

Abstract
Background:
A high consumption of fructose leads to hepatic steatosis. About 20–30% of triglycerides are synthesized via de novo lipogenesis. Some studies showed that endoplasmic reticulum stress (ERS) is involved in this process, while others showed that a lipotoxic environment directly influences ER homeostasis. Here, our aim was to investigate the causal relationship between ERS and fatty acid synthesis and the effect of X-box binding protein-1 (XBP-1), one marker of ERS, on hepatic lipid accumulation stimulated by high fructose.
Methods:
HepG2 cells were incubated with different concentrations of fructose. Upstream regulators of de novo lipogenesis (i.e., carbohydrate response element-binding protein [ChREBP] and sterol regulatory element-binding protein 1c [SREBP-1c]) were measured by polymerase chain reaction and key lipogenic enzymes (acetyl-CoA carboxylase [ACC], fatty acid synthase [FAS], and stearoyl-CoA desaturase-1 [SCD-1]) by Western blotting. The same lipogenesis-associated factors were then evaluated after exposure of HepG2 cells to high fructose followed by the ERS inhibitor tauroursodeoxycholic acid (TUDCA) or the ERS inducer thapsigargin. Finally, the same lipogenesis-associated factors were evaluated in HepG2 cells after XBP-1 upregulation or downregulation through cell transfection.
Results:
Exposure to high fructose increased triglyceride levels in a dose- and time-dependent manner and significantly increased mRNA levels of SREBP-1c and ChREBP and protein levels of FAS, ACC, and SCD-1, concomitant with XBP-1 conversion to an active spliced form. Lipogenesis-associated factors induced by high fructose were inhibited by TUDCA and induced by thapsigargin. Triglyceride level in XBP-1-deficient group decreased significantly compared with high-fructose group (4.41 ± 0.54 μmol/g vs. 6.52 ± 0.38 μmol/g, P < 0.001), as mRNA expressions of SREBP-1c (2.92 ± 0.46 vs. 5.08 ± 0.41, P < 0.01) and protein levels of FAS (0.53 ± 0.06 vs. 0.85 ± 0.05, P = 0.01), SCD-1 (0.65 ± 0.06 vs. 0.90 ± 0.04, P = 0.04), and ACC (0.38 ± 0.03 vs. 0.95 ± 0.06, P < 0.01) decreased. Conversely, levels of triglyceride (4.22 ± 0.54 μmol/g vs. 2.41 ± 0.35 μmol/g, P < 0.001), mRNA expression of SREBP-1c (2.70 ± 0.33 vs. 1.00 ± 0.00, P < 0.01), and protein expression of SCD-1 (0.93 ± 0.06 vs. 0.26 ± 0.05, P < 0.01), ACC (0.98 ± 0.09 vs. 0.43 ± 0.03, P < 0.01), and FAS (0.90 ± 0.33 vs. 0.71 ± 0.02, P = 0.04) in XBP-1s-upregulated group increased compared with the untransfected group.
Conclusions:
ERS is associated with de novo lipogenesis, and XBP-1 partially mediates high-fructose-induced lipid accumulation in HepG2 cells through augmentation of de novo lipogenesis.
Keywords: Endoplasmic Reticulum Stress, Fatty Liver, Lipogenesis, X-Box Binding Protein-1
摘要
背景:
高果糖易导致肝脏的脂质沉积,其中20-30%的甘油三酯来源于脂质从头合成。有的研究证实内质网应激(Endoplasmic reticulum stress,ERS)参与脂质沉积的过程,而有的研究发现高脂环境可诱发ERS。因此本研究的目的旨在深入探讨ERS和脂 质合成之间的关系,及ERS的重要因子X-盒结合蛋白-1(X-box binding protein-1,XBP-1)在高果糖诱发的脂质沉积中的作用。
方法:
首先收集不同浓度的果糖干预处理后的HepG2细胞,应用PCR的方法测定其脂质从头合成的上游调控因子碳水化合物反 应元件结合蛋白(carbohydrate response element-binding protein,ChREBP) 和(sterol regulatory element-binding protein 1c,SREBP- 1c),应用Western方法测定了脂质从头合成的三个关键酶乙酰-CoA 羧化酶(acetyl-CoA carboxylase,ACC)、脂肪酸合酶 (fatty acid synthase,FAS)、硬脂酰-CoA 去饱和酶(stearoyl-CoA desaturase-1,SCD-1)。然后HepG2 细胞中加入ERS诱导 剂(thapsigargin),或是ERS抑制剂牛磺熊去氧胆酸 (tauroursodeoxycholic acid,TUDCA)加入到高果糖培养的HepG2 细胞 中,再通过上述方法测定脂质从头合成的相关因子的表达水平。最后,采用细胞转染技术分别上调和下调XBP-1后,测定脂 质从头合成的相关因子的表达。
结果:
高果糖以时间和浓度依赖性增加细胞内脂质沉积,增加细胞内SREBP-1c和ChREBP的mRNA的表达水平,及 FAS、ACC和SCD-1 的蛋白表达水平,并促进了XBP-1转化为活性形式。而且这一过程可以被ERS的抑制剂TUDCA所抑制, 相反,ERS的诱导剂却促进脂质从头合成。下调XBP-1组甘油三酯的水平较高果糖组明显下降(4.41±0.54 μmol/g vs. 6.52±0.38 μmol/g, P <0.001),且脂质从头合成的上游调控因子SREBP-1c的mRNA (2.92±0.46 vs. 5.08±0.41, P <0.01)和FAS(0.53±0.06 vs. 0.85±0.05, P=0.01)、SCD-1(0.65±0.06 vs. 0.90±0.04, P=0.04) ACC(0.38±0.03 vs. 0.95±0.06, P<0.01)的蛋白水平明显下降。 与之相反的是上调XBP-1s组的甘油三酯的水平较未转染组却明显升高(4.22±0.54 μmol/g vs. 2.41±0.35 μmol/g, P<0.001),且上 游调控因子SREBP-1c的mRNA (2.70±0.33 vs. 1.00±0.00, P<0.01) 和SCD-1(0.93±0.06 vs. 0.26±0.05, P<0.01)、ACC(0.98±0.09 vs. 0.43±0.03, P<0.01)、FAS(0.90±0.33 vs. 0.71±0.02,P=0.04)的蛋白表达水平明显增加。
结论:
ERS与脂质从头合成有关,且XBP-1在高果糖诱导的HepG2细胞的脂质沉积的发挥着一定的作用,可能是与上调了脂 质从头合成有关。
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a common liver disease that has become a major health concern worldwide.[1] NAFLD, characterized by excessive lipid accumulation in the liver, is a hepatic manifestation of metabolic syndrome and includes liver disorders ranging from simple steatosis to nonalcoholic steatohepatitis with liver dysfunction.[2] Increased consumption of high-fructose foods, such as ice cream, candy, and bread, is an important pathogenic factor for hepatic steatosis.[3,4] High-fructose flux leads to enhanced hepatic triglyceride (TG) accumulation, which plays a central role in the emergence of NAFLD.[5,6,7,8] Nevertheless, the pathophysiology underlying this process is poorly understood. Some studies have shown that endoplasmic reticulum stress (ERS) signaling pathways contribute to the development of insulin resistance and hepatic steatosis in NAFLD.[9,10,11,12] X-box binding protein-1 (XBP-1) is a major transcription regulator of the unfolded protein response (UPR), mediating adaptation to ERS.[13] Here, we hypothesized that XBP-1 plays a role in the process of fructose-stimulated lipogenesis in HepG2 cells.
Hepatic steatosis has been associated with fructose, a food additive, because excessive consumption of fructose can increase hepatic lipid accumulation.[14] Fatty acids come from three sources: (1) lipolysis of TGs stored in adipose tissue, (2) uptake of dietary fatty acids from the intestinal tract, and (3) de novo lipogenesis (newly synthesized from glucose) in the liver, the last of which accounts for about 20–30% of all fatty acids in hepatocytes.[15] Previous studies in rodents have shown that fructose can facilitate de novo lipogenesis.[16,17,18] Feeding rats with fructose increased hepatic levels of upstream regulators of de novo lipogenesis (i.e., sterol regulatory element-binding protein 1c [SREBP-1c] and carbohydrate response element-binding protein [ChREBP]) and key lipogenic enzymes (i.e., fatty acid synthase [FAS], acetyl-CoA carboxylase [ACC], and stearoyl-CoA desaturase-1 [SCD-1]). To verify these findings at a cellular level, in the present study, we incubated HepG2 cells with fructose or palmitic acid (PA) to investigate their effects on different sources of accumulated lipid.
ERS refers to an accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER) lumen under conditions of impaired function, leading to an adaptive signaling response originating in the ER, namely the UPR.[13,19] Three pathways are involved in ERS: (1) the inositol-requiring enzyme-1 (IRE-1)/XBP-1 pathway, (2) the protein kinase R-like ER kinase (PERK)/eukaryotic translation initiation factor-2α (eIF-2α) pathway, and (3) the activating transcription factor-6 pathway.[13] Recent studies have demonstrated that ERS is associated with the initiation and progression of many diseases, such as metabolic dyslipidemia, insulin resistance, NAFLD, cardiovascular disease, and neurodegenerative disease.[20] Some studies have reported that misfolded proteins alter ER homeostasis, creating a lipotoxic environment within hepatocytes,[21] but other studies have demonstrated that the lipotoxic environment of NAFLD directly influences ER homeostasis and ERS activation.[22,23,24] Thus, a link between ERS and lipid accumulation has been proposed, but the exact nature of the causal relationship between ERS and fatty acid synthesis remains unknown. In this study, we identified the factors associated with de novo lipogenesis after culturing HepG2 cells with: (1) high fructose, (2) high fructose followed by the ERS inhibitor tauroursodeoxycholic acid (TUDCA), or (3) the ERS inducer thapsigargin.
XBP-1, also known as cAMP-response element-binding protein, belongs to a family of basic leucine zipper-containing proteins and can be found in two forms: unspliced XBP-1 (XBP-1u) and spliced XBP-1 (XBP-1s). XBP-1 is normally kept in its inactive form, but under ERS, the endoRNase domain of IRE-1 splices the mRNA of downstream sensor XBP-1, removing a 26-bp segment from the full-length XBP1 mRNA that generates a translational frameshift, leading to the expression of the active protein XBP-1s.[25,26,27] XBP-1s binds to intranuclear mRNA directly to regulate protein transcription, thereby affecting subsequent physiological activities.[28,29] Lee et al.'s study[30] showed for the first time that XBP-1 regulates hepatic lipid metabolism, because XBP1-knockout mice manifested hypotriglyceridemia and hypocholesterolemia. On the basis of these observations, we propose that XBP-1 serves as a key conduit for ERS-induced hepatic lipid accumulation and steatosis in response to a high-fructose stimulus. Thus, in the present study, we investigated the expression of the transcription factor XBP-1 in the human HepG2 cell line after stimulation of cells with high fructose, and the regulatory effects of XBP-1 on de novo lipogenesis in the initial stages of NAFLD by evaluating the expression of key enzymes involved in lipogenesis.
METHODS
Reagents and chemicals
Reagents: rabbit anti-SCD-1, anti-ACC, anti-IRE-1, anti-phosphorylated (p-) IRE-1, and anti-XBP-1s antibodies (Cell Signaling Technology, Beverly, MA, USA); thapsigargin (Abcam, Cambridge, UK); mouse anti-β-actin antibody (SAB Bioengineering Institute, College Park, Maryland, USA); anti-FAS antibody, goat anti-mouse secondary antibody, XBP-1 short hairpin (sh) RNA plasmid (human, sc-38627-SH) and control shRNA plasmid-A (sc-108060; Santa Cruz Biotechnology, Santa Cruz, CA, USA); and PA and fructose (Sigma Chemical, St. Louis, MO, USA). TG levels were determined using a commercially available kit (Pulilai Bioengineering Institute, Changchun, China). The ERS inhibitor TUDCA was obtained from Sichuan Hengtai Biotechnology (Sichuan, China). The plasmids pcDNA 3.1-XBP-1u and pcDNA 3.1-XBP-1s were gifts from Dr. Hao Jun (Hebei Medical University, Shijiazhuang, Hebei, China). HepG2 cells were from Bumrungrad Biomedical Technology (HUCL-0085; Jiangyin, Jiangsu, China).
Cell treatment groups
HepG2 cells were prepared with different stimulations as follows:
To investigate the effects of high fructose on lipid accumulation induced by fructose, HepG2 cells were stimulated with 0, 1, 5, or 20 mmol/L fructose for 12, 24, 48, or 72 h.
To elucidate the underlying mechanisms, HepG2 cells were treated with 20 mmol/L fructose or 0.25 mmol/L PA for 72 h.
To explore the causal relationships between ERS and lipogenesis, the ERS inhibitor TUDCA (0.2 mmol/L) was added after HepG2 cells were cultured with 20 mmol/L fructose for 24 h, and other HepG2 cells were cultured with the ERS inducer thapsigargin (600 nmol/L) for 10 h (without fructose pretreatment).
To investigate the immediate effects of XBP-1 on lipid accumulation and whether XBP-1 mediates high-fructose-induced lipid metabolism, XBP-1 expression was downregulated using cell transfection with an shRNA targeting XBP-1, and the active form XBP-1s was upregulated using cell transfection with vector pcDNA 3.1-XBP-1s.
After the different stimulations, HepG2 cells were harvested for TG measurement and Oil Red O staining. Metabolic factors involved in lipogenesis (i.e., FAS, SCD-1, and ACC) were detected using Western blotting analysis, and gene expression of the lipogenic pathway regulators SREBP-1c and ChREBP was evaluated using polymerase chain reaction (PCR).
Transient transfection
For cell transient transfection, Lipofectamine 2000 was used. Briefly, HepG2 cells were cultured in 6-well plates. XBP-1 plasmids or empty vectors transduced into HepG2 cells. Then, cells were transfected with 0.8 g vector DNA using 2 μl Lipofectamine 2000 in 2 ml serum-free Dulbecco's modified Eagle's medium (DMEM) per well. After 8 h, the medium was replaced with 10% fetal bovine serum DMEM. Cells were harvested 48 h after transfection and subsequently harvested for determination.
Determination of triglyceride levels in HepG2 cells
After the different treatments described above, HepG2 cells were collected to wash twice with phosphate-buffered saline (PBS) and then treated with radioimmunoprecipitation (RIPA) buffer for 30 min on ice. Samples were centrifuged at 13,000 ×g for 20 min at 4°C for getting the supernatant. The concentration of protein was measured using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). TG levels were measured based on an enzymatic assay from a method adapted for 96-well plates according to the manufacturer's instructions (Pulilai Bioengineering Institute).
Oil Red O staining
Cultured cells were fixed with 4% paraformaldehyde (in PBS) for 30 min and stained with 1% Oil Red O for 2 h. The stained sections were observed with an Olympus microscope and examined by a pathologist in a blinded manner.
Real-time polymerase chain reaction
Analyses on gene transcript levels were conducted by quantitative real-time polymerase chain reaction (PCR) method. After washing HepG2 cells twice with PBS, total mRNA was extracted using a standard TRIzol RNA isolation method[7] after stimulation as described above. The concentration and purity of total RNA were measured using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). RNA was reverse transcribed using the Easy Script First-Strand cDNA Synthesis Super Mix kit (TransGen Biotech, Beijing, China). Real-time PCR was performed on an ABI PRISM 7300 PCR System (Applied Biosystems, Foster City, CA, USA) using SYBR Green I GoTaq qPCR Master Mix (Promega, Madison, WI, USA). PCRs were performed in a total volume of 25 μl as follows: 95°C for 5 min, then 40 cycles of 95°C for 15 s, 58°C for 20 s, and 72°C for 30 s in the end. Each sample's gene expression was analyzed in duplicate and normalized against that of GAPDH used as an internal control. Results are expressed as relative gene expression, determined using the comparative threshold cycle (CT) method and normalized by GAPDH as an internal control.
The primers were: GAPDH: forward 5’-GGATGATGTTCTGGAGAGCC-3’ and reverse 5’-CATCACCATCTTCCAGGAGC-3’; SREBP1c: forward 5’-CTTCCGCCCTTGAGCTG-3’ and reverse 5’-CTGGTGTGTCCGTGTGG-3’; and CHREBP: forward 5’-TGCGGGATGAGATTGAGGA-3’ and reverse 5’-TCCAGTTGTGCAGCGTAC-3’.
Western blotting analysis
Proteins were extracted from HepG2 cells which were cultured in 25 cm-bottom-surface bottle. Cells were washed by 4°C PBS for 2 times firstly. Then, cells were added with 1 ml RIPA and 10 μl PMSF. The cells were collected with a cell scraper blow gently, after which in a centrifuge tube. Up steps were operated on ice. Clumps of cells were placed in 4000 ×g centrifugation at 4°C for 30 min. Supernatant of 800 μl mixture was kept at −80°C. The concentration of protein was measured using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Proteins were mixed with sodium dodecyl sulfate (SDS)-loading buffer and boiled for 10 min. Then, protein samples were separated using 10% SDS-polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride membrane, and blocked with nonfat dry milk for 2 h at 37°C. Then, membranes were probed with antibodies (primary antibodies against SCD-1, ACC, IRE-1, p-IRE-1, XBP-1s, FAS, XBP-1u, PERK, p-eIF-2α, eIF-2α, and β-actin) overnight at 4°C. Subsequently, membranes were rinsed three times with TBST, after which they were incubated with secondary antibodies at room temperature for 2 h. After incubation, membranes were washed three times with TBST and followed by detection with electrochemiluminescence method. And, β-actin was used as an internal control.
Statistical analysis
All data are represented as the mean ± standard deviation (SD) and were analyzed. Two-tailed unpaired Student's t-tests were used for between-group comparisons. The analysis of variance multiple comparison test (SPSS 11.0, SPSS Inc., Chicago, IL, USA) followed by Tukey's post hoc test was used for comparisons among groups. A value of P < 0.05 was considered statistically significant.
RESULTS
Effects of fructose on de novo lipogenesis in HepG2 cells
The lipotoxic effects of fructose in hepatocytes were investigated by culturing HepG2 cells in the presence of normal medium (N group) or medium containing different concentrations of fructose (1, 5, or 20 mmol/L; F1, F5, and F20 groups, respectively) for different times (12, 24, 48, or 72 h). Fructose increased TG levels in HepG2 cells in a time- and concentration-dependent manner. TG levels reached maximal value after 72 h incubation with 20 mmol/L fructose; thus, 20 mmol/L fructose was used in subsequent experiments [Figure 1a]. Oil red O staining showed an obvious increase in lipid droplets in HepG2 cells cultured with 20 mmol/L fructose compared with those cultured with other fructose concentrations [Figure 1b]. PA increased lipid accumulation in the liver by providing substrate for TG synthesis. To explore the different mechanisms by which fructose induced lipid accumulation, HepG2 cells were cultured in the presence of 20 mmol/L fructose or 0.2 mmol/L PA. As expected, compared with HepG2 cells incubated with PA, high (20 mmol/L) fructose increased the mRNA expression of the two upstream transcription factors SREBP1c and CHREBP [Figure 2b, all P < 0.01] and the protein contents of the three downstream enzymes ACC, FAS, and SCD-1 [Figure 2c, all P < 0.01]. However, TG levels in HepG2 cells were not significantly different between groups [Figure 2a].
Figure 1.
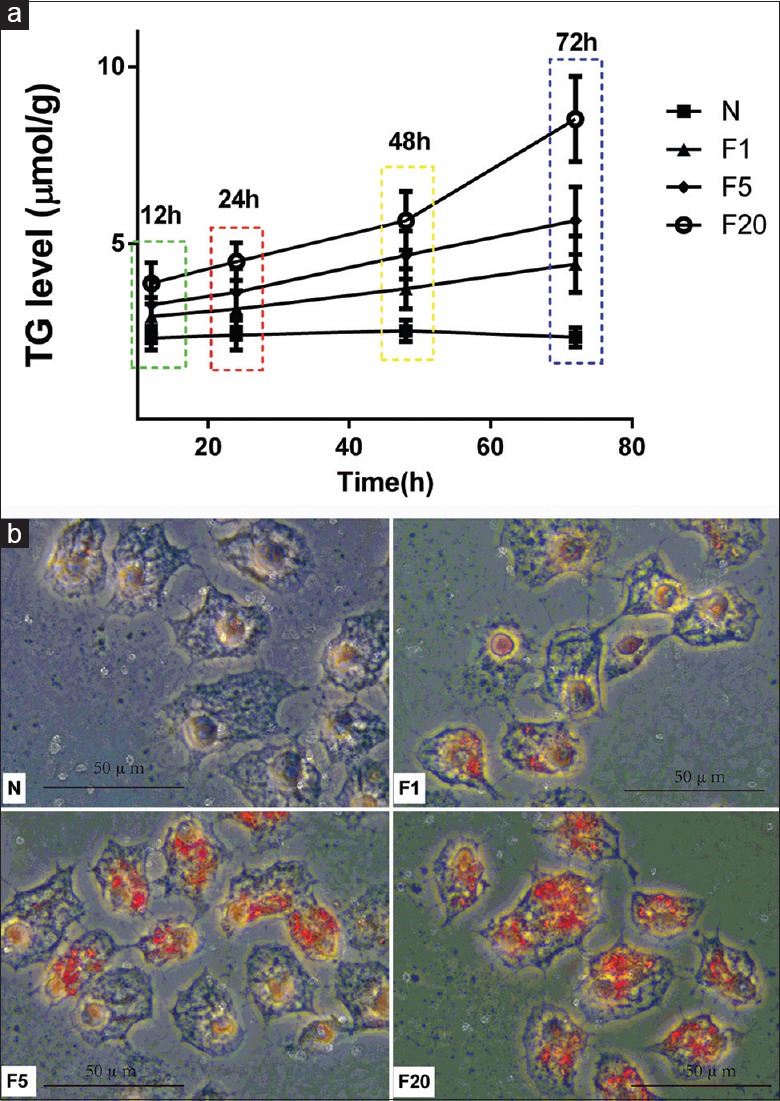
High fructose increased lipid accumulation in HepG2 cells. (a) TG was determined after incubated in different concentrations of fructose in different times. (b) Lipid droplets were increased in HepG2 cells cultured with 20 mmol/L fructose most obviously as shown by Oil red O staining after incubating 72 h. HepG2 cells were divided into four groups with different concentration of fructose, separately named as N (cultured with normal medium), F1 (cultured with 1 mmol/L fructose), F5 (cultured with 5 mmol/L fructose), and F20 (cultured with 20 mmol/L fructose). Scale bar = 50 μm. TG: Triglycerides.
Figure 2.

High fructose accelerated lipogenesis. (a) TG level in HepG2 cells of F and PA groups were significantly increased compared to N group. No change was observed between F and PA groups after incubation (n = 3), *P < 0.05 versus N group. (b) Levels of SREBP-1c, ChREBP mRNA were tested by PCR. Fold induction represents relative expression compared to that of control group. The mRNA levels of upstream transcriptional factors SREBP-1c and ChREBP in lipogenesis pathway were significantly increased in HepG2 cells in F group compared to N group, but they were lower in PA group compared to F group (n = 6), *P < 0.05 versus N group, †P < 0.05 versus F group. (c) Protein expression of critical enzymes (SCD-1, FAS, and ACC) in the lipogenic pathway in HepG2 cells were tested by Western blotting. β-actin expression is a loading control. Key enzymes of lipogenesis ACC, FAS, and SCD-1 protein levels significantly increased in HepG2 cells of F group compared to N group, and they were lower in PA group compared to F group (n = 3), *P < 0.05 versus N group, †P < 0.05 versus F group. Cells cultured with normal medium, 20 mmol/L fructose, or 0.2 mmol/L PA were assigned into N, F, or PA group respectively. Data are shown as mean ± standard deviation. TG: Triglycerides; SREBP-1c: Sterol regulatory element-binding protein 1c; ChREBP: Carbohydrate response element-binding protein; SCD-1: Stearoyl-CoA desaturase-1; FAS: Fatty acid synthase; ACC: Acetyl-CoA carboxylase; PCR: Polymerase chain reaction.
Effects of tauroursodeoxycholic acid and thapsigargin on lipogenesis
To determine the causal relationship between ERS and lipid metabolism, HepG2 cells were cultured with ordinary medium, 20 mmol/L fructose alone, 20 mmol/L fructose plus 0.2 mmol/L TUDCA, or 600 nmol/L thapsigargin alone. The fructose-induced increase in TG level and Oil Red O straining was ameliorated by TUDCA treatment of HepG2 cells (P < 0.01), whereas thapsigargin increased TG levels and Oil Red O straining [Figure 3a and 3c, P < 0.01]. Furthermore, TUDCA pretreatment decreased the fructose-induced increases in SREBP1c, CHREBP (all P < 0.01), and SCD-1, FAS, and ACC expression (all P < 0.05), whereas thapsigargin increased the expression of all factors Figure 3b and 3d]. TUDCA pretreatment of HepG2 cells blocked fructose-induced phosphorylation of IRE-1, as well as activation of XBP-1 [Figure 3e, P < 0.05]. Compared with HepG2 cells incubated with normal medium, those incubated with thapsigargin showed an increase in this arm of the UPR [Figure 3e, P < 0.05].
Figure 3.

The effect of selective ERS inhibitor-TUDCA and inducer-Tg on IRE/XBP-1 pathway and lipogenesis. (a) TG level was significantly lower in TUDCA group than F group but increased in Tg group compared to N group (n = 3), *P < 0.05 versus N group, †P < 0.05 versus F group. (b) SREBP-1c and ChREBP were significantly lower in F+ TUDCA group compared to F group and were significantly increased in Tg group compared to N group (n = 6), *P < 0.05 versus N group, †P < 0.05 versus F group. (c) Oil Red O straining was alleviated in F+ TUDCA group compared to F group and was sharpened in Tg group compared to N group. Scale bar = 50 μm. (d) Protein levels of three key enzymes (SCD-1, FAS, and ACC) were upregulated in F group compared to N group but were suppressed by TUDCA. They were significantly increased in Tg group compared to N group (n = 3), *P < 0.05 versus N group, †P < 0.05 versus F group. (e) Protein expression of p-IRE-1/IRE-1 and XBP-1s were significantly increased in HepG2 cells in F group compared to N group. They were lower in F+ TUDCA group compared to F group, but increased in Tg group (n = 3), *P < 0.05 versus N group, †P < 0.05 versus F group. Cells cultured with normal medium, 20 mmol/L fructose, 20 mmol/L fructose plus 0.2 mmol/L TUDCA, or 600 nmol/L thapsigargin were assigned into N, F, F+TUDCA, or Tg group respectively. Data are shown as mean ± standard deviation. TUDCA: Tauroursodeoxycholic acid; Tg: Thapsigargin; TG: Triglycerides; SREBP-1c: Sterol regulatory element-binding protein 1c; ChREBP: Carbohydrate response element-binding protein; SCD-1: Stearoyl-CoA desaturase-1; FAS: Fatty acid synthase; ACC: Acetyl-CoA carboxylase.
Effects of silencing X-box binding protein-1 expression on lipid deposition in HepG2 cells
In this series of experiments, the level of the active form XBP-1 (XBP-1s) was decreased after transfecting cells with an shRNA targeting XBP-1 [Figure 4a, P < 0.01]. Triglyceride level in XBP-1 shRNA group decreased significantly compared with high-fructose group [Figure 4b and 4c, 4.41 ± 0.54 μmol/g vs. 6.52 ± 0.38 μmol/g, P < 0.001]. Expression of the upstream lipogenic transcription factors SREBP-1c and CHREBP mRNA was measured by PCR. SREBP-1c was significantly downregulated in XBP-1 shRNA group (2.92 ± 0.46 vs. 5.08 ± 0.41, P < 0.01), but CHREBP was not [Figure 4d]. The protein contents of the key downstream lipogenic enzymes FAS (0.53 ± 0.06 vs. 0.85 ± 0.05, P = 0.01), SCD-1 (0.65 ± 0.06 vs. 0.90 ± 0.04, P = 0.04), and ACC (0.38 ± 0.03 vs. 0.95 ± 0.06, P < 0.01) were significantly decreased in XBP-1-deficient HepG2 cells [Figure 4e].
Figure 4.

Lipid accumulation and expression of critical enzymes in the lipogenic pathway in HepG2 cells were decreased after transfection with XBP-1s shRNA. (a) Protein expression level of XBP-1s 8 h after transfection decreased significantly (n = 3), *P < 0.05 versus N group, †P < 0.05 versus F group. (b) TG induced by high fructose was decreased after transfection of XBP-1s shRNA (n = 3), *P < 0.01 versus N group, †P < 0.01 versus F group. (c) Oil Red O straining was alleviated in F+ XBP-1s shRNA group. Scale bar = 50 μm. (d) Level of SREBP-1c and ChREBP mRNA were tested by PCR in different groups (n = 6), *P < 0.05 versus untransfection group, †P < 0.05 versus F group. (e) Expression of critical enzymes in the lipogenic pathway proteins (SCD-1, FAS, and ACC) in HepG2 cells in different groups were tested by Western blotting, β-Actin expression is a loading control. Fold induction represents relative expression level compared to that of control group (n = 3), *P < 0.05 versus untransfection group, †P < 0.05 versus F group. Cells cultured with normal medium, 20 mmol/L fructose, 20 mmol/L fructose followed by transfection with empty vector, or 20 mmol/L fructose followed by transfection with shRNA targeting XBP-1 were assigned into N, F, F+NC, or F+XBP-1 shRNA group respectively. Data are shown as mean ± standard deviation. TG: Triglycerides; SREBP-1c: Sterol regulatory element-binding protein 1c; ChREBP: Carbohydrate response element-binding protein; SCD-1: Stearoyl-CoA desaturase-1; FAS: Fatty acid synthase; ACC: Acetyl-CoA carboxylase; PCR: Polymerase chain reaction.
Effects of spliced X-box binding protein-1 overexpression on lipid synthesis in HepG2 cells
To further investigate the causal relationship between XBP-1 and de novo lipogenesis, active XBP-1s was overexpressed by transfection with a XBP-1s vector. As shown by Western blotting analysis [Figure 5a], XBP-1s protein expression was induced after transfection for 8 h with vector pcDNA 3.1-XBP-1s, but not with vector cDNA 3.1-XBP-1u (P < 0.01). Cellular TG accumulation was higher in HepG2 cells overexpressing XBP-1s than in those cultured in ordinary medium or transfected with empty vector or vector pcDNA 3.1-XBP-1u [Figure 5b and 5c], 4.22 ± 0.54 μmol/g vs. 2.41 ± 0.35 μmol/g, P < 0.001]. Quantitative real-time PCR analysis showed a significant increase in SREBP1c mRNA expression in HepG2 cells transfected with XBP-1s upregulation vector (2.70 ± 0.33 vs. 1.00 ± 0.00, P < 0.01), but CHREBP mRNA showed no change in expression [Figure 5d]. XBP-1s upregulation also significantly increased SCD-1 (0.93 ± 0.06 vs. 0.26 ± 0.05, P < 0.01), ACC (0.98 ± 0.09 vs. 0.43 ± 0.03, P < 0.01), and FAS (0.90 ± 0.33 vs. 0.71 ± 0.02, P = 0.04) protein content compared with the untransfected group [Figure 5e].
Figure 5.

Expressions of critical enzymes in the lipogenic pathway were increased in XBP-1s overexpressed HepG2 cells. (a) Protein expression level of XBP-1s increased after transfection, *P < 0.05 versus untransfection group, †P < 0.05 versus pcDNA3.1(+)-XBP-1u group. (b) TG increased after transfection of XBP-1s plasmid, (n = 3), *P < 0.05 versus untransfection group. Scale bar = 50 μm. (c) Oil Red O straining was aggravated after XBP-1s upregulation. Scale bar = 50 μm. (d) Level of SREBP-1c, ChREBP mRNA was tested by PCR in different groups, and they were induced in pcDNA 3.1-XBP-1s group (n = 6), *P < 0.05 versus untransfection group. (e) Expression of critical enzymes in the lipogenic pathway proteins (SCD-1, FAS, and ACC) in HepG2 cells in different groups was tested by Western blotting inclined compared to that of control group (n = 3), *P < 0.05 versus untransfection group. Cells cultured with normal medium, transfecting with empty vector pcDNA3.1(+), vector pcDNA3.1(+)-XBP-1u or vector pcDNA3.1(+)-XBP-1s were assigned into untransfection, pcDNA3.1(+), pcDNA3.1(+)-XBP-1u, or pcDNA3.1(+)-XBP-1s group respectively. Data are shown as mean ± standard deviation. TG: Triglycerides; SREBP-1c: Sterol regulatory element-binding protein 1c; ChREBP: Carbohydrate response element-binding protein; SCD-1: Stearoyl-CoA desaturase-1; FAS: Fatty acid synthase; ACC: Acetyl-CoA carboxylase; PCR: Polymerase chain reaction.
DISCUSSION
Chronic fructose consumption is considered as a contributing factor in the development of NAFLD.[31] There is now a great interest in the mechanism by which high fructose induces fatty liver. Acquired and accumulated excessive TGs in hepatocytes may be considered as the first step of the “two hits” in the progression of NAFLD.[32] Although considerable progress has been made in understanding the molecular mechanisms underlying hepatic steatosis,[33] satisfactory treatment modalities remain limited. The liver is the predominant organ in the body delegated to de novo lipogenesis. This process, in which fatty acids are newly synthesized from acetyl coenzyme A (converted from glucose through the tricarboxylic acid cycle), usually contributes about one-third of all TG in hepatocytes, but this contribution increases by approximately 6-fold in the abnormal state.[34] Two key transcription factors, SREBP-1c and ChREBP, and three key target enzymes, ACC, FAS, and SCD-1, participate in the lipogenesis process. In the present study, by determining TG levels and by histologic analysis, we demonstrated that fructose increased lipid accumulation in HepG2 cells in a dose-dependent manner. This study also demonstrated that factors associated with de novo TG synthesis (i.e., ACC, FAS, SCD-1, SREBP-1c, and ChREBP) were increased in HepG2 cells incubated with high fructose (20 mmol/L), compared with those incubated with PA (a saturated free fatty acid). Consequently, the findings suggest that chronic intake of fructose induces lipid accumulation through lipogenesis.
The UPR is an adaptive signaling pathway triggered in response to perturbations in ER homeostasis, conditions referred to as ERS.[13,19] Recent animal and human studies have revealed on the one hand that obesity and fatty liver are associated with ERS-associated factors[21,35,36] and that these diseases can be attenuated by chemical chaperone 4-phenylbutyric acid, Schisandra chinensis extract, or gastric bypass surgery.[21,37,38] On the other hand, lipotoxic stress leads to ERS activation in the liver,[4] so the causal relationship between ERS and lipogenesis remains to be clarified. Whereas these previous studies were based on human and animal experiments, the present study examined the role of ERS in lipogenesis at a cellular level. To investigate the role of ERS in fructose-induced lipogenesis, we first tested whether the putative chemical chaperone TUDCA (an ERS inhibitor) protected against experimental ERS in HepG2 cells cultured in the presence of fructose. Pretreatment of HepG2 cells with TUDCA suppressed fructose-induced ERS (IRE-1 and XBP-1) and de novo lipogenesis (mRNA expression of SREBP-1c and ChREBP and protein content of ACC, SCD-1, and FAS). Conversely, thapsigargin (an ERS inducer) increased the expression of PERK, eIF2α, IRE-1, XBP-1, and factors involved in lipogenesis. Although other studies have demonstrated that ERS may interfere with insulin signaling, inhibit insulin action in liver, and thereby affect lipid accumulation,[20] this study provides evidence for a direct effect of ERS on lipogenesis. On this basis, we speculate that there may be a positive feedback loop between ERS and lipogenesis, and therefore that reducing ERS is a key to reducing lipogenesis and reversing NAFLD.
IRE1/XBP-1 is one of the three classic ERS pathways associated with growth, differentiation, and cellular apoptosis.[13] Recent studies in animals and cells have shed light on the role of XBP-1 in numerous diseases, such as obesity, insulin resistance, and metabolic syndrome.[39] XBP-1 protein expression in mice was elevated after mice were fed carbohydrates, and deletion of XBP-1 resulted in marked hypocholesterolemia and hypotriglyceridemia.[30] Fatty liver induced by a high-calorie diet could be protected against by decreasing XBP-1.[12] The 3T3-L1 cells with XBP1 or IRE1α knockdown and XBP1-deficient mouse embryonic fibroblasts showed significantly weakened adipogenesis.[40] Further studies showed that selectively knocking down the XBP1 gene in the liver decreased critical genes involved in fatty acid synthesis.[30] Our study showed that in the presence of high fructose (20 mmol/L), there was significant upregulation of XBP-1s (active form) and downregulation of XBP-1u (inactive form). To elucidate the underlying mechanisms, we investigated hepatic regulation of TG synthesis by activating XBP-1. XBP-1s overexpression increased cellular TG accumulation, which was accompanied by increased mRNA expression of SREBP-1c and protein content of three key target enzymes associated with lipogenesis, ACC, FAS, and SCD-1, but not by a change in ChREBP mRNA expression. Again, silencing of XBP-1s expression effectively prevented against the lipid deposition in HepG2 cells induced by high fructose, by decreasing the transcriptional expression of de novo lipogenesis genes to alleviate further lipotoxic stress. Therefore, we explored the causal relationship between XBP-1s and lipid accumulation and found that high fructose-induced cellular lipid deposition in NAFLD was partially regulated by XBP-1s.
SREBP-1c and ChREBP are mediators of the transcriptional effects of lipogenic enzyme genes. Lipogenic genes such as FAS, ACC, and SCD-1 are activated by both SREBP-1c and ChREBP. SREBP-1c mainly targets glucokinase, the first enzyme of the glycolytic pathway, as well as FAS and ACC.[41] SREBPs are synthesized as inactive precursors bound to the membranes of the ER and thus must undergo proteolytic cleavage to liberate their N-terminal domain, which constitutes the mature transcription factor. ERS could induce cleavage of the precursor form of SREBP-1c (increasing the expression of the mature form of SREBP-1c in the nucleus) and expression of SREBP-1c target genes independent of insulin, but the role of XBP-1 is unclear.[42] Lee et al.'s research shows that the genetic deletion of XBP-1 in liver leads to a decrease in de novo lipid synthesis.[30] Our research indicates that XBP-1 is associated with SREBP-1c without ChREBP, but further experiment need to be done to research the exact mechanism. However, our study convinced that the high fructose accelerates ERS to enhance de novo lipogenesis.
Our study had several limitations. First, we investigated the effect of XBP-1 on lipogenesis in vitro; rodent experiments are needed to verify our results in vivo. Second, ERS includes three pathways, and research has shown that transcription factor-4 activation contributes to lipid accumulation; thus, XBP-1 may be only partially responsible for ERS aggravation.
The present study provides novel insight into the mechanisms involved in fructose-mediated hepatic hypertriglyceridemia and identifies ERS as a potential therapeutic target for NAFLD. Furthermore, we show that XBP-1 plays a critical role in de novo lipogenesis during NAFLD.
To sum up, exposure of HepG2 cells to fructose resulted in increased hepatic lipid deposition via acceleration of de novo lipogenesis compared with PA exposure. In addition, the findings suggest that XBP-1 is crucial for lipid synthesis. Thus, using compounds that selectively target XBP-1 may be beneficial in the treatment of NAFLD.
Financial support and sponsorship
This work was supported by a grant from the National Natural Science Funds of China (No. 81200639).
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We thank Hao Jun (Department of Pathology, Hebei Medical University, Shijiazhuang, China) for XBP-1s and XBP-1u vectors. We wish to express our the deepest thanks and appreciation to the staff of Department of Hebei Key Laboratory of Metabolic Disease for their support of this research.
Footnotes
Edited by: Qiang Shi
REFERENCES
- 1.Schattenberg JM, Schuppan D. Nonalcoholic steatohepatitis: The therapeutic challenge of a global epidemic. Curr Opin Lipidol. 2011;22:479–88. doi: 10.1097/MOL.0b013e32834c7cfc. doi: 10.1097/MOL.0b013e32834c7cfc. [DOI] [PubMed] [Google Scholar]
- 2.Postic C, Girard J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 2008;34:643–8. doi: 10.1016/S1262-3636(08)74599-3. doi: 10.1016/S1262-3636(08)74599-3. [DOI] [PubMed] [Google Scholar]
- 3.Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, et al. Nonalcoholic fatty liver disease: Predisposing factors and the role of nutrition. J Nutr Biochem. 2007;18:184–95. doi: 10.1016/j.jnutbio.2006.12.006. doi: 10.1016/j.jnutbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–64. doi: 10.1038/nrgastro.2010.41. doi: 10.1038/nrgastro.2010.41. [DOI] [PubMed] [Google Scholar]
- 5.Johnson RJ, Perez-Pozo SE, Sautin YY, Manitius J, Sanchez-Lozada LG, Feig DI, et al. Hypothesis: Could excessive fructose intake and uric acid cause type 2 diabetes? Endocr Rev. 2009;30:96–116. doi: 10.1210/er.2008-0033. doi: 10.1210/er.2008-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101:7281–6. doi: 10.1073/pnas.0401516101. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sevastianova K, Santos A, Kotronen A, Hakkarainen A, Makkonen J, Silander K, et al. Effect of short-term carbohydrate overfeeding and long-term weight loss on liver fat in overweight humans. Am J Clin Nutr. 2012;96:727–34. doi: 10.3945/ajcn.112.038695. doi: 10.3945/ajcn.112.038695. [DOI] [PubMed] [Google Scholar]
- 8.Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I, et al. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology. 2009;50:1094–104. doi: 10.1002/hep.23122. doi: 10.1002/hep.23122. [DOI] [PubMed] [Google Scholar]
- 9.Jang MK, Yun YR, Kim SH, Kim JH, Jung MH. Protective effect of gomisin N against endoplasmic reticulum stress-induced hepatic steatosis. Biol Pharm Bull. 2016;39:832–8. doi: 10.1248/bpb.b15-01020. doi: 10.1248/bpb.b15-01020. [DOI] [PubMed] [Google Scholar]
- 10.Passos E, Ascensão A, Martins MJ, Magalhães J. Endoplasmic reticulum stress response in non-alcoholic steatohepatitis: The possible role of physical exercise. Metabolism. 2015;64:780–92. doi: 10.1016/j.metabol.2015.02.003. doi: 10.1016/j.metabol.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and oxidative stress in the pathogenesis of non-alcoholic fatty liver disease. Free Radic Res. 2015;49:1405–18. doi: 10.3109/10715762.2015.1078461. doi: 10.3109/10715762.2015.1078461. [DOI] [PubMed] [Google Scholar]
- 12.Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell. 2008;15:829–40. doi: 10.1016/j.devcel.2008.10.015. doi: 10.1016/j.devcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 14.Choi Y, Abdelmegeed MA, Song BJ. Diet high in fructose promotes liver steatosis and hepatocyte apoptosis in C57BL/6J female mice: Role of disturbed lipid homeostasis and increased oxidative stress. Food Chem Toxicol. 2017;103:111–21. doi: 10.1016/j.fct.2017.02.039. doi: 10.1016/j.fct.2017.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–51. doi: 10.1172/JCI23621. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren LP, Song GY, Hu ZJ, Zhang M, Peng L, Chen SC, et al. The chemical chaperon 4-phenylbutyric acid ameliorates hepatic steatosis through inhibition of de novo lipogenesis in high-fructose-fed rats. Int J Mol Med. 2013;32:1029–36. doi: 10.3892/ijmm.2013.1493. doi: 10.3892/ijmm.2013.1493. [DOI] [PubMed] [Google Scholar]
- 17.Love S, Mudasir MA, Bhardwaj SC, Singh G, Tasduq SA. Long-term administration of tacrolimus and everolimus prevents high cholesterol-high fructose-induced steatosis in C57BL/6J mice by inhibiting de-novo lipogenesis. Oncotarget. 2017;8:113403–17. doi: 10.18632/oncotarget.15194. doi: 10.18632/oncotarget.15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura K, Fukunishi S, Yokohama K, Ohama H, Tsuchimoto Y, Asai A, et al. A long-lasting dipeptidyl peptidase-4 inhibitor, teneligliptin, as a preventive drug for the development of hepatic steatosis in high-fructose diet-fed ob/ob mice. Int J Mol Med. 2017;39:969–83. doi: 10.3892/ijmm.2017.2899. doi: 10.3892/ijmm.2017.2899. [DOI] [PubMed] [Google Scholar]
- 19.Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- 20.Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339:172–7. doi: 10.1126/science.1230721. doi: 10.1126/science.1230721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 22.Flamment M, Kammoun HL, Hainault I, Ferré P, Foufelle F. Endoplasmic reticulum stress: A new actor in the development of hepatic steatosis. Curr Opin Lipidol. 2010;21:239–46. doi: 10.1097/MOL.0b013e3283395e5c. doi: 10.1097/MOL.0b013e3283395e5c. [DOI] [PubMed] [Google Scholar]
- 23.Lee JS, Mendez R, Heng HH, Yang ZQ, Zhang K. Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. Am J Transl Res. 2012;4:102–13. [PMC free article] [PubMed] [Google Scholar]
- 24.Lake AD, Novak P, Hardwick RN, Flores-Keown B, Zhao F, Klimecki WT, et al. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol Sci. 2014;137:26–35. doi: 10.1093/toxsci/kft230. doi: 10.1093/toxsci/kft230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–6. doi: 10.1038/415092a. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 26.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, et al. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16:452–66. doi: 10.1101/gad.964702. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 28.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 29.Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 30.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–6. doi: 10.1126/science.1158042. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond) 2005;2:5. doi: 10.1186/1743-7075-2-5. doi: 10.1186/1743-7075-2- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eslam M, George J. Genetic and epigenetic mechanisms of NASH. Hepatol Int. 2016;10:394–406. doi: 10.1007/s12072-015-9689-y. doi: 10.1007/s12072-015-9689-y. [DOI] [PubMed] [Google Scholar]
- 33.Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:360–9. doi: 10.1055/s-0028-1091980. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diraison F, Beylot M. Role of human liver lipogenesis and reesterification in triglycerides secretion and in FFA reesterification. Am J Physiol. 1998;274:E321–7. doi: 10.1152/ajpendo.1998.274.2.E321. [DOI] [PubMed] [Google Scholar]
- 35.Sharma NK, Das SK, Mondal AK, Hackney OG, Chu WS, Kern PA, et al. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab. 2008;93:4532–41. doi: 10.1210/jc.2008-1001. doi: 10.1210/jc.2008-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, et al. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. doi: 10.2337/db08-1220. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang MK, Nam JS, Kim JH, Yun YR, Han CW, Kim BJ, et al. Schisandra chinensis extract ameliorates nonalcoholic fatty liver via inhibition of endoplasmic reticulum stress. J Ethnopharmacol. 2016;185:96–104. doi: 10.1016/j.jep.2016.03.021. doi: 10.1016/j.jep.2016.03.021. [DOI] [PubMed] [Google Scholar]
- 38.Mosinski JD, Pagadala MR, Mulya A, Huang H, Dan O, Shimizu H, et al. Gastric bypass surgery is protective from high-fat diet-induced non-alcoholic fatty liver disease and hepatic endoplasmic reticulum stress. Acta Physiol (Oxf) 2016;217:141–51. doi: 10.1111/apha.12640. doi: 10.1111/apha.12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bailly-Maitre B, Belgardt BF, Jordan SD, Coornaert B, von Freyend MJ, Kleinridders A, et al. Hepatic bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J Biol Chem. 2010;285:6198–207. doi: 10.1074/jbc.M109.056648. doi: 10.1074/jbc.M109.056648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sha H, He Y, Chen H, Wang C, Zenno A, Shi H, et al. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009;9:556–64. doi: 10.1016/j.cmet.2009.04.009. doi: 10.1016/j.cmet.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foretz M, Guichard C, Ferré P, Foufelle F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci U S A. 1999;96:12737–42. doi: 10.1073/pnas.96.22.12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119:1201–15. doi: 10.1172/JCI37007. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]