ and may also be influenced by crystal contacts.
Most of the protein-DNA contacts are similar in the four ternary structures (Figure S2; see below for a few exceptions). For the three structures in the C2 space group (red, magenta, and blue), there is a correlation between in vitro DNA-binding affinity and the number of H-bonds between Exd and DNA (Table S3). However, other measurements, such as the buried surface area between any two components of these ternary structures, do not correlate with affinity (Table S3). We conclude that the number of hydrogen bonds or differences in other intermolecular contacts are not sufficient to account for the differences in affinities to these four DNA sequences.
The AbdB N-Terminal Arm
We next turned our attention to the N-terminal arm (NTA) of AbdB’s homeodomain, given its importance for conferring binding specificity in other Hox-Exd-DNA complexes. In general, the W motif-Exd homeodomain contact stabilizes Hox NTAs in the minor groove of the DNA, where local minima in groove width have the potential to create electrostatically negative binding sites for basic side chains (Joshi et al., 2007; Yang et al., 2017). As with all other Hox-Exd-DNA structures, the four ternary structures solved here follow this rule (Figures 1A–1D). However, the AbdB NTAs in all four structures share a nearly identical path in the minor groove that is distinct from the path taken by Scr’s NTA when bound to either fkh250 or fkh250con (Figure 2A). Moreover, the AbdB NTA trajectory is nearly identical to that observed for the NTA of its vertebrate ortholog, HoxA9 in complex with Pbx (Figure 2A). Although all seven ternary structures (four AbdB, one HoxA9, two Scr) are similar up to the insertion of Arg5 in the minor groove, the Scr NTAs diverge N-terminal to this residue (Figure 2B). One likely reason for this difference is that Thr6 of Scr makes a direct H-bond with the phosphate backbone of the DNA, thus pulling the NTA close to the DNA backbone (Figure 2C). In both AbdB and HoxA9, this residue is a lysine and is unable to make this contact, leading to an alternative conformation of the NTA. Consistently, the NTA of Ubx, which has a Gln at position 6, has a similar conformation as observed for AbdB (Figure S3).
Figure 2. Distinct NTA Conformations of Anterior and Posterior Hox Proteins.

(A)Comparison of NTA conformations seen in four AbdB-Exd complexes and two Scr-Exd complexes.
(B)Same as (A) but showing only AbdB-Exd (red) and both Scr NTAs, highlighting the shared insertion of Arg5, which inserts into the minor groove at the same position in all of these Hox-Exd complexes.
(C)Close-up of AbdB-Exd (red) and Scr-Exd (fkh) NTAs showing that Thr6 of Scr, but not Lys6 of AbdB, makes a hydrogen bond with the phosphate backbone.
(D)Sequence alignment of Hox W-motifs and homeodomains, with position 6 highlighted in red.
(E) Data from Slattery et al. (2011), showing the selection of sequences with two minor groove width minima for anterior Hox proteins (Lab, Pb, Dfd, Scr) and only a single minor groove width minimum for posterior Hox proteins (Antp, Ubx, AbdA, AbdB). All Hox proteins preferring a second minimum have a threonine at position 6 (see D).
These findings, together with previous SELEX-seq data generated with Scr NTA mutants (Abe et al., 2015), highlight the importance of residues 4 and 6 for determining the overall conformation of the Hox NTA and, consequently, DNA binding site preferences. Notably, the NTA conformation in Scr, due to the Thr6-DNA backbone interaction, facilitates the insertion of Arg3 into the second minor groove width minimum present in fkh250 (Figure 2C) (Joshi et al., 2007). We hypothesize that the alternative conformation of AbdB’s NTA, due to differences at positions 4 and 6, makes this Hox protein less able to recognize minor groove width minima and, more generally, less sensitive to DNA shape compared to more anterior Hox proteins such as Scr.
High-Affinity Sites Have More Optimal Shapes Prior to Binding
Although the variations in NTA trajectories described above likely contribute to differences in DNA recognition by anterior (e.g., Scr) compared to posterior (e.g., AbdB) Hox proteins, they fail to account for differences in affinity that AbdB-Exd has for the red, magenta, blue, and black binding sites. Due to the role that DNA shape has in conferring both specificity and affinity for previously analyzed Hox-Exd-DNA complexes, we next turned our attention to DNA shape differences in the four ternary structures solved here. We used Curves+ (Blanchet et al., 2011) to analyze DNA shape in the four X-ray structures and the DNA-Shape tool (Zhou et al., 2013) to predict the intrinsic DNA shapes of the DNA sequences in the absence of protein binding. Due to its importance in other contexts, we focused on minor groove width.
Consistent with the overall similarities of the four AbdB-Exd-DNA structures, including the paths of their NTAs in the minor groove, the minor groove width profiles are very similar in all four ternary complexes (Figures 3A and 3D). Most strikingly, a prominent minor groove width minimum occurs at position 7 of the binding site, regardless of the DNA sequence. The side chain of AbdB’s Arg5 inserts into the minor groove at this position. Arg5 also inserts into a local minimum of minor groove width in both Scr-Exd-DNA complexes (Joshi et al., 2007). A second smaller minimum is observed at position 10 in the blue and black structures and, to a lesser extent, in the red structure, but not in the magenta structure (Figure 3D).
Figure 3. Differences in DNA Shape in the Absence and Presence of Protein Binding.

(A and B) Overlapping and highly similar DNA structures in all four crystal structures (A), and the definition of locations for measurements of minor groove width (B).
(c) Predicted minor groove width profiles for the red and magenta (top) and blue and black (bottom) binding sites. The position of AbdB Arg5 (R5) insertion in the crystal structures is indicated.
(D) Observed minor groove width profiles for the red and magenta (top) and blue and black (bottom) binding sites present in the crystal structures. The position of AbdB Arg5 (R5) insertion in the crystal structures is indicated.
In contrast to the similar minor groove width profiles seen in the four crystal structures, DNAShape predicts very different profiles for these four DNA sequences in the absence of bound protein. Most notably, the red and magenta sequences, which have the highest affinity for AbdB-Exd, have minor groove width profiles that match well with the profiles seen in the crystal structures, in particular, the minimum at position 7 is observed (Figure 3C). In contrast, this minimum is not observed in either the black or blue sequences (Figure 3C). Moreover, the black sequence, which has the lowest affinity of the four sequences for AbdB-Exd, has a local maximum of minor groove width at position 7. In addition, the weak minimum observed at position 10 in the blue and black crystal structures is predicted to be even narrower in the absence of protein binding. Based on these calculations, we conclude that the lower affinity blue and black DNA sequences significantly change their conformations upon binding to AbdB-Exd. In contrast, the red and magenta sequences appear to be preconfigured with the correct shape, likely decreasing the energetic barrier to binding.
An Additional Binary Complex in the Black DNA Crystal Supports a Role for Intrinsic DNA Shape in Determining Affinity
The asymmetric unit of the black crystal structure contains, in addition to the AbdB-Exd-black ternary complex (referred to as blackF, for black Forward), an additional binary complex in which the AbdB homeodomain is bound to DNA without Exd (Figures 4A–4C). Interestingly, compared to the AbdB-Exd dimer, the AbdB homeodomain binds on the opposite face of the DNA and in the opposite orientation, using the binding site 5´-ATTTAT (referred to as blackR, for black Reverse) (Figures 4A–4C). In general, AbdB binds blackR in a manner that is typical of homeodomain-DNA binary structures, with its third recognition helix making hydrogen bonds in the major groove and NTA in the minor groove (Figures 4C and S2). Moreover, as with all other homeodomain-DNA structures, the side chain of Arg5 inserts into the minor groove. Residues N-terminal to Arg5, such as Lys3, could not be modeled in the binary complex, consistent with the notion that interaction with Exd contributes to the stabilization of the NTA. Notably, when AbdB binds to this binding site, a cognate Exd half site (usually, 5ˊ-TGAT) is not available, and monomeric AbdB binding to the blackR site precludes the binding of AbdB-Exd in the blackF orientation.
Figure 4. Comparison between the Ternary and Binary Complexes in the Black Crystal.

(A–C) Overview of the entire asymmetric unit (A), only the ternary complex (B), and only the binary complex (C) in the black crystal. DNA sequences recognized by each complex are below the structures; blackF refers to the binding site used in the ternary complex (black), and blackR refers to the binding site used in the binary complex (green). AbdB Arg5 is in orange (B and C).
(D) Comparison of the DNA structures present in the blackR and blackF complexes.
(E) Minor groove width profiles of blackF (top) and blackR (bottom). In both cases, the predicted minor groove widths are indicated (dashed lines), as is the position of AbdB Arg5 (R5).
The presence of this alternative binding mode raises the question of how an AbdB monomer can successfully compete for binding with an AbdB-Exd heterodimer. Upon closer inspection, we noticed that AbdB’s Arg5 in the binary complex is inserted into the pre-existing minor groove width minimum at position 10 of the black binding site (Figure 4E). Consequently, the shape of the black DNA differs in the binary and ternary complexes (Figure 4D). We speculate that this preformed minor groove width minimum at position 10 creates a monomeric binding site that is sufficiently favorable to compete with the weak, conformationally sub-optimal, heterodimer binding site on the opposite strand.
Additional Features
In addition to the features described above, we highlight three additional features that may contribute to affinity differences between AbdB-Exd and cognate binding sites.
An Additional Contact between AbdB and Exd
The sequences and structures C-terminal to the third α helices of Hox homeodomains differ. In the red and magenta AbdB-Exd structures reported here, two additional residues extend the third recognition helix of the homeodomain (Figure 5A). This feature is also observed for HoxA9 and Ubx (Figure 5A). Further, in the AbdB-Exd red structure, which includes the highest affinity binding site, we identify a hydrogen bond between Lys58 of AbdB and Ser43 of Exd (Figures 5B and 5C). Significantly, this contact is also observed in the HoxA9-Pbx structure but is not observed when AbdB-Exd binds to the magenta sequence, which is also a high-affinity site, or in any of the lower affinity AbdB-Exd complexes.
Figure 5. Other Notable Interactions.

(A) Residues C-terminal to AbdB (red), Ubx (purple), and HoxA9 homeodomains extend the third helix to different extents. Scr’s C terminus (cyan) diverges from the helix, although this may in part be because only one additional residue is present.
(B) AbdB-Exd (red) has a hydrogen bond between Lys58 of AbdB and Ser43 of Exd. The fourth helix of Exd can also be observed folding back against the Exd homeodomain.
(C) The Lys58-Ser43 contact is observed in both AbdB-Exd (red) and HoxA9-Pbx structures
(D) In the AbdB-Exd (red) complex, Arg3 of Exd’s NTA inserts into the minor groove. This insertion is not observed for Exd’s NTA in the Scr-Exd complexes or for Pbx’s NTA in the HoxA9-Pbx complex (LaRonde-LeBlanc and Wolberger, 2003)..
Exd NTA-Minor Groove Interactions
Like Hox NTAs, Exd’s NTA has several basic residues, raising the possibility that these side chains also have the ability to insert into the DNA minor groove. For three of the ternary structures described here, red, magenta, and black, the Exd NTA is well-ordered, and Arg3 can be seen inserting into the minor groove (Figure 5D). This contact is not observed in the HoxA9-Pbx structure or either Scr-Exd structure.
Exd Helix 4
The Exd homeodomain ends at residue 300 of the full-length Exd sequence. All four of our crystals contain ten extra residues following the homeodomain. These residues have been shown to be disordered in solution but form an α-helix upon binding to DNA (PDB: 1PUF [LaRonde-LeBlanc and Wolberger, 2003], PDB: 1LFU [Sprules et al., 2003], PDB: 1DU6 [Sprules et al., 2000]). These residues are highly conserved among Exd orthologs, (Exd, Pbx, Ceh-20) and have been shown to increase affinity to DNA and Hoxproteins (Greenetal.,1998;LuandKamps,1996). Consistent with these previous observations, the four ternary complexes described here also show clear electron density after the end of the Exd homeodomain that is consistent with a helical conformation (Figure 5B). Although some differences areapparent (Figure S4), all four structures show this helix folding back to contact the Exd homeodomain. As a consequence, this helix may help to stabilize the recognition helix (Sprules et al., 2003) and to deepen the binding pocket for the W motif, which in turn could influence DNA binding (see Discussion). However, this additional helix is unlikely to contribute to differences in affinity because it is present in all four ternary complexes.
DISCUSSION
Intrinsic DNA Shape Predicts Binding Affinity
By comparing the X-ray crystal structures of four ternary complexes of AbdB-Exd bound to subtly different DNA binding sites, we have identified a mechanism for how differences in affinity can be encoded in DNA sequences. As discussed above, although there are a number of structural differences in the crystal structures (Figure 1), these may in part be a consequence of the different resolutions of the structures, where in general fewer side chains can be modeled in lower resolution structures. Moreover, in the red (highest affinity) complex we observed a hydrogen bond between Lys58 of AbdB and Ser43 of Exd that was not seen in any of the other AbdB-Exd complexes. Interestingly, this interaction is also present in the HoxA9-Pbx complex (LaRonde-LeBlanc and Wolberger, 2003), suggesting that this contact contributes to the affinity (and/or stabilization) of these complexes. However, because this hydrogen bond was not observed in the magenta complex, which also has a high-affinity for AbdB-Exd, the presence or absence of this contact is unlikely to play a general role in determining differences in affinity. Thus, although it remains possible that subtle structural differences contribute to these relative affinities, we found no features in the protein-protein or protein-DNA contacts in the ternary complexes that could readily account for the existence of two high-affinity and two lower affinity sites.
In contrast, we found that the four DNA sequences compared here are predicted to have distinct shapes prior to protein binding, and these differences in shape correlate well with affinity. Specifically, the two highest affinity binding sites, red and magenta, are predicted to have DNA shapes—in particular minor groove width profiles—that match well with the DNA shapes present in the crystal structures, when bound to AbdB-Exd. In contrast, the two lower affinity binding sites, blue and black, have minor groove width profiles that are very different from those observed in the crystal structures, with the lowest affinity black sequence having a shape that is most distinct from the protein-bound shape. Thus, our results can be explained in a straightforward way if we posit that affinity differences are a consequence of intrinsic differences in DNA shape prior to protein binding. It would be of interest to obtain further support for this model by calculating conformational energies for each of the relevant structures but we are not aware of any method that can reliably calculate conformational energies of DNA to the level of accuracy required to provide a quantitative correlation. However, the DNAshape method is based on Monte Carlo calculations and has been very effective in predicting minor groove widths (Azad et al., 2018; Bishop et al., 2011; Chiu et al., 2015; Joshi et al., 2007; Rohs et al., 2009, 2010; Zhou et al., 2013), leaving little doubt that, compared to the two lower affinity sequences, the two high-affinity sites have free DNA structures that are closer to those seen in their respective ternary complexes.
NTA Trajectory Differences between Anterior and Posterior Hox Proteins
In previous work using the SELEX-seq assay (Abe et al., 2015; Slattery et al., 2011), we found that anterior Hox proteins generally prefer DNA sequences with two minor groove width minima (see also Figure 2E). This contrasts with more posterior Hox proteins, which prefer DNA sequences with only a single minor groove width minimum. The minimum that is shared by all Hox binding sites allows insertion of Arg5, present in all Hox NTAs, while the anterior Hox-specific minimum allows insertion of Arg3 of Scr. Here, by comparing the NTAs of all available Hox structures (AbdB, HoxA9, Ubx, Scr, HoxB1), we find an additional striking difference between anterior and posterior Hox proteins. In particular, the NTA conformations in the minor groove differ between anterior and posterior Hox proteins (Figures 2A and S3). These differences appear to depend on different residues at position 6 of the NTA: Scr has a Thr in this position, while AbdB (and HoxA9) has a Lys in this position. Thr6 of Scr makes a hydrogen bond with the phosphate backbone, thus altering its NTA path in the minor groove (Figure 2C). We hypothesize that this difference in conformation is critical in allowing other basic side chains (e.g., Arg3) to insert into the minor groove. These observations therefore provide an explanation for why some posterior Hox proteins, such as Abdominal-A (AbdA) and Ubx, also have an Arg at position 3, yet fail to prefer sequences that have a second minor groove minimum (Abe et al., 2015; Slattery et al., 2011): the different NTA paths are either poised (in the case of anterior Hox proteins) or not (in the case of posterior Hox proteins) to correctly position the Arg3 side chain in the minor groove.
Role of Sequences C-Terminal to Hox and Cofactor Homeodomains
Although the classical homeodomain was defined, based on homology, as a 60-amino acid domain, subfamilies of homeodomains have additional conserved residues that are frequently adjacent and C-terminal to the classically defined homeodomain (Burglin and Affolter, 2016). For example, Ubx, AbdA, and many of their orthologs share a conserved motif known as UbdA, which is C-terminal and adjacent to the homeodomain. The UbdA motif has been shown to contribute to DNA binding affinity (Lelli et al., 2011; Saadaoui et al., 2011). Consistent with that view, in a recent Ubx-Exd X-ray structure the UbdA motif of Ubx extends the third alpha helix of the Ubx homeodomain and lies close to the third helix of Exd (Foos et al., 2015). Although the sequences of Ubx and AbdB differ in this region, we also observe that the third helix of AbdB’s homeodomain is extended by several residues in the red and magenta complexes. Moreover, the potential contacts seen in the Ubx structure (Foos et al., 2015) are in a similar position to the contact between Lys58 of AbdB and Ser43 of Exd that we observe in the red complex. Taken together, these observations support the conclusion that, in addition to the W-motif-TALE interaction, other direct contacts between Hox proteins and PBC cofactors contribute to complex formation and stability.
Members of the PBC family, which includes the Hox cofactors Exd and Pbx, also have an additional ~10 highly conserved residues C-terminal to the homeodomain. In the HoxA9-Pbx and HoxB1-Pbx crystal structures, this region of Pbx formed a fourth α-helix that folds back and packs against the rest of the Pbx homeodomain (LaRonde-LeBlanc and Wolberger, 2003; Piper et al., 1999). Although the degree to which Exd forms an α-helix differs between the four complexes described here, the analogous part of Exd is also observed folding back and packing against the rest of the Exd homeodomain, suggesting that this is a conserved feature of PBC proteins. The fourth helix may help to stabilize the Exd homeodomain and, given its proximity, may also help to stabilize the interaction with the HEWT motif.
Structural Insights into Posterior Dominance
Posterior dominance is a phenomenon in which posterior Hox proteins phenotypically dominate over anterior ones when they are co-expressed (Bachiller et al., 1994; Duboule, 1991; LaRonde-LeBlanc and Wolberger, 2003; Noro et al., 2011). Several of the structural features uncovered here may be relevant to this phenomenon. As noted above, the posterior Hox proteins AbdB, HoxA9, and Ubx have an extended α-helix (e.g., the UbdA motif) following their homeodomains and the potential for additional contacts with Exd/Pbx (e.g., the H-bond between Lys58 of AbdB and Ser43 of Exd). These additional contacts and helices have not been observed in any anterior Hox ternary complex and raise the possibility that additional Hox-cofactor interactions may allow posterior Hox proteins to have a higher affinity for some binding sites compared to anterior Hox proteins. Consistent with this hypothesis, the posterior Hox protein AbdA requires its UbdA motif to phenotypically dominate over the anterior Hox protein Scr (Noro et al., 2011).
Conclusions
Taken together, these observations suggest that the differences in affinity for the four AbdB-Exd binding sites characterized here are not primarily a consequence of different or additional protein-protein or protein-DNA contacts in the final ternary complex. Instead, they point to differences in intrinsic DNA shape, which are more or less favorable for binding, that determine differences in affinity. We hypothesize that the mechanism uncovered here may prove to be a general way that affinity differences are encoded in DNA sequences, especially for highly related members of transcription factor families that, like subfamilies of homeodomain proteins, share many of the same DNA-contacting residues.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification
The proteins used in the crystallizations were His-tagged at their N terminus and included the following residues:
- AbdB (residues 146–229 of isoform A, NP_650577.1):
- VGPCTPNPGLHEWTGQVSVRKKRKPYSKFQTLELEKEFLFNAYVSKQKRWELARNLQLTERQVKIWFQNRRMKNKKNSQRQANQ
- Homeodomain is in italics and W motif is in bold.
- Exd (residues 237–310 of isoform A, AAF48555.1):
- DARRKRRNFSKQASEILNEYFYSHLSNPYPSEEAKEELARKCGITVSQVS NWFGNKRIRYKKNIGKAQEEANLY
Homeodomain is in italics, and the TALE (three amino acid loop extension) motif is underlined.
The coding sequences were cloned using a combination of BP and LR GATEWAY cloning methods. The pENTR-TEV-AbdB and Exd constructs cloned using BP cloning method (Invitrogen) were verified by sequencing and transformed into destination vectors containing N-terminal His6Mbp.
A first batch of proteins (batch I) was used to grow the red and blue crystals; a second batch (batch II) to grow the magenta and black crystals. The buffers used for purification of both batches differed slightly and are specified in Table S4.
For batch I, the BL21 (DE3) were grown at 37°C until OD 0.6, induced with 0.2 mM IPTG and harvested after growing them overnight at 18°C. For batch II, the BL21 (DE3) were grown at 37°C until OD 0.6, induced with 0.5 mM IPTG and harvested after 4 hr at 37°C.
The fusion His6-Mbp-Tev- AbdB and the fusion His6-Mbp-Tev- Exd proteins were purified by Ni affinity chromatography using the equilibration and elution buffers specified in Table S4. The eluted fusion protein was incubated with TEV protease in a 1:100 ratio (TEV:fusion protein) overnight at 4°C or at room temperature for 6 hr. Batch II underwent another Ni affinity chromatography step to remove uncleaved protein and cleaved tags. The protein of interest was further purified using ion exchange column (Resource S for batch I, HiTrap SP for batch II). For batch I, pure protein obtained after gel filtration (S200) and was concentrated and stored at 80°C. Batch II was buffer exchanged directly to storage buffer after ion exchange purification (Table S4) and then frozen.
Complex Formation and Crystallization
In the case of red and blue crystals, PAGE purified blunt ended complementary DNA strands were mixed in equimolar ratio in the presence of MgCl2 (10 mM Tris pH:8.0, 10 mM MgCl2) and annealed. In the case of magenta and black, DNA was annealed and buffer exchanged to the same storage buffer as the protein of batch II, containing 50 mM MgCl2, to avoid precipitation (see Table S4), and incubated on ice. AbdB, Exd homeodomains, and DNA were mixed in 1:1:1.2 ratio at a concentration of 400:400:480 mM and incubated on ice for at least 1 hr before setting up crystal plates. Crystallization conditions were identified by sparse matrix screening, and the optimized crystallization conditions for all the complexes are provided in Table S1. All crystals were cryo protected with 20%–30% glycerol and flash frozen in liquid nitrogen.
The sequences of the DNAs used in crystallization conditions of the diffracting crystals are shown in Table S1.
Data Collection and Integration
Diffraction data were collected on the NE-CAT beamline at the Advanced Photon Source (APS) at Argonne National Laboratory (Argonne, IL). Collected images for red, blue, and magenta datasets were processed by RAPD (rapid automated processing of X-ray data, https://github.com/RAPD/RAPD) using the XDS software package (Kabsch, 1993). Black dataset was processed manually with iMosflm and merged, scaled, and truncated to 2.4 Å using Scala from the CCP4 software package (Collaborative Computational Project, Number 4, 1994; Winn et al., 2011). Data were processed to 2.4, 2.9, 3.03, and 2.4 Å for red, blue, magenta, and black complexes, respectively. Space groups were found to be C121 (C2) for red, blue, and magenta complex and P1 for the black complex (Table S2).
Structure Solution and Refinement
A polyalanine-substituted 2.4 Åresolution structure of Ubx/Exd/DNA domain (1B8I) was used as the initial model for molecular replacement using PHASER within the PHENIX suite of programs (McCoy et al., 2007). A unique solution was obtained and either phenix.refine or REFMAC was used to calculate electron density maps (Adams et al., 2004,2010; Afonine et al., 2005). Initial model building was done manually in COOT using both the 2Fo-Fc and the Fo-Fcmaps. 5% of the reflections were marked for Rfree determination throughout the refinement cycles (Collaborative Computational Project, Number 4, 1994). The final model of the AbdB-Exd structure was refined using the PHENIX suite of programs. CCP4 programs were used for analyzing the structure and PyMol was used for generating figures. A summary of refinement statistics is included in Table S2. Structures have been submitted to the Protein Data Bank with the following identifiers: red, 5ZJQ; magenta, 5ZJR; blue, 5ZJS; and black, 5ZJT.
Composite Omit Maps
The composite omit map 2mFo-DFc of the AbdB-Exd (red) complex electron density map was calculated using PHENIX with the anneal mode. This clearly showed the density of NTA region of AbdB in AbdB-Exd (red) complex structure.
To further remove bias, another composite omit map with anneal mode 2mFo-DFc was calculated using PHENIX after deleting the DNA coordinates of the red, blue, magenta, and black DNA bound to AbdB-Exd.
The composite omit maps with anneal mode are displayed in gray mesh contoured at 1.0 σ.
Competition EMSAs
EMSAs and protein purification for EMSAs were performed as described previously (Kribelbauer et al., 2017; Slattery et al., 2011). The constructs used for EMSAs also correspond to those described previously, instead of the isolated homeodomain constructs that were used for crystallization. His-tagged, full-length Exd was co-purified with the HM domain of Hth (Noro et al., 2006). The AbdB construct corresponds to residues 224-C terminus of isoform B (identical to full-length isoform A, residues 1–270) and thus also includes more than the residues used for crystallization (Slattery et al., 2011). The oligomers used are listed in Table S5.
IC50 values were calculated using ImageJ for quantification and the Python package scipy.optimize for fitting (curve_fit). The function used for the fitting was: PL = M*IC50/(IC50 + U), where PL is the percentage of labeled red probe bound in the AbdB-Exd/HM complex, U is the total concentration of unlabeled competitor probe (red, magenta, blue or black) in the well, and M is a normalization constant corresponding to the maximum percentage of bound complex in the absence of competitor (intercept term). M and IC50 were treated as free parameters to be optimized.
Supplementary Material
Highlights.
The X-ray crystal structures of four Hox-cofactor-DNA complexes were solved
Although affinity to DNA differs up to 20-fold, the ternary structures are very similar
The predicted intrinsic DNA shapes of the binding sites correlate with affinity
Hox N-terminal arm paths in the minor groove correlate with DNA shape readout
ACKNOWLEDGMENTS
We thank Judith Kribelbauer for help with protein binding assays and Gorka Cabrero, Oliver Harrison, and Alina Sergeeva for help with crystallography software and analysis. This work was supported by NIH (R01GM050291 to A.G.P., R01GM30518 to B.H., and R35GM118336 and R01GM054510 to R.S.M.).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the four structures reported in this paper are PDB: 5ZJQ (red), 5ZJS (blue), 5ZJR (magenta), and 5ZJT (black).
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.100
REFERENCES
- Abe N, Dror I, Yang L, Slattery M, Zhou T, Bussemaker HJ, Rohs R, and Mann RS (2015). Deconvolving the recognition of DNA shape from sequence. Cell 161, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Gopal K, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Pai RK, Read RJ, Romo TD, et al. (2004). Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron Radiat 11, 53–55. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkó czi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, and Adams PD (2005). A robust bulksolvent correction and anisotropic scaling procedure. Acta Crystallogr. D Biol. Crystallogr 61, 850–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad RN, Zafiropoulos D, Ober D, Jiang Y, Chiu TP, Sagendorf JM, Rohs R, and Tullius TD (2018). Experimental maps of DNA structure at nucleotide resolution distinguish intrinsic from protein-induced DNA deformations. Nucleic Acids Res. 46, 2636–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachiller D, Macías A, Duboule D, and Morata G (1994). Conservation of a functional hierarchy between mammalian and insect Hox/HOM genes. EMBO J. 13, 1930–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop EP, Rohs R, Parker SC, West SM, Liu P, Mann RS, Honig B, and Tullius TD (2011). A map of minor groove shape and electrostatic potential from hydroxyl radical cleavage patterns of DNA. ACS Chem. Biol 6, 1314–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet C, Pasi M, Zakrzewska K, and Lavery R (2011). CURVES+ web server for analyzing and visualizing the helical, backbone and groove parameters of nucleic acid structures. Nucleic Acids Res. 39, W68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürglin TR, and Affolter M (2016). Homeodomain proteins: an update. Chromosoma 125, 497–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu TP, Yang L, Zhou T, Main BJ, Parker SC, Nuzhdin SV, Tullius TD, and Rohs R (2015). GBshape: a genome browser database for DNA shape annotations. Nucleic Acids Res. 43, D103–D109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 (1994). The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Crocker J, Abe N, Rinaldi L, McGregor AP, Frankel N, Wang S, Alsawadi A, Valenti P, Plaza S, Payre F, et al. (2015). Low affinity binding site clusters confer hox specificity and regulatory robustness. Cell 160, 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker J, Noon EP, and Stern DL (2016). The soft touch: low-affinity transcription factor binding sites in development and evolution. Curr. Top. Dev. Biol 117, 455–469. [DOI] [PubMed] [Google Scholar]
- Duboule D (1991). Patterning in the vertebrate limb. Curr. Opin. Genet. Dev 1, 211–216. [DOI] [PubMed] [Google Scholar]
- Farley EK, Olson KM, and Levine MS (2015a). Regulatory principles governing tissue specificity of developmental enhancers. Cold Spring Harb. Symp. Quant. Biol 80, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, and Levine MS (2015b). Suboptimization of developmental enhancers. Science 350, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foos N, Maurel-Zaffran C, Maté MJ, Vincentelli R, Hainaut M, Berenger H, Pradel J, Saurin AJ, Ortiz-Lombardía M, and Graba Y (2015). A flexible extension of the Drosophila ultrabithorax homeodomain defines a novel Hox/PBC interaction mode. Structure 23, 270–279. [DOI] [PubMed] [Google Scholar]
- Green NC, Rambaldi I, Teakles J, and Featherstone MS (1998). A conserved C-terminal domain in PBX increases DNA binding by the PBX homeodomain and is not a primary site of contact for the YPWM motif of HOXA1. J. Biol. Chem 273, 13273–13279. [DOI] [PubMed] [Google Scholar]
- Guertin MJ, and Lis JT (2010). Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet. 6, e1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi R, Passner JM, Rohs R, Jain R, Sosinsky A, Crickmore MA, Jacob V, Aggarwal AK, Honig B, and Mann RS (2007). Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell 131, 530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (1993). Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr 26, 795–800. [Google Scholar]
- Kribelbauer JF, Laptenko O, Chen S, Martini GD, Freed-Pastor WA, Prives C, Mann RS, and Bussemaker HJ (2017). Quantitative analysis of the DNA methylation sensitivity of transcription factor complexes. Cell Rep. 19, 2383–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRonde-LeBlanc NA, and Wolberger C (2003). Structure of HoxA9 and Pbx1 bound to DNA: Hox hexapeptide and DNA recognition anterior to posterior. Genes Dev. 17, 2060–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli KM, Noro B, and Mann RS (2011). Variable motif utilization in homeotic selector (Hox)-cofactor complex formation controls specificity. Proc. Natl. Acad. Sci. USA 108, 21122–21127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, and Kamps MP (1996). Structural determinants within Pbx1 that mediate cooperative DNA binding with pentapeptide-containing Hox proteins: proposal for a model of a Pbx1-Hox-DNA complex. Mol. Cell. Biol 16, 1632–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony S, and Pugh BF (2015). Protein-DNA binding in high-resolution. Crit. Rev. Biochem. Mol. Biol 50, 269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merabet S, and Mann RS (2016). To be specific or not: the critical relationship between Hox and TALE proteins. Trends Genet. 32, 334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noro B, Culi J, McKay DJ, Zhang W, and Mann RS (2006). Distinct functions of homeodomain-containing and homeodomain-less isoforms encoded by homothorax. Genes Dev. 20, 1636–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noro B, Lelli K, Sun L, and Mann RS (2011). Competition for cofactor-dependent DNA binding underlies Hox phenotypic suppression. Genes Dev. 25, 2327–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passner JM, Ryoo HD, Shen L, Mann RS, and Aggarwal AK (1999). Structure of a DNA-bound Ultrabithorax-Extradenticle homeodomain complex. Nature 397, 714–719. [DOI] [PubMed] [Google Scholar]
- Piper DE, Batchelor AH, Chang CP, Cleary ML, and Wolberger C (1999). Structure of a HoxB1-Pbx1 heterodimer bound to DNA: role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell 96, 587–597. [DOI] [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, and Honig B (2009). The role of DNA shape in protein-DNA recognition. Nature 461, 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R, Jin X, West SM, Joshi R, Honig B, and Mann RS (2010). Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem 79, 233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadaoui M, Merabet S, Litim-Mecheri I, Arbeille E, Sambrani N, Damen W, Brena C, Pradel J, and Graba Y (2011). Selection of distinct Hox-Extradenticle interaction modes fine-tunes Hox protein activity. Proc. Natl. Acad. Sci. USA 108, 2276–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, Zhou T, Rohs R, Honig B, Bussemaker HJ, and Mann RS (2011). Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell 147, 1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Zhou T, Yang L, Dantas Machado AC, Gordân R, and Rohs R (2014). Absence of a simple code: how transcription factors read the genome. Trends Biochem. Sci 39, 381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprules T, Green N, Featherstone M, and Gehring K (2000). Conformational changes in the PBX homeodomain and C-terminal extension upon binding DNA and HOX-derived YPWM peptides. Biochemistry 39, 9943–9950. [DOI] [PubMed] [Google Scholar]
- Sprules T, Green N, Featherstone M, and Gehring K (2003). Lock and key binding of the HOX YPWM peptide to the PBX homeodomain. J. Biol. Chem 278, 1053–1058. [DOI] [PubMed] [Google Scholar]
- Weinberg RL, Veprintsev DB, and Fersht AR (2004). Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol 341, 1145–1159. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Orenstein Y, Jolma A, Yin Y, Taipale J, Shamir R, and Rohs R (2017). Transcription factor family-specific DNA shape readout revealed by quantitative specificity models. Mol. Syst. Biol 13, 910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Yang L, Lu Y, Dror I, Dantas Machado AC, Ghane T, Di Felice R, and Rohs R (2013). DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 41, W56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.